

Abstract
Background
Alternative (M2) macrophage activation is associated with tumor development in many tumor types, including those in the lung. Herein the biological consequences of forcing classical (M1) or alternative (M2) macrophage activation on lung tumor development are examined.
Materials and Methods
Urethane-induced lung tumor multiplicity and size were compared in IFN-γ-/- mice which lack M1 macrophage activation, IL-4Rα-/- mice which lack M2 macrophage activation, and wild-type BALB/cJ (background strain of the IFN-γ-/- and IL-4Rα-/- mice) mice. Tumor-associated macrophage (TAM) and bone marrow derived monocyte (BDMC) activation were each examined.
Results
The TAMs and BDMCs in the IFN-γ-/- mice exhibited M2 activation, and their lung tumors were significantly larger than those in the wild-type mice. In contrast, urethane-treated IL-4Rα-/- mice, whose TAMs and BDMCs were M1 activated, developed smaller tumors than the wild-type mice.
Conclusions
Altered innate immunity can diminish or accelerate lung tumor progression in response to defective cytokine signaling.
Keywords: Macrophages, lung cancer, cytokines, activation state
Inflammatory cells contribute both pro- and anti-tumorigenic actions. Understanding how macrophage phenotypes change during neoplastic progression and the function of these biochemical alterations could engender new molecularly-targeted therapeutic strategies. Cancer and chronic inflammation are linked (1), with tumor-associated macrophages (TAMs) associated with a poor prognosis in human lung cancer (2-4). TAMs help orchestrate tumor development, and their removal from the tumor micro-environment inhibits lung tumorigenesis (5).
Two distinct macrophage activation phenotypes, M1 and M2, are regulated by the relative abundance of TH1 (interferon-γ, IFN-γ) and TH2 (interleukins 4 and 13, IL-4 and IL-13) cytokines, and macrophages can shuttle back and forth between these activation pathways if cytokine administration is sequentially varied in vitro (6;7). Macrophages classically (M1) activated by exposure to IFN-γ or lipopolysacchride (LPS) produce interleukins 6 and 12 (IL-6 and IL-12), and up-regulate inducible nitric oxide synthase (iNOS), nitric oxide (NO) and citrulline. Alternative (M2) activation induced by exposure to IL-4 and IL-13, which bind to a common receptor subunit, IL-4Rα (8), produces anti-inflammatory IL-10 and up-regulates expression of the mannose receptor and arginase I, as well as other macrophage-specific genes. This leads to polyamine and proline production and depletion of the iNOS substrate, arginine (9-13).
The TH2 cytokines that regulate TAM activation are associated with many neoplasias, including non-small cell lung (NSCLC) (14), breast (15), and ovarian cancer (16;17). We have shown that macrophages responding to TH2 cytokine signals become alternatively (M2) activated during pre-malignancy after the mouse lung carcinogen, urethane, has been administered to sensitive Strain A/J mice (18). In addition, these lung tumors produced systemic signals that induced bone marrow derived macrophages (BDMCs) to assume an activation state identical to the alveolar macrophages, allowing circulating macrophages to infiltrate the lungs as M2 macrophages. Herein the tumorigenic consequences of manipulating M1 and M2 activation by subjecting IFN-γ-/- and IL-4Rα-/- mice to chemical carcinogenesis was examined to determine the consequences of these altered macrophage populations in the tumor microenvironment.
Materials and Methods
Mice
IFN-γ-/- and IL-4Rα-/- breeder pairs on BALB/c strain background, purchased from the Jackson Laboratory (Bar Harbor, ME, USA), were bred and housed on hardwood bedding with 12 h light/dark cycles, and fed standard rodent diet (Harlan Teklad, Maddison, WI, USA) at the Center for Laboratory Animal Care at the University of Colorado Denver according to a protocol approved by the University of Colorado Institution of Animal Care and Use Committee. Male and female wild-type control BALB/c mice (4 to 6 weeks of age) also purchased from Jackson Laboratory were similarly housed.
Urethane-induced lung tumor model
IFN-γ-/-, IL-4Rα-/-, and BALB/c wild-type male and female mice received 1 mg urethane (ethyl carbamate; Sigma, St. Louis, MO, USA)/g body wt (dissolved in sterile 0.9% NaCl) by intraperitoneal (i.p.) injection once a week for 6 weeks (19). Chronic urethane exposure induces the formation of several tumors in BALB/c mice, a strain with intermediate susceptibility to lung carcinogenesis (19). The animals were sacrificed 16, 24, or 32 weeks after the first urethane exposure.
Tumor dissection and quantification
Mice (5/gender/time-point) were sacrificed by lethal i.p. pentobarbital injection at the above time-points. The lungs were removed and tumors separated from adjacent uninvolved lung tissue under a dissecting light microscope. Individual tumors were counted and their diameters measured using digital calipers.
Pulmonary macrophage harvest by bronchoalveolar lavage (BAL)
Mice (5/time-point) were sacrificed by lethal i.p. pentobarbital injection, their tracheas cannulated, and lungs lavaged three times with 1 ml PBS containing 0.6 mM EDTA. Inflammatory cell infiltration was determined by pooling the lavaged samples from each animal and counting the cells using a hemocytometer. Twenty thousand cells were cytospun onto glass slides (4 slides/sample) for differential cell counts based on cell morphology (determined by Wright/Giemsa staining) and immunofluorescent staining. Infiltrating cells were classified as monocytes/macrophages, lymphocytes, neutrophils or eosinophils (18).
Preparation of BDMCs
One femur was removed from each mouse, and bone marrow cells harvested by flushing 1ml of sterile PBS through the bone marrow cavities with a 25 5/8 gauge syringe (18). Cell numbers were determined and differential cell counts performed as above.
Immunofluorescence analysis of macrophages and BDMCs
BAL macrophage and BDMC slides (3 slides/sample type) were fixed in -20°C methanol for 3 min. Immunofluorescence was performed as previously described (18) using iNOS as a marker of M1 activation, arginase I expression to detect M2 macrophage activation, and the macrophage markers F4/80 or CD-68 to detect alveolar macrophages and BDMCs, respectively. A digital deconvolution microscopy imaging system (Carl Zeiss Microimaging, Inc., Thornwood, NY, USA) attached to a Zeiss Axioplan 2 epi-fluorescence upright microscope was used to image fluorescent staining (18) at a 630× final magnification. Macrophages and BDMCs displaying no detectable arginase I or iNOS staining were designated as having an M0 phenotype. Co-localization of proteins was detected in cells fluorescently tagged with more than one stain.
Immunohistochemistry (IHC)
Lung sections were prepared and immunohistochemistry performed as described previously (18;20), with a minimum of three individual lungs/treatment group examined for histological analysis. Images were obtained using an Olympus BX-41 microscope equipped with a digital camera (Olympus Imaging America, Inc., Center Valley, PA, USA) and analyzed with Spot Advanced software (Diagnostic Instruments, Inc., Sterling Heights, MI, USA) at 400× final magnification. For each mouse, 3 slides with at least four individual tumor fields/slide were stained with the macrophage marker F4/80, and tumor-infiltrating macrophages (TIMs; macrophages within the tumors) counted.
Statistics
Data are presented as means ± SEM. Differences at specific time-points were examined using Student's unpaired t-test. One-way ANOVA with Newman-Keuls post-hoc analysis was used to compare the results from more than two groups with p < 0.05 considered significant.
Results
Influence of manipulating macrophage activation during tumorigenesis influences tumor size and multiplicity
The tumors from the IFN-γ-/- mice were 1.3-fold larger than the tumors in the urethane-treated wild-type BALB/c mice throughout tumor development and 1.6-fold larger than the tumors in the IL-4Rα-/- mice (Figure 1A). The absence of IL-4Rα expression had the opposite effect (Figure 1A). The tumors in the IL-4Rα-/- mice were 23% smaller than those in the wild type mice and 42% smaller than the tumors in the IFN-γ-/- mice 32 weeks after urethane treatment. The urethane-treated wild-type and IL-4Rα-/- mice developed similar numbers of tumors, with multiplicities increasing linearly over time. The IFN-γ-/- mice had the same number of tumors as the wild-type and IL-4Rα-/- mice 16 and 24 weeks post-urethane, but fewer at 32 weeks (Figure 1B).
Figure 1.

Influence of genetic ablation of IFN-γ and IL-4 on urethane-induced tumor development and BAL macrophage infiltration. Tumor diameter (A), tumor multiplicity (B), and BAL macrophage concentration (C) in IL-4Rα-/- (Δ) and IFN-γ-/- (○) mice compared to wild-type controls (■) as a function of time after urethane treatment. *p<0.05, **p<0.001 vs. wild-type; data represents the mean ± SEM.
Lung tumor-induced macrophage infiltration is attenuated in M1 and M2-deficient mice
The number of infiltrating macrophages in the BAL increased as the tumors developed in the BALB/c mice (Figure 1C), as reported previously for A/J mice (18). At 16 weeks after the initial carcinogen treatment, the tumor-bearing IL-4Rα-/- mice had the same number of BAL macrophages as the tumor-bearing wild-type mice, while the tumor-bearing IFN-γ-/- mice had more. By 24 and 32 weeks after the first urethane injection, however, the wild type mice had significantly more BAL macrophages than either knockout strain (Figure 1C).
Effect of ablation of IFN-γ or IL-4Rα on M2 or M1 macrophage activation during mouse lung tumorigenesis
Macrophages from naïve, age-matched, wild-type, IL-4Rα-/-, and IFN-γ-/- mice were M0, displaying neither arginase I (M2) nor iNOS (M1) expression (data not shown). The loss of IFN-γ prevented the BAL macrophages and BDMCs from becoming classically (M1) activated (Figure 2), while the absence of IL-4Rα prevented alternative (M2) activation. The macrophage activation data was consistent throughout tumor development, so therefore only the 32 week time-points are shown in Figure 2. The tumor-bearing, wild-type BALB/c mice contained lung TAMs and BDMCs that stained positively for arginase I (blue) but not for iNOS (red) after urethane administration, indicating M2 activation (Figure 2A). The TAMs and BDMCs expressed arginase I (blue) but extremely low levels of iNOS (M2) in the IFN-γ-/- mice (Figure 2B). The IL-4Rα-/- TAMs and BDMCs expressed negligible arginase I, but iNOS expression was detected (red), indicating an M1 activation state (Figure 2C).
Figure 2.
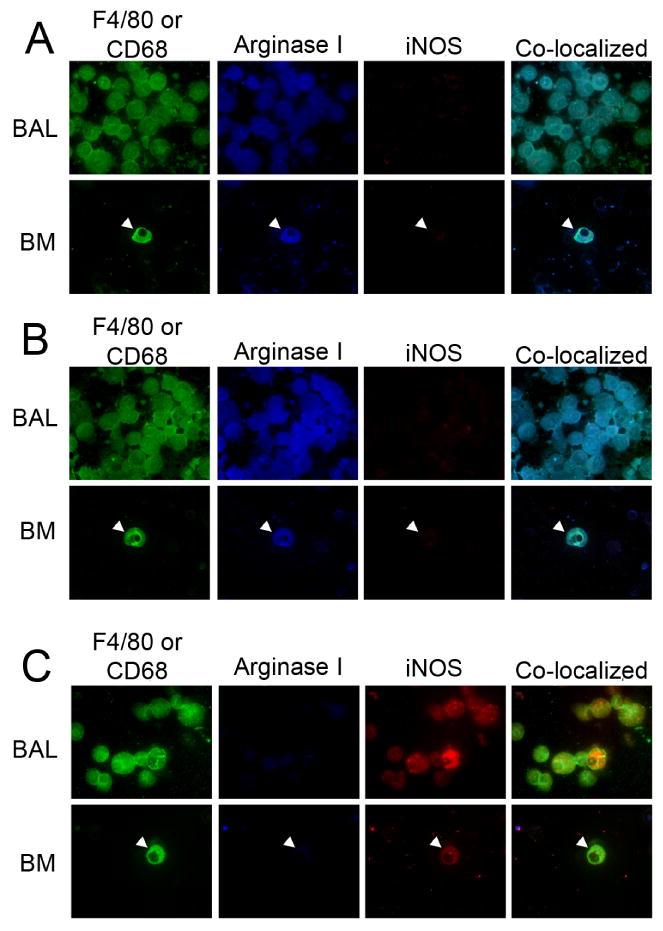
TAM and BDMC activation during urethane-induced tumorigenesis in IFN-γ-/- and IL-4Rα-/- mice. Tumor-associated BAL macrophages (F4/80): green, BDMCs (BM. CD-68): green, arginase I: blue, iNOS: red, 32 weeks after the first urethane injection in (A) wild-type controls, (B) IFN-γ-/- mice and (C) IL-4Rα-/- mice. Co-localization of F4/80 and iNOS or arginase I indicates that the cells staining for iNOS and arginase I are indeed macrophages. Final magnification 630×.
IL-4Rα deficiency and macrophage infiltration into the tumor parenchyma
The TAMs were located adjacent to the lung tumors in the wild-type, IFN-γ-/- and IL-4Rα-/- mice as determined by histological examination (Figure 3A, right column), consistent with our earlier observations in wild type A/J mice (18). However, large numbers of TIMs were present within the lung tumors of the IL-4Rα-/- mice (Figure 3A left column). The wild-type mice had a few TIMs near the tumor periphery, associated with the leading edge of the tumor (Figure 3A, left column). The TIM population in the IL-4Rα-/- mice increased as the tumors progressed (data not shown), congregating in large clusters as well as presenting as individual cells within the tumor parenchyma. TAMs were observed outside the tumor edges in the IFN-γ-/- mice. In addition a single TIM located within a large blood vessel within the tumor parenchyma was detected (Figure 3A, left column). Forcing M1 pulmonary macrophage activation decreased the number of TAMs (Figure 1C), but increased the number of TIMs. The number and location of the TIMs inversely correlated with tumor diameter. The large tumors in the IFN-γ-/- mice had no TIMs, while the mid-size tumors in the wild-type mice had few TIMs, and the small tumors in the IL-4Rα-/- mice had many TIMs.
Figure 3.

The presence of TIMs in IL-4Rα-/- and IFN-γ-/- mice. (A) Macrophages (F4/80) stained dark brown in uninvolved lung tissue adjacent to tumors (TAMs, right column) and within the tumor parenchyma (TIMs, left column) at 32 weeks after urethane induction. Final magnification 400×. (B) TIMs at 32 weeks. **p<0.001 compared to wild-type and IFN-γ-/- mice. Data represents the mean ± SEM. (C) iNOS immunohistochemical staining in TIMs (circled) 32 weeks after urethane treatment in IL-4Rα-/- mice.
TIMs from IL-4Rα-/- mice
Because BAL only provides knowledge about macrophages within the alveolar air spaces that readily detach from the septae and are easily lavaged, TIMs are not likely to be present in lavage fluid. Therefore, immunostaining for iNOS was performed to determine the activation state of the TIMs in the IL-4Rα-/- mice. The TIMs at 16 and 32 weeks stained positively for iNOS, as did the TAMs, confirming the immunofluorescence analysis of the BAL cells (Figure 3C).
Discussion
The urethane-induced pulmonary tumors were larger than the tumors from wild-type mice when the macrophages could not be classically activated because of IFN-γ deficiency. Both the TAMs and BDMCs in this strain remained M2 throughout tumorigenesis, suggesting that M2 macrophage activation enhances tumor growth. Because the BDMC activation mirrored that of the BAL macrophages, deficiencies in these cytokine pathways did not affect the ability of the lung tumors or their TAMs to activate bone marrow macrophages. However, the macrophages and BDMCs in the tumor-bearing, wild-type mice were also M2, but differences in tumor size and tumor numbers were apparent. Loss of IFN-γ affects not only macrophage activation but may also have negatively impacted T cell function, providing an environment advantageous for tumor growth. Rag-2 deficient mice which cannot initiate V(D)J rearrangement and thus lack functional lymphocytes are more susceptible to 3-methylcholanthrene (MCA) induced sarcoma development (21). Also, mice made insensitive to IFN-γ by ablation of IFN-γR1 or signal transducer and activator of transcription 1 (STAT1) develop more MCA-induced flank tumors, implying that IFN-γ maintains proper lymphocyte functioning (22). In the present study, the macrophages from the wild-type mice at 32 weeks did not express iNOS, the functional consequence of IFN-γ mediated effects in macrophages. We hypothesize that M1-activated macrophages retard the clonal expansion of initiated cells, while M2 macrophages stimulate lung tumor growth rate.
The loss of IL-4/IL-13 signaling upon deleting IL-4Rα influenced tumor development, yielding smaller tumors and stimulating macrophage infiltration into the tumor parenchyma. The TAMs, TIMs and BDMCs in these mice were M1 activated, and M1-activated TIMs may significantly inhibit tumor growth. Loss of IL-4Rα affects the course of development in other diseases as well. Macrophage-specific ablation of IL-4Rα protein in mice (23) showed increased M1 activation through TH1 cytokine up-regulation of iNOS and NO and decreased IL-10 expression, producing a cytotoxic environment that decreased survival during Schistosoma mansoni infection (24). Reduced expression of IL-4Rα in macrophages also decreased cutaneous Leishmania major disease progression and increased M1 cytokine signals, as occurred in the present study in the tumor-bearing IL-4Rα-/- mice. When IL-4Rα was ablated in CD4+ T cells, mice were more resistant to infection with L. major compared to wild-type mice who developed ulcerating lesions (25).
Related conclusions have been made about the role of IL-4/IL-13 in other cancer models. When M2 macrophages were specifically targeted for T cell recognition by vaccination against legumain, an asparaginyl endopeptidase over-expressed specifically in TAMs, the decreased number of TAMs correlated with reduced breast tumor growth (15). Binding IL-4/IL-13 to IL-4Rα activates the STAT6 pathway. STAT6 deficient mice contained more M1 macrophages and exhibited 60% less metastasis of 4T1 mammary carcinoma cells compared to wild-type mice (26). The loss of M2 activation in the IL-4Rα-/- mice affirmed studies in other systems in which IL-4 and IL-13 induce alternative activation through binding to a common IL-4Rα receptor subunit. The macrophages and BDMCs in the IL-4Rα-/- mice were classically activated which is consistent with decreased lung tumor mass (Figure 1B) if M1 activation impedes tumorigenesis.
By comparing wild-type and null mice, the roles for macrophage activation can be differentiated. Interestingly, even with genetically mandated losses of INF-γ or IL-4/IL-13 signaling, the mice still recruited macrophages to the tumor microenvironment, albeit not as many. Altered abilities to become classically or alternatively activated did not dictate whether macrophages can be recruited to the lungs. The IL-4Rα-/- and wild-type mice had similar tumor multiplicities and contained TIMs, indicating a role for IFN-γ and TH1 cytokines in regulating the anatomic sites to which macrophages migrate. Whether the TIMs reduced tumor growth, possibly by digesting tumor cells or reversing growth inhibitory signals, is not yet known. No quantitative differences between the tumors in the wild-type and IL-4Rα-/- mice in their proliferation (by Ki67 immunostaining), apoptosis (by TUNEL analysis), or angiogenesis (by CD31 immunostaining; data not shown) were found. With a lack of counterbalancing signaling through IL-4Rα, the IFN-γ-stimulated macrophages displayed enhanced phagocytosis of tumor cells (27), consistent with a hypothesis that centrally located M1-activated TIMs digest tumor cells to cause tumor shrinkage. The IFN-γ-/- and wild-type mice had similar TAM and BDMC activation states, and IL-4Rα-/- and IFN-γ-/- caused similar perturbations in the ability to recruit macrophages. As with other reciprocal signaling relationships, the balance between IFN-γ and IL-4/IL-13 activation pathways is critical to maintaining a disease-free environment. Since IFN-γ and IL-4Rα are expressed and functional in other cell types, genetic ablation of IFN-γ and IL-4Rα does not yield as interpretable a phenotype as deletion mutants targeted to macrophages. Nevertheless, these results imply that inhibiting IL-4/IL-13 signaling or enhancing that of IFN-γ could have therapeutic potential.
Acknowledgments
This work was supported by USPHS grants CA33497 and CA132552.
References
- 1.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson SK, Kerr KM, Chapman AD, Kennedy MM, King G, Cockburn JS, Jeffrey RR. Immune cell infiltrates and prognosis in primary carcinoma of the lung. Lung Cancer. 2000;27:27–35. doi: 10.1016/s0169-5002(99)00095-1. [DOI] [PubMed] [Google Scholar]
- 3.Koukourakis MI, Giatromanolaki A, Kakolyris S, O'Byrne KJ, Apostolikas N, Skarlatos J, Gatter KC, Harris AL. Different patterns of stromal and cancer cell thymidine phosphorylase reactivity in non-small-cell lung cancer: impact on tumour neoangiogenesis and survival. Br J Cancer. 1998;77:1696–1703. doi: 10.1038/bjc.1998.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takanami I, Takeuchi K, Kodaira S. Tumor-associated macrophage infiltration in pulmonary adenocarcinoma: association with angiogenesis and poor prognosis. Oncology. 1999;57:138–142. doi: 10.1159/000012021. [DOI] [PubMed] [Google Scholar]
- 5.Bauer AK, Dwyer-Nield LD, Keil K, Koski K, Malkinson AM. Butylated hydroxytoluene (BHT) induction of pulmonary inflammation: a role in tumor promotion. Exp Lung Res. 2001;27:197–216. doi: 10.1080/019021401300053948. [DOI] [PubMed] [Google Scholar]
- 6.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–6173. [Google Scholar]
- 7.Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005;175:342–349. doi: 10.4049/jimmunol.175.1.342. [DOI] [PubMed] [Google Scholar]
- 8.Kotowicz K, Callard RE, Friedrich K, Matthews DJ, Klein N. Biological activity of IL-4 and IL-13 on human endothelial cells: functional evidence that both cytokines act through the same receptor. Int Immunol. 1996;8:1915–1925. doi: 10.1093/intimm/8.12.1915. [DOI] [PubMed] [Google Scholar]
- 9.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 10.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 11.Montaner LJ, da Silva RP, Sun J, Sutterwala S, Hollinshead M, Vaux D, Gordon S. Type 1 and type 2 cytokine regulation of macrophage endocytosis: differential activation by IL-4/IL-13 as opposed to IFN-gamma or IL-10. J Immunol. 1999;162:4606–4613. [PubMed] [Google Scholar]
- 12.Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol. 1999;163:3771–3777. [PubMed] [Google Scholar]
- 13.Nair MG, Cochrane DW, Allen JE. Macrophages in chronic type 2 inflammation have a novel phenotype characterized by the abundant expression of Ym1 and Fizz1 that can be partly replicated in vitro. Immunol Lett. 2003;85:173–180. doi: 10.1016/s0165-2478(02)00225-0. [DOI] [PubMed] [Google Scholar]
- 14.Huang M, Wang J, Lee P, Sharma S, Mao JT, Meissner H, Uyemura K, Modlin R, Wollman J, Dubinett SM. Human non-small cell lung cancer cells express a type 2 cytokine pattern. Cancer Res. 1995;55:3847–3853. [PubMed] [Google Scholar]
- 15.Luo Y, Zhou H, Krueger J, Kaplan C, Lee SH, Dolman C, Markowitz D, Wu W, Liu C, Reisfeld RA, Xiang R. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest. 2006;116:2132–2141. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Pluddemann A, Charles K, Gordon S, Balkwill FR. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J Immunol. 2006;176:5023–5032. doi: 10.4049/jimmunol.176.8.5023. [DOI] [PubMed] [Google Scholar]
- 17.Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, Van RN, Husseinzadeh N, Farland-Mancini MM, Drew AF. Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res. 2007;67:5708–5716. doi: 10.1158/0008-5472.CAN-06-4375. [DOI] [PubMed] [Google Scholar]
- 18.Redente EF, Orlicky DJ, Bouchard RJ, Malkinson AM. Tumor signaling to the bone marrow changes the phenotype of monocytes and pulmonary macrophages during urethane-induced primary lung tumorigenesis in A/J mice. Am J Pathol. 2007;170:693–708. doi: 10.2353/ajpath.2007.060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malkinson AM, Beer DS. Major effect on susceptibility to urethan-induced pulmonary adenoma by a single gene in BALB/cBy mice. J Natl Cancer Inst. 1983;70:931–936. [PubMed] [Google Scholar]
- 20.O'Donnell EP, Zerbe LK, Dwyer-Nield LD, Kisley LR, Malkinson AM. Quantitative analysis of early chemically-induced pulmonary lesions in mice of varying susceptibilities to lung tumorigenesis. Cancer Lett. 2006;241:197–202. doi: 10.1016/j.canlet.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 24.Herbert DR, Holscher C, Mohrs M, Arendse B, Schwegmann A, Radwanska M, Leeto M, Kirsch R, Hall P, Mossmann H, Claussen B, Forster I, Brombacher F. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20:623–635. doi: 10.1016/s1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- 25.Radwanska M, Cutler AJ, Hoving JC, Magez S, Holscher C, Bohms A, Arendse B, Kirsch R, Hunig T, Alexander J, Kaye P, Brombacher F. Deletion of IL-4Ralpha on CD4 T cells renders BALB/c mice resistant to Leishmania major infection. PLoS Pathog. 2007;3:e68. doi: 10.1371/journal.ppat.0030068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–645. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- 27.Dinapoli MR, Calderon CL, Lopez DM. The altered tumoricidal capacity of macrophages isolated from tumor-bearing mice is related to reduce expression of the inducible nitric oxide synthase gene. J Exp Med. 1996;183:1323–1329. doi: 10.1084/jem.183.4.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]