

Abstract
Hermansky-Pudlak syndrome (HPS; MIM 203300) is a genetically heterogeneous disorder characterized by oculocutaneous albinism, prolonged bleeding and pulmonary fibrosis due to abnormal vesicle trafficking to lysosomes and related organelles, such as melanosomes and platelet dense granules1–3. In mice, at least 16 loci are associated with HPS4–6, including sandy (sdy; ref. 7). Here we show that the sdy mutant mouse expresses no dysbindin protein owing to a deletion in the gene Dtnbp1 (encoding dysbindin) and that mutation of the human ortholog DTNBP1 causes a novel form of HPS called HPS-7. Dysbindin is a ubiquitously expressed protein that binds to α- and β-dystrobrevins, components of the dystrophin-associated protein complex (DPC) in both muscle and nonmuscle cells8. We also show that dysbindin is a component of the biogenesis of lysosome-related organelles complex 1 (BLOC-1; refs. 9–11), which regulates trafficking to lysosome-related organelles and includes the proteins pallidin, muted and cappuccino, which are associated with HPS in mice. These findings show that BLOC-1 is important in producing the HPS phenotype in humans, indicate that dysbindin has a role in the biogenesis of lysosome-related organelles and identify unexpected interactions between components of DPC and BLOC-1.
We previously showed7 that the sdy mutant mouse is a valid model for human HPS and localized the gene sdy to mouse chromosome 13. Here we genotyped 20 microsatellite markers in 1,250 progeny of sdy backcrosses to localize sdy to the 2.2-cM interval (42.73–43.29 Mb) between D13Mit244 and D13Mit267 (Fig. 1). We identified the sdy interval within a 28-Mb scaffold (Celera Discovery System) containing two known genes, Jmj and Dtnbp1 (Fig. 1b). We used PCR products of D13Mit179 and the Dtnbp1 cDNA as probes to generate a BAC contig covering the sdy interval (Fig. 1b).
Figure 1.

High-resolution genetic and physical maps of the sdy gene region of mouse chromosome 13. (a) High-resolution genetic map of the sdy gene region. Numbered D13Mit markers are listed above (without the prefix). The centromere is represented by a solid circle. (b) High-resolution physical map based on the BAC contig and Celera database. Two known genes in the sdy critical region are in bold with arrows indicating directions of transcription and relative sizes. A BAC contig (with RPCI-23 library designations) spans the sdy genetic interval (∼570 kb). Arrows, proportional in length to the BAC sizes and oriented with the SP6 end at the arrowhead and the T7 end opposite, represent BACs.
Northern-blot analysis and sequencing of RT–PCR products of Jmj identified no abnormalities in sdy mutants, but truncated genomic PCR products (Fig. 2a) and mRNA (Fig. 2b) of Dtnbp1 were apparent in sdy tissues. Sequencing of RT–PCR products showed that exons 6 and 7 (156 bp) of Dtnbp1 were deleted in mutant mice, resulting in the loss of 52 amino acids from position 119–172 of the dysbindin protein (Supplementary Fig. 1 online). Genomic sequencing showed that this results from a large deletion (38,129 bp) from nucleotide 3,701 of intron 5 to nucleotide 12,377 of intron 7. This deletion was not found in twelve other inbred mouse strains (Fig. 2a), including coisogenic DBA/2J, indicating that it was not a strain-specific polymorphism. This in-frame deletion creates a 1.5-kb mutant dysbindin transcript (Fig. 2b) and abolishes expression of the 51-kDa dysbindin8 protein in sdy/sdy mice (Fig. 2c). Expression of dysbindin is restored in sdy/sdy transgenic mice containing BAC54F9 (Fig. 2c). Platelet serotonin levels of six of these transgenics were normal (>1.12 μg per 109 platelets), whereas all five sdy/sdy litter-mates without BAC54F9 had platelet serotonin levels of <0.06 μg per 109 platelets. sdy/sdy progeny containing the BAC transgene had darker coats than sdy/sdy progeny lacking the transgene. These results confirmed that mutation of dysbindin causes the sdy phenotype and that dysbindin is important for normal platelet dense granule and melanosome biogenesis.
Figure 2.

Dtnbp1 mRNA is smaller and dysbindin is undetectable in tissues of sdy mutant mice. (a) Genomic PCR. We amplified genomic DNA from control DBA/2J, sdy/+, sdy/sdy and various inbred strains by a duplex PCR method targeting exon 7 of Dtnbp1 and designed to produce PCR products of 472 bp from wild-type DNA and 274 bp from sdy/sdy DNA. sdy arose in the DBA/2J strain. (b) Northern-blot analysis. We hybridized total RNA (20 μg) from kidney, brain and heart of wild-type (DBA/2J), heterozygous (sdy/+) and homozygous (sdy/sdy) mice with Dtnbp1 (upper panels) and Gapd (lower panels) cDNA probes. Dtnbp1 mRNA in sdy tissues was 1.5 kb in size compared with 1.65 kb in control DBA/2J; mRNA of heterozygous sdy/+ mice contains both ∼1.5-kb and 1.65-kb Dtnbp1 mRNAs. (c) Western-blot analysis. We resolved kidney extracts of control DBA/2J, sdy/+ and sdy/sdy mutant mice together with transgenic progeny containing (+) or lacking (−) BAC54F9, which contains the entire Dtnbp1 genomic region, in denaturing gels, blotted gels and incubated them with polyclonal antibody to dysbindin 3111A (top). The blots were reprobed with antibody to Rab4 as a loading control (bottom). Immunoblot analyses of brain, heart, liver and skeletal muscle (data not shown) also showed lack of expression of dysbindin in sdy/sdy mutants.
We screened for mutations in the 10 exons of the human ortholog DTNBP1 in 22 unrelated non-Puerto Rican individuals with HPS who did not have mutations in HPS1, HPS3, HPS4, HPS5 or HPS6. We found six non-pathological polymorphisms (162A→G, IVS5+21G→C, IVS6-8—9delTT, IVS8+2C→T, 814C→T, 874A→G) in these individuals and the nonsense mutation 307C→T (resulting in the amino acid substitution Q103X; Supplementary Fig. 2 online) in the homozygous state in a 48-year-old Portuguese woman with oculocutaneous albinism, ease of bruising and a bleeding tendency. Bleeding time was 13 min, and platelet aggregation indicated a storage-pool deficiency. This woman has mild shortness of breath on exertion and reduced lung compliance but otherwise normal pulmonary function and high-resolution computed-tomography (CT) chest scans with no muscle weakness or ataxia. Her parents were first cousins.
Destabilization of the pallidin protein in the muted mutant (mu) and of the muted protein in the pallid mutant (pa) has been shown, consistent with their association in the BLOC-1 complex10,11. We confirmed these results (Fig. 3) and observed similar destabilization of dysbindin in kidney extracts of both pa and mu mutants and destabilization of both muted and pallidin proteins in sdy kidney extracts. These observations suggested that dysbindin, like pallidin, muted and cappuccino, is a component of the BLOC-1 complex. We also observed the reported10,11 smaller reductions in the amounts of the muted and pallidin proteins in the reduced pigment (rp) mutant, although we saw no change in levels of the dysbindin protein in rp kidney extracts. We observed no changes or minimal changes in the amounts of α1- or β-dystrobrevin in tissues of any HPS mutant (data not shown) or in the amounts of pallidin, muted or dysbindin in 11 other mouse HPS mutants (Fig. 3).
Figure 3.

Destabilization of the dysbindin, pallidin and muted proteins in extracts of three HPS mutants (sdy, pa and mu). We separated kidney extracts from 16 mouse HPS or HPS-related mutants and 4 control strains (DBA/2J, mu/+, C57BL/6J and C3H/HeJ) on denaturing gels and probed them with antibodies to dysbindin, muted and pallidin as indicated at left. Dystrobrevins were similarly analyzed in the brain as expression of α-dystrobrevin is weak in kidney. Inbred strain DBA/2J served as the control for sdy/sdy, mu/+ for mu/mu, C3H/HeJ for sut/sut, ash/ash and ru6J/ru6J and C57BL/6J for the remaining mutants. Antibodies to Rab4 and α-tubulin served as loading controls.
To further identify proteins that directly interact with dysbindin, we used yeast two-hybrid analyses. We found interaction between dysbindin and pallidin and confirmed the interaction8 between dysbindin and β-dystrobrevin (Fig. 4a). We also found that β-dystrobrevin interacted with muted but did not directly interact with either pallidin or pearl (also called Ap3b1) and that the muted and pallidin BLOC-1 proteins interacted (Fig. 4a). We confirmed the association of dysbindin, pallidin, muted and β-dystrobrevin by coimmunoprecipitation (Fig. 4b). We found dysbindin and pallidin in common fractions in both size-exclusion chromatography (Fig. 5a) and sedimentation velocity (Fig. 5b) analyses, consistent with coresidence in BLOC-1. In contrast, the bulk of α1- and β-dystrobrevins and another DPC component, Dp71 (a dystrophin isoform found in liver12), were separable from the BLOC-1 components dysbindin and pallidin. Together, these experiments indicate that dysbindin is a subunit of BLOC-1 and that the dystrobrevins are not subunits of BLOC-1 although they interact with BLOC-1 components. The dystrobrevins and dysbindin may have been separable in cytosolic liver extracts (Fig. 5) because the intact DPC complex was not extracted from liver membranes.
Figure 4.
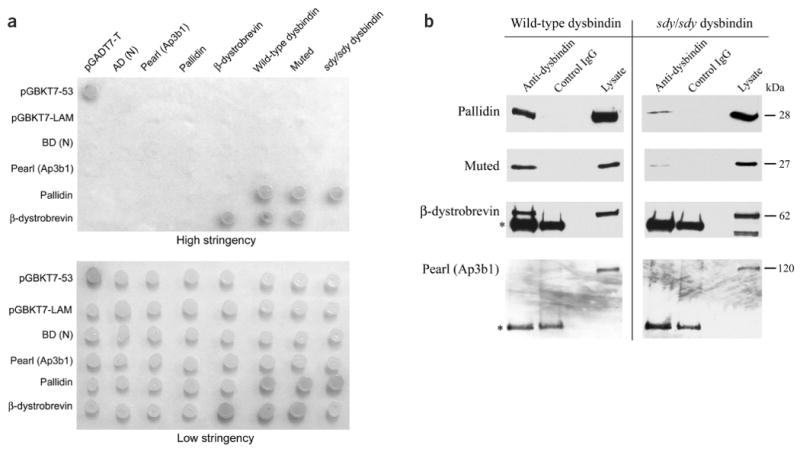
Association of dysbindin with muted, pallidin and β-dystrobrevin. (a) Yeast two-hybrid interactions. We transformed yeast strain AH109 with constructs expressing the entire coding regions of the indicated proteins fused to binding domains (left) or activation domains (top) and spotted cotransformants on plates containing high-stringency and low-stringency media. All combinations grew vigorously on low-stringency plates, indicating that no construct was lethal to the cells. Cotransformants pGBKT7-53 and pGADT7-T are positive controls for interacting proteins; pGBKT7-LAM and pGADT7-T are negative controls. AD (N) and BD (N) represent ‘empty’ activation and binding domain constructs, respectively. sdy/sdy dysbindin is the sdy mutant form of dysbindin with the 52-amino-acid deletion. We were unable to test for interactions among wild-type dysbindin, sdy/sdy dysbindin and muted because they autoactivated in the binding domain. sdy/sdy dysbindin did not interact with dystrobrevin but did interact with pallidin. (b) Coimmunoprecipitation shows direct association of dysbindin with components of BLOC-1. We cotransfected COS-7 cells with either dysbindin or sdy/sdy dysbindin (the sdy mutant form of dysbindin with the 52-amino-acid deletion) and Myc-pallidin, Myc-muted, β-dystrobrevin or Myc-pearl. Proteins were immunoprecipitated with antibody to dysbindin or an unrelated polyclonal antibody (control IgG) and detected with an antibody to Myc. Muted and pallidin were detected when immunoprecipitated with dysbindin but not with an unrelated IgG, indicating that both muted and pallidin form a complex with dysbindin. The cell lysate is shown for comparison. In control experiments, β-dystrobrevin coimmunoprecipitated with dysbindin as described previously8 but pearl did not. Pallidin and muted immunoprecipitated with sdy/sdy dysbindin, but to a lesser extent. β-dystrobrevin, wild-type dysbindin and pearl did not coimmunoprecipitate with sdy/sdy dysbindin. The asterisk indicates the IgG heavy chain that is detected by the secondary antibody.
Figure 5.
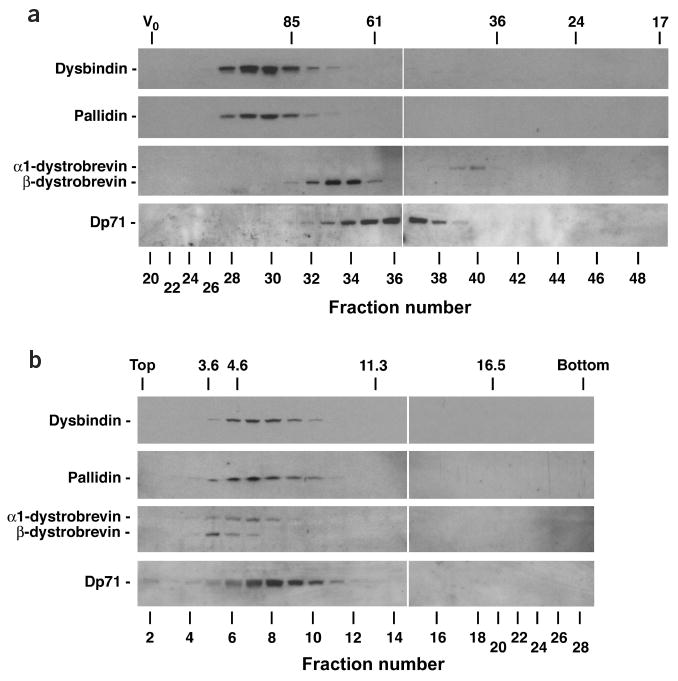
Dysbindin and pallidin are components of a common protein complex in mouse liver. (a) Size exclusion chromatography. We fractionated mouse liver cytosol on a calibrated Superose 6 column and analyzed the resulting fractions by immunoblotting with antibodies to dysbindin, pallidin, α1-dystrobrevin, β-dystrobrevin and the dystrophin isoform Dp71 (ref. 12). The exclusion volume (V0) and the elution positions of standard proteins (Stokes radii given in Angstroms) are indicated at the top. (b) Sedimentation velocity analysis. We fractionated mouse liver cytosol by ultracentrifugation on a 5–20% (w/v) sucrose gradient and analyzed the resulting fractions by immunoblotting as in a. Fractions 1 and 28 correspond to the top and bottom ends of the gradient, respectively. The positions of standard proteins (sedimentation coefficients given in Svedberg units) are indicated at the top. Taken together, the size-exclusion chromatography and sedimentation velocity analyses indicate a native molecular mass of ∼230 kDa (221–247 kDa) and a frictional ratio of ∼2.4 for mouse BLOC-1. Both numbers are in close agreement with those calculated for bovine BLOC-1 (ref. 11), indicating that mouse BLOC-1 is similarly a large asymmetric protein complex.
To characterize the impact of the sdy mutation on melanosome biogenesis, we carried out electron microscopic analysis of eye sections (Fig. 6). Relative to control DBA/2J mice, sdy mice had much fewer melanosomes in the retinal pigment epithelium and choroid, and choroidal melanosomes were generally smaller and irregular, further illustrating the importance of dysbindin in melanosome biogenesis.
Figure 6.
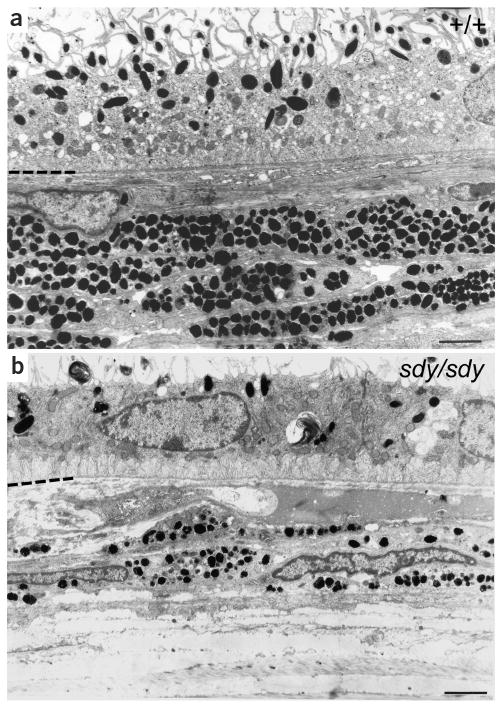
Retinal melanosomes are deficient and abnormal in sdy mice. Retinal sections of DBA/2J (a) and sdy/sdy (b) mice. In each case, the hyphenated partial line at the left indicates the interface between the retinal pigment epithelium (above) and the choroid (below). Scale bars = 2 μm.
The human disorders HPS-1 (ref. 13), HPS-2 (ref. 14), HPS-3 (ref. 15), HPS-4 (ref. 16), HPS-5 (ref. 17) and HPS-6 (ref. 17) result from mutations in genes that encode both known vesicle trafficking proteins and novel proteins. In the mouse, seven additional genes4,5,9,18–21 associated with HPS have been identified. We found that mutations in DTNBP1/Dtnbp1 cause HPS-7/sdy, providing the first evidence to our knowledge that a mutation affecting a BLOC-1 complex component causes HPS in humans. The phenotypes of the four BLOC-1 mutants (pallid, muted, cappuccino and sandy) are the most severe of the mouse HPS mutants6,22, emphasizing the importance of BLOC-1 in the biogenesis of lysosome-related organelles, and their similar phenotypes6,22 are consistent with their encoded proteins forming a common complex. One interpretation of the fact that dystrobrevin interacts with components (dysbindin and muted) of the BLOC-1 complex is that DPC components are involved in vesicle trafficking in nonmuscle tissues. Alternatively, dysbindin may be independently part of both BLOC-1 and the DPC complex, with its vesicle trafficking function in BLOC-1 distinct from its DPC function. In muscle, the DPC complex connects the actin cytoskeleton with the extracellular matrix23. Interaction of the pallidin subunit of the BLOC-1 complex with actin11 suggests the BLOC-1 complex and the actin cytoskeleton similarly interact to control biogenesis of lysosome-related organelles.
Methods
Mice and backcrosses
We obtained mutant sdy and coisogenic DBA/2J mice from The Jackson Laboratory and subsequently bred them at Roswell Park Cancer Institute. We established a backcross between homozygous sdy mice and PWK wild-type mice (subspecies of Mus musculus musculus) as described24. sdy mutant mice have no obvious muscle abnormalities. Despite the reported genetic association of haplotypes involving the gene encoding dysbindin with schizophrenia25,26, preliminary tests of sdy mice, which included the prepulse inhibition test, have identified no behavioral abnormalities, in agreement with the fact that there are no apparent behavioral abnormalities in the individual with HPS-7. All procedures (mouse protocol 125M) were reviewed and approved by the Roswell Park Institutional Animal Care and Use Committee and adhered to the principles of the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Genetic mapping, physical mapping and BAC sequencing
We generated high-resolution genetic and BAC-based physical maps of the sdy critical region as described27. Altogether, we typed 1,250 backcross progeny at 6 weeks of age for the sdy phenotype and for crossovers in the region surrounding sdy using the flanking microsatellite markers D13Mit138 (proximal) and D13Mit39 (distal). Because coat color alone was unreliable, we classified the sdy phenotype by determination of flashing, on ultraviolet illumination, of platelet dense granules loaded with the fluorescent dye mepacrine28. We further genotyped informative mice for markers D13Mit164, D13Mit165, D13Mit244, D13Mit179, D13Mit64, D13Mit245, D13Mit277, D13Mit267, D13Mit180, D13Mit181, D13Mit155, D13Mit308, D13Mit91 and D13Mit92 to derive a genetic map spanning the sdy interval.
We selected BACs by screening the RPCI-23 BAC library (Roswell Park Cancer Institute) with probes in the sdy interval, including microsatellite markers and mouse Dtnbp1 cDNA and based BAC alignment on the Celera database and direct sequencing of BAC ends. We sequenced BAC 257B17 completely at the University of Oklahoma Genome Center using standard optimized BAC shotgun sequencing, closure and finishing methods29. We annotated BAC sequences based on GenBank nr and dbest homologies using the BLAST programs after masking repetitive sequences.
Mutation screening of Dtnbp1 in sdy mice and DTNBP1 in individuals with HPS
We isolated total RNA from mouse tissues with RNAzol B reagent (Tel-Test) and carried out reverse transcription of RNA with the Superscript Preamplification System for RT–PCR (Gibco BRL). Primers 13282F and 13282R (sequences available on request) amplified a region spanning exons 5–10 of mouse Dtnbp1 cDNA. We designed 22 sets of primers (sequences available on request) to amplify overlapping regions of introns 4–8 in genomic DNA and used primer IN5F4 (sequence available on request) as a sequencing primer for the breakpoint between intron 5 and intron 7.
We used primers (sequences available on request) spanning the 10 exons of DTNBP1 for mutational analysis and subjected PCR products to single-strand conformation polymorphism/heteroduplex analysis30 and then sequencing. The homozygous Q103X mutation in DTNBP1 in the woman with HPS-7 was not observed in genomic DNA of 61 normal white controls. These experiments were approved by the Colorado Multiple Institutional Review Board. Informed consent was obtained from all individuals.
We tested genomic DNA samples of various inbred mouse strains (The Jackson Laboratory) for the deletion of exon 7 in Dtnbp1 found in sdy/sdy mice with a duplex PCR procedure (primer sequences available on request) designed to produce PCR products of 472 bp (from exon 7, deleted in sdy) with normal DNA and 274 bp (spanning introns 5–7) with sdy DNA.
Western-blot analysis
We rapidly homogenized and froze fresh mouse tissues in Protease Inhibitor Cocktail Set I (Calbiochem) and stored extracts at −20 °C. We boiled extracts in Laemlli sample buffer with 5% mercaptoethanol and separated 40 μg of extract protein on 12% SDS–PAGE gels or 4–20% gradient gels (Gradipore) and blotted gels to a PVDF membrane. We probed the blots with rabbit polyclonal antisera to dysbindin8 (1:250 dilution), muted11, pallidin11 and dystrobrevin8. We purchased rabbit polyclonal antibody to Rab4 (D-20) from Santa Cruz Biotechnology and mouse monoclonal antibody to α-tubulin (Clone B512) from Sigma. Film was developed after treatment with the ECL Plus kit (Amersham; ref. 20).
Electron microscopy
We fixed eye tissues for 18 h at 4 °C in 3% glutaraldehyde and 0.1 M phosphate buffer, pH 7.2, and embedded and stained them with uranyl acetate and lead citrate20.
Coimmunoprecipitation of dysbindin with other proteins associated with HPS
We transfected COS-7 cells with different combinations of expression constructs encoding dysbindin8, dysbindin lacking amino acids 120–171 (sdy/sdy dysbindin) in pCMVTag-4A, β-dystrobrevin8, pallidin in pCMV-Myc (Clontech), muted in pCMV-Myc and pearl (Ap3b1) in pCMVTag-3B (Stratagene) using Fugene-6 (Roche). Twenty-four hours after transfection we scraped the cells into 4 ml of RIPA buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.4, 1% v/v Triton X-100, 0.5% w/v sodium deoxycholate, 1 mM EGTA), clarified the supernatant by centrifugation and precipitated proteins with 5 μg of antibody for 6 h at 4 °C. We captured immune complexes by overnight incubation with goat anti-rabbit MagnaBind beads (Pierce), washed them extensively in RIPA buffer and resuspended them in 200 μl of SDS/urea buffer (4 M urea, 3.8% w/v SDS, 20% v/v glycerol, 75 mM Tris pH 6.8, 5% v/v 2-mercaptoethanol). We resolved proteins on 4–15% SDS–PAGE gradient gels (BioRad) and detected immunoprecipitated proteins with antibody to Myc 9E10 (1:200, pallidin and muted; Covance), antibody to β-dystrobrevin12 β521 (1:200) or a goat polyclonal antibody to Myc (1:100, pearl; Santa Cruz). We included controls in which the antibody to dysbindin was replaced by an unrelated rabbit polyconal antibody in parallel on the same lysates.
Yeast two-hybrid interactions
We used the Matchmaker Gal4 Two-Hybrid System 3 kit (CLONTECH) for two-hybrid analyses. We fused cDNA sequences containing the entire protein coding regions of mouse wild-type and sdy/sdy dysbindin (from kidney of sdy mice), β-dystrobrevin, pallidin and muted in-frame to the DNA binding (pGBKT7) and activation (pGADT7) domains of the Gal4 transcription factor and verified resulting fusion constructs by sequencing. We cotransformed fusion constructs into Saccharomyces cerevisiae strain AH109 grown at high and low stringency as described17, incubated plates at 30 °C for 5 d and monitored growth and development of blue color.
Size-exclusion chromatography and sedimentation velocity analyses
We fractionated mouse liver cytosol by size exclusion chromatography on Superose 6 columns and linear 5–20% sucrose gradients as described11 and analyzed these fractions by western blotting.
BAC rescue of sdy/sdy in transgenic mice
We injected BAC RP23-54F9 into pronuclei derived from C3H/HeJ × C57BL/6J F1 females and transferred the transgene to the sdy/sdy background by mating with DBA/2J sdy/sdy mice to produce F1 progeny. We mated those F1 progeny that contained BAC RP23-54F9 to sdy/sdy mice to produce F2 progeny, which were typed by PCR techniques (primer sequences available on request) for the presence of the BAC transgene, the Dtnbp1 deletion and polymorphisms in microsatellite markers closely linked to Dtnbp1. We compared platelet serotonin concentrations7 and coat colors in sdy/sdy F2 progeny containing and lacking the BAC transgene.
Supplementary Material
Acknowledgments
We thank D. Reddington, L. Zhen, Y. Jiang, D. Poslinski, D. Tabaczynski, M.K. Ellsworth, J. Tan, H. Chen and X. Hu for technical assistance. M. Pagan contributed to the construction of the sdy genetic map. This work was supported in part by grants from the US National Institutes of Health (R.T.S., R.W.E., R.A.S., E.C.D. and B.A.R.) and by the National Cancer Institute, US Department of Health and Human Services (N.G.C. and N.A.J.). D.J.B. is a Wellcome Trust Senior Fellow. This research used core facilities supported in part by Cancer Center Support Grant to Roswell Park Cancer Institute funded by the National Cancer Institute.
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
Competing Interests Statement: The authors declare that they have no competing financial interests.
References
- 1.Dell'Angelica EC, Mullins C, Caplan S, Bonifacino JS. Lysosome-related organelles. FASEB J. 2000;14:1265–1278. doi: 10.1096/fj.14.10.1265. [DOI] [PubMed] [Google Scholar]
- 2.Spritz RA. Multi-organellar disorders of pigmentation: intracellular traffic jams in mammals, flies and yeast. Trends Genet. 1999;15:337–340. doi: 10.1016/s0168-9525(99)01785-0. [DOI] [PubMed] [Google Scholar]
- 3.Huizing M, Anikster Y, Gahl WA. Hermansky-Pudlak syndrome and Chediak-Higashi syndrome: disorders of vesicle formation and trafficking. Thromb Haemost. 2001;86:233–245. [PubMed] [Google Scholar]
- 4.Novak EK, et al. The regulation of platelet-dense granules by Rab27a in the ashen mouse, a model of Hermansky-Pudlak and Griscelli syndromes, is granule-specific and dependent on genetic background. Blood. 2002;100:128–135. doi: 10.1182/blood.v100.1.128. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki T, et al. The mouse organellar biogenesis mutant buff (bf) results from a mutation in Vps33a, a homologue of yeast vps33 and Drosophila carnation (car) Proc Natl Acad Sci USA. 2003;100:1146–1150. doi: 10.1073/pnas.0237292100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swank RT, Novak EK, McGarry MP, Rusiniak ME, Feng L. Mouse models of Hermansky-Pudlak syndrome: a review. Pigment Cell Res. 1998;11:60–80. doi: 10.1111/j.1600-0749.1998.tb00713.x. [DOI] [PubMed] [Google Scholar]
- 7.Swank RT, Sweet HO, Davisson MT, Reddington M, Novak EK. Sandy: a new mouse model for platelet storage pool deficiency. Genet Res. 1991;58:51–62. doi: 10.1017/s0016672300029608. [DOI] [PubMed] [Google Scholar]
- 8.Benson MA, Newey SE, Martin-Rendon E, Hawkes R, Blake DJ. Dysbindin, a novel coiled-coil-containing protein that interacts with the dystrobrevins in muscle and brain. J Biol Chem. 2001;276:24232–24241. doi: 10.1074/jbc.M010418200. [DOI] [PubMed] [Google Scholar]
- 9.Ciciotte SL, et al. Cappuccino, a mouse model of Hermansky-Pudlak syndrome, encodes a novel protein that is part of the pallidin-muted complex (BLOC-1) Blood. 2003;101:4402–4407. doi: 10.1182/blood-2003-01-0020. [DOI] [PubMed] [Google Scholar]
- 10.Moriyama K, Bonifacino JS. Pallidin is a component of a multi-protein complex involved in the biogenesis of lysosome-related organelles. Traffic. 2002;3:666–677. doi: 10.1034/j.1600-0854.2002.30908.x. [DOI] [PubMed] [Google Scholar]
- 11.Falcon-Perez JM, Starcevic M, Gautam R, Dell'Angelica EC. BLOC-1, a novel complex containing the pallidin and muted proteins involved in the biogenesis of melanosomes and platelet-dense granules. J Biol Chem. 2002;277:28191–28199. doi: 10.1074/jbc.M204011200. [DOI] [PubMed] [Google Scholar]
- 12.Blake DJ, Hawkes R, Benson MA, Beesley PW. Different dystrophin-like complexes are expressed in neurons and glia. J Cell Biol. 1999;147:645–658. doi: 10.1083/jcb.147.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh J, et al. Positional cloning of a gene for Hermansky-Pudlak syndrome, a disorder of cytoplasmic organelles. Nat Genet. 1996;14:300–306. doi: 10.1038/ng1196-300. [DOI] [PubMed] [Google Scholar]
- 14.Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the β 3A subunit of the AP-3 adaptor. Mol Cell. 1999;3:11–21. doi: 10.1016/s1097-2765(00)80170-7. [DOI] [PubMed] [Google Scholar]
- 15.Anikster Y, et al. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet. 2001;28:376–380. doi: 10.1038/ng576. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki T, et al. Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat Genet. 2002;30:321–324. doi: 10.1038/ng835. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Q, et al. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky-Pudlak syndrome types 5 and 6. Nat Genet. 2003;33:145–153. doi: 10.1038/ng1087. [DOI] [PubMed] [Google Scholar]
- 18.Kantheti P, et al. Mutation in AP-3 delta in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron. 1998;21:111–122. doi: 10.1016/s0896-6273(00)80519-x. [DOI] [PubMed] [Google Scholar]
- 19.Detter JC, et al. Rab geranylgeranyl transferase alpha mutation in the gunmetal mouse reduces Rab prenylation and platelet synthesis. Proc Natl Acad Sci USA. 2000;97:4144–4149. doi: 10.1073/pnas.080517697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Q, et al. The gene for the muted (mu) mouse, a model for Hermansky-Pudlak syndrome, defines a novel protein which regulates vesicle trafficking. Hum Mol Genet. 2002;11:697–706. doi: 10.1093/hmg/11.6.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang L, Kuo YM, Gitschier J. The pallid gene encodes a novel, syntaxin 13-interacting protein involved in platelet storage pool deficiency. Nat Genet. 1999;23:329–332. doi: 10.1038/15507. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen T, et al. Melanosome morphologies in murine models of Hermansky-Pudlak syndrome reflect blocks in organelle development. J Invest Dermatol. 2002;119:1156–1164. doi: 10.1046/j.1523-1747.2002.19535.x. [DOI] [PubMed] [Google Scholar]
- 23.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 24.Gardner JM, et al. The mouse pale ear (ep) mutation is the homologue of human Hermansky-Pudlak syndrome. Proc Natl Acad Sci USA. 1997;94:9238–9243. doi: 10.1073/pnas.94.17.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwab SG, et al. Support for association of schizophrenia with genetic variation in the 6p22.3 gene, dysbindin, in sib-pair families with linkage and in an additional sample of triad families. Am J Hum Genet. 2003;72:185–190. doi: 10.1086/345463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Straub RE, et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71:337–348. doi: 10.1086/341750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki T, et al. The gene mutated in cocoa mice, carrying a defect of organelle biogenesis, is a homologue of the human Hermansky-Pudlak syndrome-3 gene. Genomics. 2001;78:30–37. doi: 10.1006/geno.2001.6644. [DOI] [PubMed] [Google Scholar]
- 28.Reddington M, et al. Immature dense granules in platelets from mice with platelet storage pool disease. Blood. 1987;69:1300–1306. [PubMed] [Google Scholar]
- 29.Bodenteich A, Chissoe S, Wang YF, Roe BA. Automated DNA Sequencing and Analysis Techniques. Academic; London: 1994. Shotgun cloning as the strategy of choice to generate templates for high throughput dideoxynucleotide sequencing; pp. 42–50. [Google Scholar]
- 30.Lee ST, Park SK, Lee KH, Holmes SM, Spritz RA. A non-radioactive method or simultaneous detection of single-stand conformation polymorphisms (SSCPs) and heteroduplexes. Mol Cells. 1995;5:668–672. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.