

Abstract
Background
In 1994, an International Task Force proposed criteria for the clinical diagnosis of ARVC/D which facilitated recognition and interpretation of the frequently non-specific clinical features of ARVC/D. This enabled confirmatory clinical diagnosis in index cases through exclusion of phenocopies, and provided a standard upon which clinical research and genetic studies could be based. Structural, histological, electrocardiographic, arrhythmic, and familial features of the disease were incorporated into the criteria, subdivided into major and minor according to the specificity of their association with ARVC/D. At that time, clinical experience with ARVC/D was dominated by symptomatic index cases and sudden cardiac death victims: the overt and/or severe end of the disease spectrum. Consequently, the 1994 criteria were highly specific but lacked sensitivity for early and familial disease.
Methods and Results
Revision of the diagnostic criteria provides guidance on the role of emerging diagnostic modalities and advances in the genetics of ARVC/D. The criteria have been modified to incorporate new knowledge and technology to improve diagnostic sensitivity, but with the important requisite of maintaining diagnostic specificity. The approach classifying structural, histological, electrocardiographic, arrhythmic, and genetic features of the disease as major and minor criteria has been maintained. In this modification of the Task Force Criteria, quantitative criteria are proposed and abnormalities are defined based on comparison with normal subject data.
Conclusions
The diagnosis of ARVC/D based on modification of the original Task Force criteria is a working framework to improve the diagnosis and management of this condition.
Keywords: cardiomyopathy, diagnosis, echocardiography, electrocardiography, magnetic resonance imaging
Introduction
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is predominantly a genetically determined heart muscle disorder that is characterized pathologically by fibrofatty replacement of the right ventricular myocardium.1 In the early stage of the disease, structural changes may be absent or subtle and confined to a localized region of the right ventricle (RV), typically the inflow tract, outflow tract, or apex of the RV, the “triangle of dysplasia”.2 Progression to more diffuse RV disease and left ventricular (LV) involvement is common.3 Predominant LV disease is also recognized, typically of the posterior lateral wall.4 Post mortem diagnosis may require extensive sampling and transillumination.5 Disease expression is variable. In the early ‘concealed’ phase, individuals are often asymptomatic, but may nonetheless be at risk of sudden cardiac death, notably during exertion.6 In the overt ‘electrical’ phase, individuals present with symptomatic arrhythmias and RV morphological abnormalities are readily discernible by conventional imaging. Later, diffuse disease may result in biventricular heart failure while ventricular arrhythmias may or may not be present. The ultimate phenotype may resemble dilated cardiomyopathy. Clinical manifestations vary with age and stage of disease.7
ARVC/D is considered to be familial with autosomal dominant inheritance, although there are recessive forms (e.g. Naxos Disease, Carvajal Syndrome), which are associated with a cutaneous phenotype.8,9 Genetic variations have been found in the desmosomes that are responsible for cell to cell binding.10,11 (Figure 1) Seven genes have been identified that are associated with ARVC/D: plakoglobin (JUP),12 desmoplakin (DSP),13 plakophilin-2 (PKP2),14 desmoglein-2 (DSG2),15,16 desmocollin-2 (DSC2),17,18 transforming growth factor beta-3 (TGFβ3),19 and TMEM4320 Mutations in RYR2 coding the ryanodine receptor have been reported as a subtype of ARVC/D with a notable paucity of structural RV disease but with fibrofatty replacement of RV myocytes on histopathological examination. At present, catecholaminergic polymorphic ventricular tachycardia is generally considered as a distinctly separate disorder from ARVC/D, although interpretation of the clinical criteria used to make this diagnosis can be ambiguous.11 Preliminary observations suggest that the mechanical defect of the desmosomes alters function of the gap junction. ECG changes and arrhythmias may develop prior to histological evidence of myocyte loss or clinical evidence of RV dysfunction.21,22 It has been proposed that similar clinical phenotypes occur based on disruption of a “final common pathway” by mutations in genes encoding proteins in the defined desmosomal pathway.23 Recognition of the genetic basis of ARVC/D facilitates examination of the pathogenesis in relation to arrhythmogenesis and disease progression.24
Figure 1.
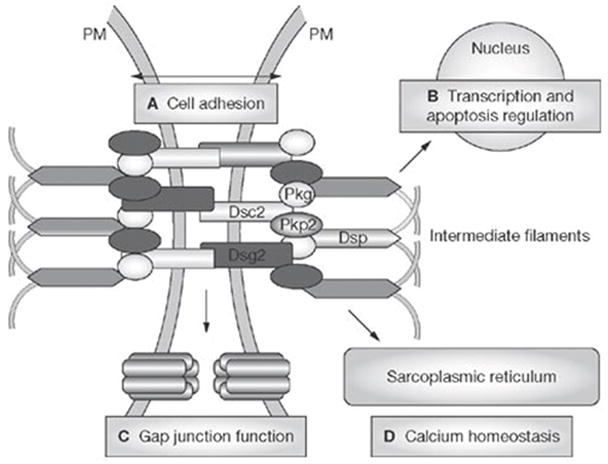
The cardiac desmosome and proposed roles of the desmosome in (A) supporting structural stability through cell-cell adhesion, (B) regulating transcription of genes involved in adipogenisis and apoptosis, and maintaining proper electrical conductivity through regulation of (C) gap junctions and (D) calcium homeostasis. Abbreviations: Dsc2, desmocollin-2; Dsg2, desmoglein-2; Dsp, desmoplakin; Pkg, plakoglobin; Pkp2, plakophilin-2; PM, plasma membrane. Used with permission Awad MM, et al11
It has been suggested that patients with ARVC/D may be predisposed or susceptible to viral myocarditis which could lead to a decrease in cardiac function and accelerate progression of the disease.25–27 The link between ARVC/D and myocarditis is still undefined.
Background
The original 1994 International Task Force criteria for the clinical diagnosis of ARVC/D were based on structural, histological, electrocardiographic, arrhythmic, and familial features of the disease.28 (Table 1) Abnormalities were subdivided into major and minor according to the specificity of their association with ARVC/D. Electrocardiographic abnormalities such as complete and incomplete right bundle branch block (RBBB) were excluded because of their lack of specificity. Right precordial T wave inversion, though well recognized in ARVC/D, was considered a minor criterion owing to its presence in other conditions, including anterior ischemia and right ventricular hypertrophy. Arrhythmia of right ventricular origin, another cardinal feature of ARVC/D, was designated a minor criterion because of its occurrence in other diseases, particularly idiopathic right ventricular outflow tract tachycardia. Furthermore, the 1994 criteria focused on right ventricular disease manifestations, and stipulated the absence or only mild left ventricular involvement because of the need to exclude common disorders such as ischemic heart disease and dilated cardiomyopathy.
Table 1.
| ORIGINAL TASK FORCE CRITERIA | REVISED TASK FORCE CRITERIA | |
|---|---|---|
| I. Global and/or Regional Dysfunction and Structural Alterations | I. Global and/or Regional Dysfunction and Structural Alterations* | |
|
|
|
| Regional RV akinesia, dyskinesia or aneurysm. | ||
| Severe dilatation and reduction of right ventricular ejection fraction with no (or only mild) LV impairment. | And one of the following (end diastole): | |
| Parasternal long axis view RVOT (PLAX) | ≥ 32 mm | |
| Corrected for body size (PLAX/BSA) | ≥ 19 mm/m2 | |
| Localized right ventricular aneurysms (akinetic or dyskinetic areas with diastolic bulging). | Parasternal short axis view RVOT (PSAX) | ≥ 36 mm |
| Corrected for body size(PSAX/BSA) | ≥ 21 mm/m2 | |
| or | ||
| Severe segmental dilatation of the right ventricle. | ||
| Fractional area change (FAC) | ≤ 33% | |
|
||
| Regional RV akinesia or dyskinesia or dyssynchronous RV contraction | ||
| And one of the following: | ||
| Right ventricular end diastolic volume (RVEDV/BSA) | ≥110 ml/m2 male | |
| ≥100 ml/m2 female | ||
| OR | ||
| Right ventricular ejection fraction (RVEF) | ≤40% | |
|
||
| Regional RV akinesia, dyskinesia or aneurysm | ||
|
|
|
| Regional RV akinesia or dyskinesia | ||
| Mild global right ventricular dilatation and/or ejection fraction reduction with normal left ventricle. | And one of the following (end diastole): | |
| Parasternal long axis view RVOT (PLAX) | ≥ 29 - < 32 mm | |
| Mild segmental dilatation of the right ventricle. | Corrected for body size (PLAX/BSA) | ≥ 16 - < 19 mm/m2i |
| Regional right ventricular hypokinesia. | Parasternal short axis view RVOT (PSAX) | ≥ 32 - < 36 mm |
| Corrected for body size (PSAX/BSA) | ≥ 18 - < 21 mm/m2 | |
| or | ||
| Fractional area change (FAC) | > 33% - ≤ 40% | |
|
||
| Regional RV akinesia or dyskinesia or dyssynchronous RV contraction | ||
| And one of the following: | ||
| Right ventricular end diastolic volume/BSA | ≥100 - < 110 ml/m2 male | |
| ≥90 - < 100 ml/m2 female | ||
| OR | ||
| Right ventricular ejection fraction (RVEF) | > 40% - ≤ 45% | |
| II. Tissue Characterization of Wall | II. Tissue Characterization of Wall | |
|
|
|
| Fibrofatty replacement of myocardium on endomyocardial biopsy. | Residual myocytes <60% by morphometric analysis, (or < 50% if estimated), with fibrous replacement of the RV free wall myocardium in at least 1 sample, with or without fatty replacement of tissue on endomyocardial biopsy. | |
|
||
| Residual myocytes 60 – 75% by morphometric analysis, (or 50 to 65% if estimated), with fibrous replacement of the RV free wall myocardium in at least 1 sample, with or without fatty replacement of tissue on endomyocardial biopsy. | ||
| III. Repolarization Abnormalities | III. Repolarization Abnormalities | |
|
|
|
| Inverted T waves in right precordial leads (V2 and V3) (people aged >12 years, in absence of right bundle branch block). | Inverted T waves in right precordial leads (V1, V2 and V3) or beyond in individuals > 14 years of age (in the absence of complete right bundle branch block QRS ≥ 120 msecs). | |
|
||
| Inverted T waves in leads V1 and V2 in individuals > 14 years of age (in the absence of complete right bundle branch block), or in V4, V5, or V6. | ||
| Inverted T waves in leads V1, V2, V3 and V4 in individuals > 14 years of age in the presence of complete right bundle branch block. | ||
| IV. Depolarization/Conduction Abnormalities | IV. Depolarization/Conduction Abnormalities | |
|
|
|
| Epsilon waves or localized prolongation (>110 ms) of the QRS complex in right precordial leads (V1 – V3). | Epsilon wave (reproducible low amplitude signals between end of QRS complex to onset of the T wave) in the right precordial leads (V1 to V3) | |
|
|
|
| Late potentials (signal-averaged ECG). | Late potentials by signal averaged ECG in at least one of three parameters in the absence of a QRS duration of ≥110 msecs on the standard ECG. | |
| Filtered QRS duration (fQRS) | ≥114 msecs | |
| Duration of terminal QRS < 40 μV (LAS) | ≥38 msecs | |
| RMS voltage of terminal 40 msecs | ≥20 μV | |
| Terminal activation duration of QRS ≥ 55ms measured from the nadir of the S wave to the end of the QRS, including R', in V1, V2 or V3, in the absence of complete right bundle branch block. | ||
| V. Arrhythmias | V. Arrhythmias | |
|
|
|
| Left bundle branch block type ventricular tachycardia (sustained and nonsustained) (ECG, Holter, exercise testing). | Non-sustained or sustained VT of left bundle branch morphology with superior axis (negative or indeterminate QRS in II, III, AVF and positive in AVL) | |
|
||
| Non sustained or sustained VT of right ventricular outflow configuration, LBBB morphology with inferior axis (positive QRS in II, III, AVF and negative in AVL) or of unknown axis. | ||
| Frequent ventricular extrasystoles (>1000/24 hours) (Holter). | Greater than 500 ventricular extrasystoles/24 hours by Holter | |
| VI. Family History | VI. Family History | |
|
|
|
| Familial disease confirmed at necropsy or surgery. | ARVC/D confirmed in a first-degree relative who meets current task force criteria. | |
| ARVC/D confirmed pathologically at autopsy or surgery in a first degree relative. | ||
| Identification of a pathogenic mutation † categorized as associated or probably associated with ARVC/D in the patient under evaluation. | ||
|
|
|
| Family history of premature sudden death (<35 years) due to suspected right ventricular dysplasia. | History of ARVC/D in a first degree relative in whom it is not possible or practical to determine if the family member meets current task force criteria. | |
| Familial history (clinical diagnosis based on present criteria). | Premature sudden death (<35 years) due to suspected ARVC/D in a first degree relative. | |
| ARVC/D confirmed pathologically or by current Task Force Criteria in second degree relative. | ||
Hypokinesis is not included in this or subsequent definitions of RV regional wall motion abnormalities for the proposed modified criteria.
A pathogenic mutation is a DNA alteration associated with ARVC/D that alters or is expected to alter the encoded protein, is unobserved or rare in a large non ARVC/D control population and either alters or is predicted to alter the structure or function of the protein or has demonstrated linkage to the disease phenotype in a conclusive pedigree.
| Diagnostic terminology for original criteria | Diagnostic terminology for revised criteria |
| This diagnosis is fulfilled by the presence of two major, or one major plus two minor criteria or four minor criteria from different groups |
|
At the time of the publication of the original Task Force guidelines, clinical experience with ARVC/D was dominated by symptomatic index cases and sudden cardiac death victims: the overt and/or severe end of the disease spectrum. Consequently, the 1994 criteria were highly specific but they lacked sensitivity for early and familial disease.29–31
Over the past fifteen years additional ECG markers have been proposed.32–34 In addition there has been recognition of the genetic basis of disease with the potential for mutation analysis, and growing experience with quantification of imaging criteria of ARVC/D as well as the introduction of newer techniques such as contrast-enhanced echocardiography, three dimensional echocardiography, cardiovascular magnetic resonance (CMR) with late enhancement, and electroanatomic voltage mapping.35–40 Left ventricular involvement may occur early in the course of the disease with some frequency. 4,41
Since publication of the Task Force Criteria cardiovascular evaluation of the relatives of ARVC/D index cases and, more recently, genotype-phenotype association studies have highlighted the shortcomings of the criteria in the diagnosis of familial disease.7 Modifications of the original criteria have been proposed to facilitate clinical diagnosis in first-degree relatives who often have incomplete penetrance of the disease.42 According to these recommendations, in the context of proven ARVC/D in a first degree relative, the diagnosis of familial ARVC/D is based on the documentation of one of the following in a family member:
T wave inversion in right precordial leads, V1, V2 and V3 in individuals over the age of 14 years.
Late potentials by SAECG.
Ventricular tachycardia of LBBB morphology on ECG, Holter monitor, or during exercise testing or >200 PVCs in 24 hours.
Either mild global dilatation and/or reduction in RV ejection fraction with normal LV or mild segmental dilatation of the RV or regional RV hypokinesis.
Revision of the diagnostic criteria is important to provide guidance on the role of emerging diagnostic modalities and to recognize advances in the genetics of ARVC/D. The criteria have been modified to incorporate new knowledge and technology to improve diagnostic sensitivity, but with the important requisite of maintaining diagnostic specificity, and include quantitative parameters for Task Force Criteria particularly for the imaging studies. (Table 1) The approach of classifying structural, histological, electrocardiographic, arrhythmic, and genetic features of the disease as major and minor criteria has been maintained.
Methods
A limitation of the previous Task Force Criteria was the reliance on subjective criteria for assessing ventricular structure and function and for evaluation of myocardial histology. In this modification of the Task Force Criteria, quantitative criteria are proposed and abnormalities are defined based on comparison with normal subject data. (Table 1) The data from 108 probands with newly diagnosed ARVC/D, age 12 years or older, who were enrolled in the National Institutes of Health supported Multidisciplinary Study of Right Ventricular Dysplasia,43 were compared with that of normal subjects. (See appendix) The criteria were selected on the basis of analysis of sensitivity and specificity from ROC curves. For analysis of each test (i.e. echocardiogram, MRI etc.) proband data was excluded if that test was crucial for the diagnosis of the individual patient. This was done to eliminate bias in estimating the sensitivity and specificity of that particular test. In general, when determining the sensitivity and specificity of a new screening test it is recommended that none of the screening test elements are used in making the primary diagnosis; this principle also holds when establishing diagnostic criteria.
Results
There were 44 proband MRIs compared with 462 normals; 69 proband echocardiograms compared to 450 normals, 69 proband SAECGS compared with 103 normals, and 68 proband Holters compared with 398 normals The minor criteria for echocardiography were selected where specificity and sensitivity are equal (sensitivity equals specificity). (Table 2) The major criteria were selected as the value that yielded 95% specificity. Sensitivity and specificity for the MRI criteria were made independently for each sex and considering both RVEDV/BSA (size) and RVEF (function) simultaneously using the OR logistical function. If either RV size or function are positive in conjunction with RV wall motion abnormality, then the subject would be classified as having a major criteria for the MRI. The sensitivity of RV size alone or function alone ranged from 41 to 50% for major criteria and 31 to 41% for minor criteria with specificity of 96 to 100%. Using the OR logistical function improved the sensitivity of the MRI to 79 – 89% for major criteria and 68 – 78% for minor criteria.
Table 2.
Sensitivity and specificity of proposed right ventricular imaging criteria*
| Echocardiogram | Sensitivity | Specificity | ||
|---|---|---|---|---|
| MAJOR | ||||
| Parasternal long axis view RVOT (Diastole) (PLAX) | ≥ 32 mm | 75% | 95% | |
| Corrected for body size (PLAX/BSA) | ≥ 19 mm/m2 | |||
| Parasternal short axis view RVOT (Diastole) ((PSAX) | ≥ 36 mm | 62% | 95% | |
| Corrected for body size (PSAX/BSA) | ≥ 21 mm/m2 | |||
| Fractional area change (FAC) | ≤ 33% | 55% | 95% | |
| MINOR | ||||
| Parasternal long axis view RVOT (Diastole) (PLAX) | ≥ 29 mm | 87% | 87% | |
| Corrected for body size (PLAX/BSA) | ≥ 16 - ≤ 18 mm/m2i | |||
| Parasternal short axis view RVOT (Diastole) ((PSAX) | ≥ 32 mm | 80% | 80% | |
| Corrected for body size (PSAX/BSA) | ≥ 18 - ≤ 20 mm/m2 | |||
| Fractional area change (FAC) | ≤40% | 76% | 76% | |
| Magnetic resonance imaging | Either size or function or both** | |||
| MAJOR | ||||
| Right ventricular end diastolic volume (RVEDV/BSA) | ≥110 ml/m2 males |  |
76% | 90% ♂ |
| OR | ≥100 ml/m2 female | 68% | 98% ♀ | |
| Right ventricular ejection fraction (RVEF) | ≤40% | |||
| MINOR | ||||
| Right ventricular end diastolic volume (RVEDV/BSA) | ≥100 ml/m2 males |  |
79% | 85% ♂ |
| OR | ≥90 ml/m2 female | 89% | 97% ♀ | |
| Right ventricular ejection fraction (RVEF) | ≤45% | |||
All the major and minor criteria listed in this table are in addition to the requirement that regional wall motion abnormalities must also be present
The sensitivity and specificity for males and females are the same as listed if, in addition to the stated wall motion criteria, there is either abnormal RV size or function or both.
PLAX Parasternal long axis view, PSAX Parasternal short axis view, BSA Body surface area, FAC Fractional area change, RVEF Right ventricular ejection fraction, RVEDV Right ventricular end diastolic volume
The original task force criteria list late potentials as a minor criteria. It has become common practice to state that the SAECG is positive if two of the three parameters, filtered QRS duration (f QRS), root-mean square voltage of the terminal 40 msecs (RMS 40) of the QRS, or duration of the terminal QRS signal less than 40μV (LAS) are abnormal, although this is not based on evidence. Analysis of each of the single parameters of the SAECG with late potentials using a 40 – 250Hz filter had a sensitivity ranging from 58 to 60% with a specificity of 94 to 96%. Two of three parameters had a sensitivity of 66% and specificity of 95% adding little advantage with regard to sensitivity and specificity. Using any one of the 3 SAECG parameters has a sensitivity of 74% and specificity of 92%.
A definitive diagnosis of ARVC/D is based on histological demonstration of transmural fibrous and/or fibrofatty replacement of RV myocardium at either biopsy, (Figure 2) necropsy or surgery.5,44 In most patients, however, assessment of transmural myocardium is not possible. In addition, diagnosis based on right ventricular endomyocardial biopsy specimens is limited because the segmental nature of the disease causes false negatives. Use of electroanatomic voltage mapping to identify pathological areas for biopsy sampling may improve the yield.45 RV free wall biopsy has a slight risk of perforation while the more accessible interventricular septum rarely exhibits histological changes. Nevertheless, endomyocardial biopsy may identify other conditions (e.g. myocarditis, sarcoidosis, endomyocardial fibrosis), while the recognition of myocyte loss with fibrous and/or fibrofatty replacement can be a valuable diagnostic feature.46
Figure 2.
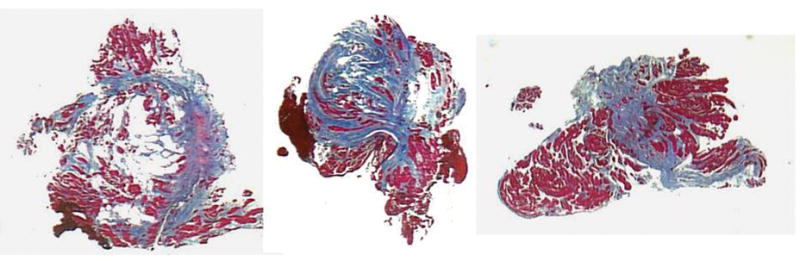
Endomyocardial biopsy findings in a proband affected by a diffuse form of ARVC/D. All three biopsy samples are from different regions of the RV free wall. There is extensive fibro-fatty tissue replacement with myocardial atrophy that is a major criterion, i.e. residual myocytes <60% by morphometric analysis, or < 50% if estimated. (Contributed by C. Basso, Padua, Italy)
The identification of disease causing genes has led to the recognition of a broader spectrum of disease expression within families, including individuals who have predominantly LV disease, manifest clinically by inferolateral T wave changes, ventricular ectopy or VT with RBBB morphology and epicardial and/or mid-myocardial late enhancement by MRI.4,7,38,39,41 The importance of familial disease highlights a role for mutation analysis of probands with cascade screening of relatives that offers an alternative strategy to serial non-invasive cardiovascular evaluation of families. A positive diagnosis in a family member changes the probability of disease in an individual suspected of the disease to 1:2 from 1:1000 – 1:5000. This is reflected in the criteria for confirmed disease in a first degree relative being a major criterion for diagnosis.42
Discussion
The diagnosis of ARVC/D relies on the demonstration of structural, functional, and electrophysiological abnormalities that are caused by or reflect the underlying histological changes. Technical advances in MRI and 2D echo have improved the capability to image the RV with reproducible measurements of volume and systolic function which permits classification of severity and differentiation from normality.47 (Table 2). Previous diagnostic reliance on subjective assessment of RV wall thinning and wall motion abnormalities and fatty infiltration of the myocardium by MRI has proven problematic.48,49 Recognition of significant fatty involvement without concomitant fibrosis of the RV in normal individuals renders this unique MRI capability of limited value. Late enhancement on MRI permits myocardial tissue characterization in the left ventricle. It can be difficult to be certain of late enhancement for characterization of RV myocardium due to the thin wall of the RV and possible confusion with fat.50
There also have been recent developments to quantitate the extent of right ventricular wall motion abnormalities by angiography using computer based analysis as well as to determine right ventricular volumes51,52 In addition, there is commercial software available to determine RV volumes and ejection fraction.53 The right ventricular angiogram obtained in multiple views is considered to be a reliable imaging test to assess wall motion abnormalities but requires considerable experience. Standardized protocols for performance of these diagnostic studies (ECG, SAECG, echocardiogram, right ventricular angiogram, and MRI) are available on www.arvd.org.
Repolarization abnormalities are early and sensitive markers of disease expression in ARVC/D. T wave inversion in V1, V2 and V3 and beyond in individuals over the age of 14 who are otherwise healthy is observed in only 4% of healthy women and 1% of men in this age group. Therefore it is reasonably specific in this population and considered a major diagnostic abnormality in ARVC/D.54 Depolarization delay in right precordial leads is also common in ARVC/D.33,34 Evaluation of the duration of terminal QRS activation (Figure 3) incorporates slurring of the S wave, as well as R prime, into a single measure of terminal activation duration (TAD).34 Depolarization abnormalities cannot be evaluated in the presence of typical complete RBBB with terminal delay in leads I and V6. However, T wave inversion in V1, V2, V3 and V4 is uncommon in patients with RBBB who do not have ARVC/D and are frequently seen in those who do have the disease. Conventional definitions are used for ventricular arrhythmias. An abnormal signal averaged electrocardiogram is based on time domain criteria with cutoffs generated from ROC curves.55,56 The sensitivity and specificity of any one of the time domain criteria is similar to that of any 2 or 3 of these criteria; therefore any one of the criteria are proposed as a criteria for this modality. The presence of LBBB VT with an inferior axis (R wave positive in II and III, and negative in AVL) is typical of focal RVOT tachycardia.57 Similar features may be seen in patients with ARVC/D, but usually coexist with anterior T wave inversion and ventricular arrhythmias of varying morphologies. The presence of ventricular ectopy increases with age but >200 ventricular premature beats in 24 hours in an adult below age 50 suggests underlying myocardial disease.58
Figure 3.
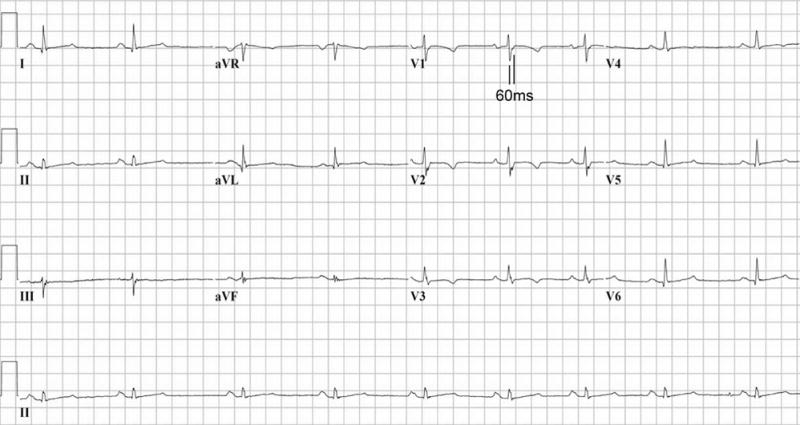
ECG from proband with T wave inversion in V1 – V4 and prolongation of the terminal activation duration ≥ 55 msecs measured from the nadir of the S wave to the end of the QRS complex in V1. (Contributed by MGPJ Cox, Utrecht, The Netherlands)
The revised criteria were applied post hoc to 108 newly diagnosed probands enrolled in the Multidisciplinary Study of Right Ventricular Dysplasia, a study supported by the National Institute of Health. They had been carefully evaluated including assessment of diagnostic tests by expert core laboratories.43 Of the 73 probands with final classification as affected, 71 remain affected and 2 were reclassified as borderline. The change from affected to borderline in the 2 was due to the echocardiogram fulfilling only minor criteria in one and only mild hypokinesis in the angiogram of the other. Of the 28 probands classified as borderline (met some but not all of the original task force criteria, i.e. one major and one minor or three minor), five remain borderline and 16 were reclassified by the new criteria as affected. Seven became unaffected (did not meet the proposed modified task force criteria).
Of seven probands previously classified as unaffected, 4 remained unaffected, 1 became affected and 2 became borderline. Therefore, the effect of the revised criteria is to increase the sensitivity of the classification, primarily in probands previously classified as borderline.
Nine of 28 probands classified as borderline by original criteria, had gene variants consistent with ARVC/D. The sensitivity of the revised criteria is not perfect as exemplified by the observation that if the genetic criteria are ignored, the proposed criteria classified 2 as unaffected and 3 remained borderline, and 4 became affected. Including the proposed genetic criteria resulted in all 9 being classified as affected.
The modified task force criteria have been applied to two different sets of phenotyped-genotyped cohorts, (Protonotarios, N et al, and Cox, MDGJ, et al, unpublished data). In both studies there was an increase in sensitivity without loss of specificity. Additionally, the ECG criteria have been applied to known ARVC/D cohorts and have shown an increase in diagnostic value.59
The diagnosis of ARVC/D based on modification of the original Task Force criteria is a working framework to improve the diagnosis and management of this condition. Awareness is growing that ARVC/D is the well recognized form of a broad disease spectrum that includes left dominant and biventricular subtypes. Lack of specific diagnostic guidelines contributes to under recognition of non classic disease. Future revisions of the Task Force Criteria may fill this gap by incorporating features such as ventricular tachycardia of right bundle branch morphology, subepicardial or midmyocardial late enhancement on magnetic resonance imaging, and global or regional left ventricular dysfunction in patients presenting with arrhythmia rather than heart failure. With the identification of disease causing genes, there is the potential for diagnostic mutation analysis and improved pedigree evaluation with better understanding of the natural history, pathogenesis and development of targeted therapies. Individuals who carry newly discovered disease causing genes, but who have incomplete or no disease expression will be recognized; their natural history and appropriate management remains to be determined.
Limitations
The reference values for the normals have been determined from select populations from centers with expertise in the diagnostic test (see appendix) since data on a large cohort of normals studied by all the modalities was not available from any one center. Therefore the reference values may not apply to all ethnic populations or those younger than age 12.
Supplementary Material
Acknowledgments
Funding Sources
This project was supported by NIH research grant R13 HL086825 funded by the National Heart Lung & Blood Institute and the Office of Rare Disorders, and supported in part by research grants U01-HL65594, U01-HL65652, U01-HL65691 and K23-HL093350 from the National Heart, Lung, and Blood Institute of the National Institutes of Health, Bethesda, MD, and by research grant QLG1 CT 2000 0901 5th Framework Programme from the European Commission, Brussels, Belgium. Additional funding was obtained from the International Society for Holter and Noninvasive Electrocardiography, and donations from the Peter French Memorial Foundation, United Desert Charities, the Podolsky Family Foundation, and private donors Mr. and Mrs. L. Becker, Mr. and Mrs. H. Danz, Mr. K. Dorn, Mr. T. Livolsi, Mr. and Mrs. L. Long, and Mr. and Mrs. H. Wilmerding.
Footnotes
Clinical Trial Registration Information: http://www.clinicaltrials.gov/ct2/show/NCT00024505
Clinical Trial Registration: URL http://www.clinicaltrials.gov Unique Identifier NCT00024505
Disclosures
The authors have no conflicts to disclose.
Contributor Information
Frank I Marcus, University of Arizona.
William J. McKenna, The Heart Hospital.
Duane Sherrill, University of Arizona
Cristina Basso, University of Padua Medical School.
Barbara Bauce, University of Padua Medical School.
David A. Bluemke, National Institutes of Health, Clinical Center.
Hugh Calkins, Johns Hopkins Hospital.
Domenico Corrado, University of Padua Medical School.
Moniek G.P.J. Cox, University Medical Center Utrecht.
James P Daubert, Strong Memorial Hospital.
Guy Fontaine, Hopital Bicetre.
Kathleen Gear, Sarver Heart Center.
Richard Hauer, University Medical Center Utrecht.
Andrea Nava, University of Padua Medical School.
Michael H. Picard, Massachusetts General Hospital.
Nikos Protonotarios, Yannis Protonotarios Medical Centre.
Jeffrey E. Saffitz, Beth Israel Deaconess Medical Center.
Danita M. Yoerger Sanborn, Massachusetts General Hospital.
Jonathan S Steinberg, St. Luke’s-Roosevelt Hospital Center, New York, NY.
Harikrishna Tandri, Johns Hopkins Hospital.
Gaetano Thiene, University of Padua Medical School.
Jeffrey A. Towbin, Cincinnati Children’s Hospital.
Adalena Tsatsopoulou, Yannis Protonotarios Medical Centre.
Thomas Wichter, Marienhospital Osnabrueck.
Wojciech Zareba, University of Rochester Medical Center.
References
- 1.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 2.Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia. A report of 24 adult cases. Circulation. 1982;65:384–98. doi: 10.1161/01.cir.65.2.384. [DOI] [PubMed] [Google Scholar]
- 3.Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lunquist C, Wlodarska EK, Fontaine G, Camerini F. Spectrum of clinicopathologic manifestation of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997;6:1512–1520. doi: 10.1016/s0735-1097(97)00332-x. [DOI] [PubMed] [Google Scholar]
- 4.Norman M, Simpson N, Mogensen J, Shaw A, Hughes S, Syrris P, Sen-Chowdhry S, Rowland E, Crosby A, McKenna WJ. Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation. 2005;112:636–642. doi: 10.1161/CIRCULATIONAHA.104.532234. [DOI] [PubMed] [Google Scholar]
- 5.Basso C, Thiene G. Autopsy and endomyocardial biopsy findings. In: Marcus FI, Nava, Thiene G, editors. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Springer Verlag; Milan, Italy: 2007. pp. 29–44. [Google Scholar]
- 6.Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–133. doi: 10.1056/NEJM198801213180301. [DOI] [PubMed] [Google Scholar]
- 7.Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–1720. doi: 10.1161/CIRCULATIONAHA.106.660241. [DOI] [PubMed] [Google Scholar]
- 8.Protonotarios N, Tsatsopoulou A, Anastasakis A, Sevdalis E, McKoy G, Stratos K, Gatzoulis K, Tentolouris K, Spiliopoulou C, Panagiotakos D, McKenna W, Toutouzas P. Genotype-phenotype assessment in autosomal recessive arrhythmogenic right ventricular cardiomyopathy (Naxos disease) caused by a deletion in plakoglobin. J Am Coll Cardiol. 2001;38:1477–1484. doi: 10.1016/s0735-1097(01)01568-6. [DOI] [PubMed] [Google Scholar]
- 9.Carvajal-Huerta L. Epidermolytic-palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol. 1998;39:418–421. doi: 10.1016/s0190-9622(98)70317-2. [DOI] [PubMed] [Google Scholar]
- 10.Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2005;16:927–935. doi: 10.1111/j.1540-8167.2005.40842.x. [DOI] [PubMed] [Google Scholar]
- 11.Awad MM, Calkins H, Judge DP. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008;5:258–267. doi: 10.1038/ncpcardio1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKoy G, Protonotarius P, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffrey S, McKenna WJ. Identification of a detection in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar kertoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- 13.Norgett EE, Hatsell SJ, Carvahal-Huerta L, Cabezas JR, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–2766. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- 14.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr G, Thierfelder L. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 15.Awad MM, Dalal D, Cho E, Amat-Alarcon N, James C, Tichnell C, Tucker A, Russel SD, Bluemke DA, Dietz HC, Calkins H, Judge DP. DSG2 muations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Hum Genet. 2006;79:136–142. doi: 10.1086/504393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilichou K, Nava A, Basso C, Beffagna GB, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli G, Rampazzo A. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- 17.Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;79:978–984. doi: 10.1086/509122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heuser A, Plovie ER, Ellinor PT, Grossmann KS, Shin JT, Wichter T, Basson CT, Lerman BB, Sasse-Klaassen S, Thierfelder L, MacRae CA, Gerull B. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2006;79:1081–1088. doi: 10.1086/509044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A. Regulatory mutations in transforming growth factor-β3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–373. doi: 10.1016/j.cardiores.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Merner ND, Hodgkinson KA, Haywood AFM, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young TL. Arrhythmogenic right ventricular cardiomyopathy is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:1–13. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saffitz JE. Cell Adhesion Pathology. In: Marcus FI, Nava A, Thiene G, editors. In arrhythmogenic right ventricular dysplasia/cardiomyopathy. Springer Verlag; Milan, Italy: 2007. pp. 45–52. [Google Scholar]
- 22.Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A, Squarcioni CP, McKenna WJ, Thiene G, Basso C, Brousse N, Fontaine G, Saffitz JE. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease) Heart Rhythm. 2004;1:3–11. doi: 10.1016/j.hrthm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 23.Vatta M, Marcus F, Towbin JA. Arrhythmogenic right ventricular cardiomyopathy: A “final common pathway” that defines clinical phenotype. Eur Heart J. 2007;28:529–530. doi: 10.1093/eurheartj/ehl530. [DOI] [PubMed] [Google Scholar]
- 24.Tsatsopoulou A, Protonotarios N, McKenna WJ. Arrhythmogenic right ventricular dysplasia, a cell-adhesion cardiomyopathy: insights into disease pathogenesis from preliminary genotype-phenotype assessment. Heart. 2006;92:1720–3. doi: 10.1136/hrt.2005.081679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calabrese F, Angelini A, Thiene G, Basso C, Nava A, Valente M. No detection of enteroviral genome in the myocardium of patients with arrhythmogenic right ventricular cardiomyopathy. J Clin Pathol. 2000;53:382–387. doi: 10.1136/jcp.53.5.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fontaliran F, Fontaine G, Brestescher C, Labrousee J, Vilde F. Signification del infiltrats lymphoplasmocytaires dans la dyplasia ventriculare droite arhythmogene. Arch Mal Cœur. 1995;88:1021–8. [PubMed] [Google Scholar]
- 27.Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy: dysplasia, dystrophy or myocarditis? Circulation. 1996;94:983–991. doi: 10.1161/01.cir.94.5.983. [DOI] [PubMed] [Google Scholar]
- 28.McKenna WJ, Thiene G, Nava A, Fontaliron F, Blomstrom-Lundquist G, Fontaine G, Camerini F. On behalf of the Task Force of the working group myocardial and pericardial disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology, Diagnosis of arrhythmogenic right ventricular dysplasia cardiomyopathy. Br Heart J. 1994;71:215–218. doi: 10.1136/hrt.71.3.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antoniades L, Tsatsopoulou A, Anastasakis A, Syrris P, Asimaki A, Panagiotakos D, Zambartas C, Stefanadis C, McKenna WJ, Protonotarios N. Arrhythmogenic right ventricular cardiomyopathy caused by deletions in plakophilin-2 and plakoglobin (Naxos Disease) in families from Greece and Cyprus: genotype-phenotype relations, diagnostic features and prognosis. Eur Heart J. 2006;27:2008–2016. doi: 10.1093/eurheartj/ehl184. [DOI] [PubMed] [Google Scholar]
- 30.Ward D, Syrris P, Sen-Chowdhry S, McKenna WJ. Diagnosis; task force criteria including modifications for family members. In: Marcus FI, Nava A, Thiene G, editors. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Springer Verlag; Milan, Italy: 2007. pp. 87–96. [Google Scholar]
- 31.Marcus FI, Sherrill D. Strengths and weaknesses of the task force criteria -proposed modifications in arrhythmogenic right ventricular cardiomyopathy/dysplasia. In: Marcus FI, Nava A, Thiene G, editors. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Springer Verlag; Milan, Italy: 2007. pp. 97–104. [Google Scholar]
- 32.Peters S, Trummel M. Diagnosis of arrhythmogenic right ventricular dysplasia-cardiomyopathy: value of standard ECG revisited. Ann Noninvasive Electrocardiol. 2003;8:238–45. doi: 10.1046/j.1542-474X.2003.08312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nasir K, Bomma C, Tandri H, Roguin A, Dalal D, Prakasa K, Tichnell C, James C, Spevak PJ, Marcus F, Calkins H. Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity; a need to broaden diagnostic criteria. Circulation. 2004;110:1527–1534. doi: 10.1161/01.CIR.0000142293.60725.18. [DOI] [PubMed] [Google Scholar]
- 34.Cox MGPJ, Nelen MR, Wilde AAM, Wiesfeld AC, van der Smagt J, Loh P, Cramer MJ, Doevendans PA, van Tintelen JP, de Bakker JMT, Hauer RNW. Activation delay and VT parameters in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Toward improvement of diagnostic ECG criteria. J Cardiovasc Electrophysiol. 2008;19:775–781. doi: 10.1111/j.1540-8167.2008.01140.x. [DOI] [PubMed] [Google Scholar]
- 35.Yoerger DM, Marcus F, Sherrill D, Calkins H, Towbin JA, Zareba W, Picard M. Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia. J Am Coll Cardiol. 2005;45:860–865. doi: 10.1016/j.jacc.2004.10.070. [DOI] [PubMed] [Google Scholar]
- 36.Prakasa KR, Dalal D, Wang J, Bomma C, Tandri H, Dong J, James C, Tichnell C, Russell SD, Spevak P, Corretti M, Bluemke DA, Calkins H, Abraham TP. Feasibility and variability of three dimensional echocardiography in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol. 2006;97:703–709. doi: 10.1016/j.amjcard.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 37.Tandri H, Friedrich MG, Calkins H, Bluemke DA. MRI of arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Cardiovasc Magn Reson. 2004;6:557–563. doi: 10.1081/jcmr-120030583. [DOI] [PubMed] [Google Scholar]
- 38.Sen-Chowdhry S, Prasad SK, Syrris P, Wage R, Ward D, Merrifield R, Smith GC, Firmin DN, Pennell DJ, McKenna WJ. Cardiovascular magnetic resonance in arrhythmogenic right ventricular cardiomyopathy revisited: comparison with task force criteria and genotype. J Am Coll Cardiol. 2006;48:2132–2140. doi: 10.1016/j.jacc.2006.07.045. [DOI] [PubMed] [Google Scholar]
- 39.Sen-Chowdhry S, Prasad SK, McKenna WJ. Complementary role of echocardiography and cardiac magnetic resonance in the non-invasive evaluation of suspected arrhythmogenic right ventricular cardiomyopathy. J Interv Card Electrophysiol. 2004;11:15–17. doi: 10.1023/B:JICE.0000035923.16175.78. [DOI] [PubMed] [Google Scholar]
- 40.Corrado D, Basso C, Leoni L, Tokajuk B, Bauce B, Frigo G, Tarantini G, Napodano M, Turrini P, Ramondo A, Daliento L, Nava A, Buja G, Iliceto S, Thiene G. Three-dimensional electroanatomic voltage mapping increases accuracy of diagnosing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2005;111:3042–50. doi: 10.1161/CIRCULATIONAHA.104.486977. [DOI] [PubMed] [Google Scholar]
- 41.Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–2187. doi: 10.1016/j.jacc.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 42.Hamid MS, Norman M, Quraishi A, Firoozi S, Thaman R, Gimeno JR, Sachdev B, Rowland E, Elliott PM, McKenna WJ. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol. 2002;40:1445–1450. doi: 10.1016/s0735-1097(02)02307-0. [DOI] [PubMed] [Google Scholar]
- 43.Marcus FI, Zareba W, Calkins H, Towbin JA, Basso C, Bluemke DA, Estes NAM, Picard MH, Sanborn DY, Thiene G, Wichter T, Cannom D, Wilber DJ, Scheinman M, Duff H, Daubert J, Talajic M, Krahn A, Sweeney M, Garan H, Sakaguchi S, Lerman BB, Kerr C, Kron J, Steinberg JS, Sherrill D, Gear K, Brown M, Severski P, Polonsky S, McNitt S. Arrhythmogenic right ventricular cardiomyopathy/dysplasia, clinical presentation and diagnostic evaluation: results from the North American Multidisciplinary Study. Heart Rhythm. 2009;6:984–992. doi: 10.1016/j.hrthm.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basso C, Burke M, Fornes P, Gallagher PJ, de Gouveia RH, Sheppard M, Thiene G, van der Wal A. On behalf of the Association for European Cardiovascular Pathology. Guidelines for autopsy investigation of sudden cardiac death. Virchows Arch. 2008;452:11–18. doi: 10.1007/s00428-007-0505-5. [DOI] [PubMed] [Google Scholar]
- 45.Avella A, D’Amati G, Pappalardo A, Re F, Silenzi PF, Laurenzi F, De Girolamo P, Pelargonia G, Messina G, Xecchi P, Zachara E, Tondo C. Diagnostic value of endomyocardial biopsy guided by electroanatomic voltage mapping in arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Cardiovasc Electrophysiol. 2008;19:1127–1134. doi: 10.1111/j.1540-8167.2008.01228.x. [DOI] [PubMed] [Google Scholar]
- 46.Basso C, Ronco F, Marcus F, Abudureheman A, Rizzo S, Frigo AC, Bauce B, Maddalena F, Nava A, Corrado D, Grigoletto F, Thiene G. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: an in vitro validation of diagnostic criteria. Eur Heart J. 2008;29:2760–71. doi: 10.1093/eurheartj/ehn415. [DOI] [PubMed] [Google Scholar]
- 47.Tandri H, Daya SK, Nasir K, Bomma C, Lima JAC, Calkins H, Bluemke DA. Normal reference values for the adult right ventricle by magnetic resonance imaging. Am J Cardiol. 2006;98:1660–1664. doi: 10.1016/j.amjcard.2006.07.049. [DOI] [PubMed] [Google Scholar]
- 48.Bluemke DA, Krupinski EA, Ovitt T, Gear K, Unger E, Axel L, Boxt LM, Casolo G, Ferrari VA, Funaki B, Globits S, Higgins CB, Julsrud P, Lipton N, Mawson J, Nygren A, Pennell DJ, Stillman A, White RD, Wichter T, Marcus F. MRI Imaging of arrhythmogenic right ventricular cardiomyopathy: morphological findings and intraobserver reliability. Cardiology. 2003;99:153–162. doi: 10.1159/000070672. [DOI] [PubMed] [Google Scholar]
- 49.Bomma C, Rutberg J, Tandri H, Nasir K, Roguin A, Tichnell C, Rodriguez R, Kasper E, Spevak P, Bluemke DA, Calkins H. Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Cardiovasc Electrophysiol. 2004;15:300–306. doi: 10.1046/j.1540-8167.2004.03429.x. [DOI] [PubMed] [Google Scholar]
- 50.Tandri H, Saranathan M, Rodriguez R, Martinez C, Bomma C, Nasir K, Rosen B, Lima JA, Calkins H, Bluemke DA. Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging. J Am Coll Cardiol. 2005;45:98–103. doi: 10.1016/j.jacc.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 51.Indik JH, Dallas WJ, Ovitt T, Wichter T, Gear K, Marcus FI. Do patients with right ventricular outflow tract ventricular arrhythmias have a normal right ventricular wall motion? Cardiology. 2005;104:10–15. doi: 10.1159/000086047. [DOI] [PubMed] [Google Scholar]
- 52.Indik JH, Wichter T, Gear K, Dallas WJ, Marcus FI. Quantitative assessment of angiographic right ventricular wall motion in arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) J Cardiovasc Electrophysiol. 2008;19:39–45. doi: 10.1111/j.1540-8167.2007.00974.x. [DOI] [PubMed] [Google Scholar]
- 53.Wellnhofer E, Ewert P, Hug J, Hui W, Kretschmar O, Chavengsuk D, Kuhne T, Abdul-Khalig H, Nagel E, Lange PE, Fleck E. Evaluation of new software for angiographic determination of right ventricular volumes. Int J Cardiovasc Imaging. 2005;21:575–585. doi: 10.1007/s10554-005-1797-7. [DOI] [PubMed] [Google Scholar]
- 54.Marcus FI. Prevalence of T-Wave Inversion beyond V1 in young normal individuals and usefulness for the diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Am. J. Cardiol. 2005;95:1070–1071. doi: 10.1016/j.amjcard.2004.12.060. [DOI] [PubMed] [Google Scholar]
- 55.Marcus FI, Zareba W, Sherrill D. Evaluation of the normal values for signal-averaged electrocardiogram. J Cardiovasc Electrophysiol. 2007;18:231–233. doi: 10.1111/j.1540-8167.2006.00685.x. [DOI] [PubMed] [Google Scholar]
- 56.Kamath GS, Zareba W, McKenna WJ, Gear K, Sherrill D, Marcus F, Steinberg JS. Value of signal averaged electrocardiogram for the diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm. 2008;5(Supp):S38. doi: 10.1016/j.hrthm.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ainsworth CD, Skanes AC, Klein GJ, Gula LJ, Yee R, Krahn AD. Differentiating arrhythmogenic right ventricular cardiomyopathy from right ventricular outflow tract ventricular tachycardia using multilead QRS duration and axis. Heart Rhythm. 2006;3:416–423. doi: 10.1016/j.hrthm.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 58.DePaula SR, Antelmi I, Vincenzi MA, Andre CD, Artes R, Grupi CJ, Mansur AJ. Cardiac arrhythmias and atrioventricular block in a cohort of asymptomatic individuals without heart disease. Cardiology. 2007;108:111–116. doi: 10.1159/000095950. [DOI] [PubMed] [Google Scholar]
- 59.Cox MGPJ, van der Smagt JJ, Wilde AM, Wiesfeld ACP, Atsma DE, Nelen MR, Rodrieguez LM, Loh P, Cramer MJ, Doevendans PA, van Tintelen JP, de Bakker JMT, Hauer RNW. New ECG Criteria in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circ Arrhythmia Electrophysiol. doi: 10.1161/CIRCEP.108.832519. published online Jul 7, 2009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.