

Abstract
CsA (cyclosporin A) is a hydrophobic undecapeptide that inhibits CyPs (cyclophilins), a family of PPIases (peptidylprolyl cis–trans isomerases). In some experimental models, CsA offers partial protection against lethal cell injury brought about by transient ischaemia; this is believed to reflect inhibition of CyP-D, a mitochondrial isoform that facilitates formation of the permeability transition pore in the mitochondrial inner membrane. To evaluate this further, we have targeted CsA to mitochondria so that it becomes selective for CyP-D in cells. This was achieved by conjugating the inhibitor to the lipophilic triphenylphosphonium cation, enabling its accumulation in mitochondria due to the inner membrane potential. In a cell-free system and in B50 neuroblastoma cells the novel reagent (but not CsA itself) preferentially inhibited CyP-D over extramitochondrial CyP-A. In hippocampal neurons, mitochondrial targeting markedly enhanced the capacity of CsA to prevent cell necrosis brought about by oxygen and glucose deprivation, but largely abolished its capacity to inhibit glutamate-induced cell death. It is concluded that CyP-D has a major pathogenic role in ‘energy failure’, but not in glutamate excitotoxicity, where cytoprotection primarily reflects CsA interaction with extramitochondrial CyPs and calcineurin. Moreover, the therapeutic potential of CsA against ischaemia/reperfusion injuries not involving glutamate may be improved by mitochondrial targeting.
Keywords: cyclophilin (CyP), cyclosporin A (CsA), glutamate toxicity, hippocampal neuron, ischaemia, necrosis
Abbreviations: CsA, cyclosporin A; CyP, cyclophilin; CyP-D+, cell line overexpressing CyP-D; DCM, dichloromethane; DMEM, Dulbecco's minimal essential medium; DMF, dimethylformamide; ESI–MS, electrospray ionization MS; FBS, fetal bovine serum; Fmoc, fluoren-9-ylmethoxycarbonyl; HBSS, Hanks balanced salt solution; I/R, ischaemia/reperfusion; LDA, lithium diisopropylamide; L-NAME, NG-nitro-L-arginine-methyl ester; mtCsA, mitochondrially targeted CsA; NBA, Neurobasal A; NBQX, 2,3-dihydro-6-nitro-7-sulfamoylbenzoquinoxaline; NMDA, N-methyl-D-aspartate; OGD, oxygen and glucose deprivation; PPIase, peptidylprolyl cis–trans isomerase; PT, permeability transition; PyBOP, benzotriazol-1-yl-tris-pyrrolidinophosphonium hexafluorophosphate; SMBz-CsA, [sarcosine-3(4-methylbenzoate)]-CsA; THF, tetrahydrofuran; TMRE, tetramethylrhodamine ethyl ester; TPP+, triphenylphosphonium
INTRODUCTION
Ischaemic diseases, notably myocardial infarction and stroke, are the leading cause of death and disability throughout the world. In general, early restoration of blood flow is essential to restrict tissue damage. Paradoxically, however, as the duration of ischaemia increases, cells become adversely sensitive to restored blood supply, giving rise to additional damage on reperfusion (reperfusion injury). Resolving the critical cellular changes that underlie I/R (ischaemia/reperfusion) injury is a major goal in developing effective therapies.
A mitochondrial pore model for I/R injury was derived from two lines of evidence: first, the commonality between factors that induce formation of the PT (permeability transition) pore and recognized cellular changes in I/R, principally cellular Ca2+ overload, oxidative stress and depleted adenine nucleotides [1,2]; secondly, inhibition of both pore formation and cell necrosis under pseudo-I/R by CsA (cyclosporin A) [3–5]. The non-specific PT pore [6], with an estimated internal diameter of 2 nm [1], allows free flow of low Mr solutes across the inner membrane. Under these conditions, mitochondria carry out ATP hydrolysis, rather than synthesis. Once ATP hydrolysis exceeds the glycolytic capacity of the cell to maintain ATP, irreversible injury leading to cell necrosis ensues. The PT pore model of I/R injury is supported by whole organ and animal studies. In particular, CsA can attenuate reperfusion-induced cell necrosis in heart [7], brain [8] and other tissues. Use of 2-deoxyglucose [9] and calcein [10] reveals that the mitochondrial inner membrane does become non-selectively permeable on reperfusion, and that CsA can prevent the permeability changes. Genetic ablation of CyP (cyclophilin)-D, a PT pore component and a target of CsA (see below), markedly reduces infarct size in heart [11] and brain [12] following transient ischaemia.
CyP-D is a PPIase (peptidylprolyl cis–trans isomerase) that is located in the mitochondrial matrix [13] and is inhibited by CsA. CyP-D overexpression promotes PT pore formation [14], whereas CyP-D ablation attenuates it [15]. It is believed that, under pathological conditions, CyP-D catalyses or stabilizes a proline-dependent conformational switch in an inner membrane protein to form the non-selective PT pore. Conceivably, the CyP-D-sensitive protein is a transport protein that loses its selective transport properties when exposed to high Ca2+ concentrations and oxidative stress. Reconstitution studies indicate that the adenine nucleotide translocase can deform into a non-selective pore under the influence of Ca2+ and CyP-D [16,17], but mitochondria from mice lacking the two translocase isoforms do display PT pore activity, albeit with much reduced Ca2+ sensitivity [18], and additional (or possibly alternative) inner membrane proteins that can interact with CyP-D, e.g. the phosphate carrier [19], have been proposed. Irrespective of the identity of the pore protein itself, however, there is little doubt that CsA inhibits PT pore formation by inhibiting CyP-D.
However, despite this evidence that CyP-D has a key role in I/R injury, cytoprotection by CsA in experimental models is highly variable (and frequently marginal) and, in recent pilot trials, CsA yielded modest protection [20]. This suggests that CyP-D has a restricted pathogenic function and/or that CsA sensitivity is not a precise indicator of CyP-D involvement in cell death, possibly due to interaction with other CyPs; the cytosolic CyP-A and the endoplasmic reticulum CyP-B also have known links to cell viability (see the Discussion section).
In order to shed light on these issues, we have developed a novel CsA derivative that is effectively CyP-D selective in cells because it accumulates in the mitochondrial compartment. Application of the mtCsA (mitochondrially targeted CsA) to hippocampal neurons indicates that CyP-D has a major pathogenic role in energy failure, but only a minor role in glutamate excitotoxicity. Moreover, the cytoprotective capacity of CsA against the adverse consequences of energy failure is markedly improved by mitochondrial targeting.
EXPERIMENTAL
Preparation of recombinant CyP-D and CyP-A
Recombinant rat CyP-D was prepared and purified as described previously [14]. For CyP-A, the coding sequence in rat was PCR-amplified with the addition of BamH1 and EcoR1 restriction sites, and cloned between the same sites of pGEX-4T-1 in Escherichia coli DH5α cells. Transformed cells were grown for 5 h at 21 °C. The GST (glutathione transferase)–CyP-A fusion protein was extracted, purified on a GSH–Sepharose column (Sigma–Aldrich), and then cleaved with thrombin to release CyP-A. The CyP-A was purified on a cation exchange column (Mono S®; Amersham Pharmacia) and by a gel filtration column (Superdex-75; Amersham Pharmacia) to give a single band on SDS/PAGE.
Interactions of cyclosporins with CyPs and calcineurin
Dissociation constants for CyP–cyclosporin interactions were measured as inhibitor constants, Ki. PPIase assays were conducted at 15 °C in 20 mM Hepes, pH 7.5, containing 100 mM NaCl using N-succinyl-alanyl-alanyl-prolyl-4-nitroanilide as the test peptide as described previously [14]. The peptide contains a mixture of cis- and trans-alanine–proline isomers, of which only the trans-conformer is hydrolysed by chymotrypsin at the C-terminal amide bond to release chromophore. Existing trans-isomer is cleaved within the mixing time, hence further cleavage requires cis–trans isomerization, which is measured. CyPs were preincubated with cyclosporins for 5 min before addition of chymotrypsin and 60 μM peptide (containing approx. 35 μM cis-peptide) to start the reaction. Cyclosporins inhibit by competing at the active site with substrate [21]. Accordingly, kinetic data were analysed by the Henderson equation for a tight-binding, competitive inhibitor [22], which can be written:
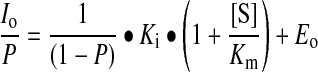 |
where Eo and Io are the total concentrations of enzyme and inhibitor (cyclosporin) respectively, Ki is the enzyme inhibitor dissociation constant, Km is the Michaelis constant and [S] is the substrate concentration. P is the fractional inhibition, equal to {1−(vi/vo)}, where vi and vo are the reaction velocities in the presence and absence of inhibitor respectively. The Km value for the cis-peptide used (980 μM; [21]) is much higher than its concentration in the assay (<35 μM). As Km>>[S], the equation may be simplified:
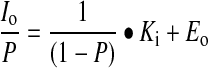 |
and plots of Io/P against 1/(1−P) are linear with slope equal to Ki.
Interaction of CyP–cyclosporin complexes with calcineurin [also known as PPP3CA (protein phosphatase 3, catalytic subunit, α isoform)] was evaluated from inhibition of the phosphatase activity of calcineurin as measured by the release of inorganic phosphate from the RII phosphopeptide (Biomol International).
Experiments with isolated mitochondria
Mitochondria were isolated from rat livers as described previously [1]. All animal experimentation conformed to U.K. Home Office legislation. PT pore opening was monitored by the associated swelling of the mitochondria [9] as measured by the decrease in absorbance at 540 nm. Mitochondria (2 mg of protein) were suspended in 3 ml of buffer (10 mM Hepes, pH 7.2, containing 120 mM KCl, 2 mM KH2PO4, 3 mM succinate, 1 μM rotenone and 5 μM EGTA) with the recombinant CyP-A and test cyclosporins, and maintained under continuous stirring at 25 °C. After 5 min, calcium chloride was slowly infused (at 10 μM/min) to a final concentration of 50 μM. Changes in preincubation time with CsA or mtCsA from 5 min to 2 min or 10 min did not change the degree of PT pore inhibition. In a parallel incubation, mitochondria were sedimented at the time indicated and the CyP-A activity of the supernatant determined (0.5 ml of supernatant per ml of reaction volume).
Neuronal cultures and assays
B50 cells from a rat neuronal cell line and a clone stably overexpressing CyP-D were cultured on coverslips in DMEM (Dulbecco's minimal essential medium) containing 10% (v/v) FBS (fetal bovine serum) as described previously [14]. Uptake of TMRE (tetramethylrhodamine ethyl ester) was measured by incubating the cells at 25 °C in basic medium (24 mM Hepes, pH 7.4, containing 140 mM NaCl, 4 mM KCl, 1 mM MgSO4, 1 mM CaCl2, 1 mM KH2PO4 and 11 mM glucose) containing 50 nM TMRE. Fluorescence images were obtained at an excitation wavelength of 530 nm and emission wavelength of >595 nm with an Olympus IX-70 fluorescence microscope with a ×60 oil objective, Micromax 1401E CCD (charge-coupled-device) camera and Metamorph software (Molecular Devices). For nitroprusside treatment, cells were incubated in basic medium containing 100 μM sodium nitroprusside for 40 min and then returned to DMEM. After 5 h, cells were extracted and the extracts were assayed for caspase-3 activity using the fluorescent derivative of the caspase-3/7 selective substrate, Ac-DEVD (N-acetyl-Asp-Glu-Val-Asp-)-AFC (7-amino-4-trifluoromethylcoumarin), as described previously [23]. For antisense suppression of CyP-A, cells were incubated with 1 μM phosphorothioate oligodeoxynucleotide (5′-CATGGCTTCCACAATGCT) for 48 h [23].
Hippocampal neurons were prepared from 2–4-day-old Sprague–Dawley rats as mixed cultures with glial cells. Dissected hippocampi were incubated in HBSS (Hanks balanced salt solution) containing 0.1% trypsin for 5 min at 37 °C, and were washed twice in HBSS. Hippocampi were then dissociated in HBSS containing 1 mg/ml BSA, 5% (v/v) FBS and 8 mM MgCl2. Dissociated cells were sedimented, suspended in NBA (Neurobasal A) medium (Gibco) supplemented with 0.5 mM glutamine, 2% (v/v) B27 supplement (Gibco) and 5% (v/v) FBS, seeded onto coverslips and incubated under 95% air/5% CO2 in the same medium with an antimitotics mix (1 μM of each of 5-fluor-2′-deoxyuridine, uridine and 1-β-D-arabinofuranosylcytosine). Medium without antimitotics was introduced after 3 days.
For OGD (oxygen and glucose deprivation) experiments, coverslips with hippocampal neurons were seated to form the base of a small, capped chamber mounted on the microscope stage. The chamber contained an inlet and outlet for continuous gassing, input and output tubes for changing the incubation medium, and a heating element to maintain the temperature at 36 °C. Pseudo-ischaemic conditions were imposed by omitting glucose and displacing air with nitrogen in the experimental chamber. Cells were incubated under 95% N2/5% CO2 with a pregassed solution of 145 mM NaCl, 26 mM NaHCO3, 5 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 4 μM ethidium homodimer, 2 μM Hoechst 33342 and the cyclosporins as indicated. After 30 min the gassing was switched to 95% air/5% CO2 and the medium replaced with NBA medium containing 4 μM ethidium homodimer. Hippocampal neurons were identified under brightfield illumination and then correlated with their respective nuclei (just above the focal plane of glia nuclei) from the Hoechst fluorescence. Necrosis was quantified from the nuclear staining by ethidium homodimer. For treatment with glutamate, cultures were incubated under 95% air/5% CO2 in a solution of Locke's medium (5 mM Hepes, 150 mM NaCl, 5 mM KCl, 25 mM NaHCO3, 2.3 mM CaCl2 and 6 mM glucose) with the cyclosporins as indicated. After 10 min, 1 mM glutamate was added. After a further period, as indicated, cells were returned to NBA medium containing Hoechst 33342 and ethidium homodimer, and necrosis was quantified 15 min later. Further inhibitors and antagonists [FK506, L-NAME (NG-nitro-L-arginine methyl ester), MK801, or NBQX (2,3-dihydro-6-nitro-7-sulfamoylbenzoquinoxaline); Sigma–Aldrich] were added as stated in the legend of Figure 8.
Figure 8. Protection against glutamate-induced and OGD-induced necrosis in hippocampal neurons.

(A) Glutamate-induced necrosis. Hippocampal neurons were incubated in Locke's medium containing either no addition (−), 1 μM CsA, 1.3 μM mtCsA, 1 μM FK506, 250 μM L-NAME, 1 μM FK506 and 1.3 μM mtCsA (mtCsA+FK506), 50 μM MK801 or 100 μM NBQX. After 10 min, 1 mM glutamate was added and cell necrosis was determined 3 h later. (B) OGD-induced necrosis. Neurons were subjected to oxygen and glucose deprivation for 30 min, and then glucose-replete normoxia for 90 min, before necrosis was quantified. Inhibitors were added to both anoxic and normoxic media at concentrations as in (A). Results are means±S.E.M. of five cell preparations with >20 cells per preparation. *P<0.05; **P<0.01 with respect to no addition.
Statistical analyses were made using a one-way ANOVA test with a post-test of Dunnett and results are presented as means±S.E.M.
Synthesis of cyclosporin analogues
A scheme of the syntheses is given in Figure 1.
Figure 1. Chemical synthesis of the CsA analogues, and their binding by CyPs.

The reagents used in each step are: i) lithium diisopropylamide, trimethylsilyl chloride, 4-bromomethylbenzoate; ii) LiOH, methanol; iii) Fmoc-diaminohexane, PyBOP; iv) piperidine, DMF; and v) 5-(carboxypentyl)triphenylphosphonium bromide, PyBOP. Compound (1) is CsA, compound (2) SMBz-CsA and compound (4) mtCsA. The lower table shows the dissociation constants [determined as inhibitor constants (Ki) as described for Figure 2] for the binding of CsA and the CsA analogues to CyP-D and CyP-A.
Synthesis of compound (2), SMBz [Sarcosine-3(4-methylbenzoate)]-CsA
To a stirred solution of CsA (1.00g, 0.83 mmol) in dry THF (tetrahydrofuran, 25 ml) under nitrogen at 0 °C was added dropwise fresh LDA (lithium diisopropylamide; 4.6 mmol, 2.3 ml, 2 M in THF). To the resultant deep-brown suspension was added, dropwise, trimethylsilylchloride (0.83 mmol, 0.1 ml) to give a clear brown solution. The mixture was stirred at 0 °C for 10 min. Then further LDA (7.1 mmol, 3.5 ml, 2 M in THF) was added dropwise and the reaction stirred for 30 min at 0 °C. A solution of 4-bromomethylbenzoate (1.3 g, 5.8 mmol) in dry THF (10 ml) was added dropwise to give a pale yellow solution which was stirred for a further 1 h at room temperature (22 °C). The reaction was quenched with saturated NH4Cl (aq) (10 ml) followed by 2 M HCl and then diluted with DCM (dichloromethane) (20 ml). The separated aqueous layer was extracted with DCM (2×20 ml). The combined organic phases were washed with 2 M HCl (aq) (2×20 ml), saturated NH4Cl (aq) (2×20 ml) and saturated NaCl (2×20 ml) and then dried (MgSO4). The volatiles were removed in vacuo to leave a dark brown oil residue. Purification by flash chromatography eluting with 6% (v/v) methanol in DCM gave a yellow solid residue (0.930 g) as a mixture of the unreacted CsA and the alkylated ester product. This was used in the next reaction without further purification.
Saponification was next performed. To a stirred solution of the yellow solid residue (0.93 g) in THF/methanol (1:1, 20 ml) at 0 °C was added dropwise a solution of LiOH·H2O (500 mg) in water (10 ml). The reaction was allowed to gradually warm-up to room temperature over 18 h. Then DCM (20 ml) was added. The resultant solution was acidified with 2 M HCl (aq) (pH 3). The separated aqueous layer was extracted with DCM (3×30 ml). The combined organic extracts were washed with saturated 2 M HCl (aq) (2×30 ml) and saturated NaCl (2×30 ml) and then dried (MgSO4). The volatiles were removed in vacuo to leave a solid residue as a mixture of the acid [Figure 1, compound (2)] and unreacted CsA. The acid was separated from the CsA by flash column chromatography through an amine column eluting with a mixture of methanol/DCM/NH3 (aq) (1:8:1) to give the acid as a salt. After stirring the salt in DCM (20 ml) and 2 M HCl (aq) (20 ml) for 10 min, extraction by DCM (3×20 ml), concentration, and purification by flash column chromatography gave the title compound, (2) (0.30 g, 0.24 mmol, 27%) as a yellow solid.
Fast atom bombardment MS in positive ionization mode: calculated m/z C70H117N11O14 (M+Na) 1358.86787; found m/z (M+Na) 1358.86447.
Synthesis of compound (3)
To a stirred solution of the acid compound (2) (109 mg, 0.08 mmol) in dry THF (3.0 ml) was added N-Fmoc (fluoren-9-ylmethoxycarbonyl)-1,6-diaminohexane hydrobromide (68.5mg, 0.16 mmol), PyBOP (benzotriazol-1-yl-tris-pyrrolidinophosphonium hexafluorophosphate; 84.5 mg, 0.16 mmol) and triethylamine (0.25 mmol, 0.4 ml) under nitrogen at room temperature and the resultant mixture was stirred for 24 h. Then DCM (5 ml) followed by saturated NH4Cl (aq) (5 ml) were added. The mixture was extracted with DCM (2×3 ml) and dried (MgSO4). The volatiles were removed in vacuo to leave a brown oil residue. Purification by chromatography gave the Fmoc-protected derivative (100 mg) as a yellow solid. This was used without further purification.
Fmoc-deprotection was next performed. A solution of the Fmoc compound (100 mg) was stirred in 20% piperidine in DMF (dimethylformamide; 4 ml) under argon for 24 h. The volatiles were removed al vacuo to leave a yellow oil. The oil was purified by flash column chromatography on silica gel eluting with 6% methanol in DCM followed by methanol/DCM/NH3 (aq) (1:8:1) to afford the title compound (3) (70 mg, 0.05 mmol, 85%) as a yellow solid.
ESI–MS (electrospray ionization MS) in positive ionization mode: calculated m/z C76H131N13O13 (M+1) 1435.00, (M+2) 718; found m/z (M+1) 1435.53, (M+2) 718.76.
Synthesis of compound (4), mtCsA
To a stirred solution of the amine compound (3) (65 mg, 0.05 mmol) in dry THF (3 ml) under argon at room temperature was added in one portion PyBOP (35.5 mg, 0.07 mmol), 5-(carboxypentyl)triphenylphosphonium bromide (32 mg, 0.07 mmol) and triethylamine (0.15 mmol, 0.05 ml) and the resultant mixture stirred for 24 h at room temperature. The volatiles were removed al vacuo to leave a yellow oil. The oil was purified by flash column chromatography on silica gel eluting with 6% (v/v) methanol in DCM followed by methanol/DCM/NH3 (aq) (1:8:1) to afford the title compound mtCsA, compound (4) (55 mg, 0.03 mmol, 70%) as a white solid.
ESI–MS in positive ionization mode: calculated m/z C100H155N13O14P+ (M+1) 1793.8199; found m/z (M+1) 1794.8270, (M+2) 898.
RESULTS
Synthesis of CsA derivatives and their interactions with CyPs and calcineurin
CsA is a lipophilic cyclic undecapeptide (Figure 1). The highly conserved nature of the CsA-binding site in CyPs offers little scope for CyP-D-selective cyclosporins (see the Discussion). Therefore we investigated the possibility of rendering CsA effectively CyP-D-selective in cells by targeting it to mitochondria, where CyP-D is located. The strategy was to conjugate CsA to the lipophilic TPP+ (triphenylphosphonium) cation so that the positively charged conjugate is accumulated electrophoretically by mitochondria in response to the negative-inside inner membrane potential. This principle has been widely applied in targeting antioxidants to mitochondria [24]. Seebach et al. [25] demonstrated that CsA may be specifically alkylated at the sarcosine-3 position if several equivalents of a strong base are used to generate a polyanion, and the same method was utilized here, as shown in Figure 1. Briefly, CsA [Figure 1; compound (1)] was treated with LDA/trimethylsilyl chloride and then with LDA to generate the sarcosine-3 anion. This was alkylated with 4bromomethylbenzoate and the crude product saponified with lithium hydroxide to give the acid [Figure 1; compound (2)]. The side chain was elongated by coupling with Fmoc-diaminohexane and Fmoc cleavage yielded the amine [Figure 1; compound (3)]. This was coupled with 5-(carboxypentyl)triphenylphosphonium bromide to give the target triphenylphosphonium salt, mtCsA [Figure 1; compound (4)].
The effects of these modifications of CsA on interactions with CyP-D and CyP-A were investigated by measuring PPIase inhibition. Approx. 7 nM (total) CsA yielded 50% inhibition of CyP-D (Figure 2A). However, this underestimated the true CsA-binding affinity as the assays had a similar concentration of CyP-D, at 8 nM. Accordingly, inhibition was analysed using the Henderson equation for a tight-binding inhibitor (see the Experimental section), which gave an inhibitory (dissociation) constant for CsA and CyP-D of 3 nM (Figure 2A, inset). Additions to position-3 impaired binding slightly for SMBz-CsA (Figure 1), but strongly for mtCsA, where the binding affinity to CyP-D was approx. 30-fold lower than for CsA (Ki of 93 nM; Figure 2B). The binding affinities of CsA, SMBz-CsA and mtCsA to CyP-A were similar to those with CyP-D (Figure 1).
Figure 2. CyP-D inhibition by CsA and mtCsA.

The PPIase activity of recombinant CyP-D was determined in the presence and absence of the given concentrations of (A) CsA and (B) mtCsA. Each data point is the average of two determinations differing by <7%. Insets show results when replotted according to the simplified Henderson equation as described in the Experimental section yielding values for Ki (slope).
In addition to inhibiting CyPs, CsA forms a complex with CyP-A that, in turn, inhibits the Ca2+/calmodulin-dependent serine/threonine protein phosphatase, calcineurin [26], thereby enlarging considerably CsA's sphere of action [27]. It was important therefore to establish how the position-3 modification affected the capacity of the complex to inhibit calcineurin. To enable comparisons, the concentrations of CsA, SMBz-CsA and mtCsA (at 0.9, 1 and 4 μM respectively) in the test incubations were chosen to establish the same concentrations (720 nM) of their respective complexes with CyP-A (calculated using the Ki values of CyP-A with CsA, SMBz-CsA and mtCsA; Figure 1). Figure 3 shows that, whereas CyP-A alone produced a small (20%) activation of calcineurin, the CsA–CyP-A complex inhibited calcineurin by approx. 70%. In contrast, the SMBz-CsA–CyP-A and mtCsA–CyP-A complexes produced no inhibition. Conjugation to position-3 of the CsA ring evidently prevents formation of the ternary CyP–cyclosporin–calcineurin complex, which is known to require interactions between calcineurin and positions 3–7 of the ring (see the Discussion).
Figure 3. Complexes of CyP-A with SMBz-CsA and mtCsA do not inhibit calcineurin.

The phosphatase activity of calcineurin was measured in the presence and absence of CyP-A, CsA, SMBz-CsA and mtCsA at concentrations to give 720 nM complexes (see the Experimental section).
Evaluating the CyP-D selectivity of mtCsA in a mixed in vitro system
We first investigated whether mtCsA would select for intramitochondrial CyP-D and the PT pore, rather than extramitochondrial CyPs, using a test system comprising isolated mitochondria and externally added, recombinant, CyP-A. PT pore opening was induced by addition of a high concentration of Ca2+, and was monitored by the resultant mitochondrial swelling, as the inner membrane became freely permeable to low-Mr solutes. As Ca2+ influx into mitochondria is electrophoretic, the inner membrane potential, ΔϕM, becomes dissipated during rapid Ca2+ uptake {and then restored when uptake is complete (e.g. see [14])}. As dissipation of ΔϕM would compromise accumulation of the positively charged mtCsA in mitochondria, Ca2+ was infused slowly into the test incubations to limit the rate of Ca2+ uptake and thereby avoid membrane depolarization (this was confirmed using a tetraphenylphosphonium electrode, as described in Li et al. [14], and CsA to block PT pore opening; results not shown). CsA and its derivatives inhibited pore opening (Figures 4A, 4C and 4E). Estimation of the degrees of inhibition, assessed by the decreases in absorbance (at the time marked by the broken lines in Figure 4) indicated that approx. 0.1 μM CsA and 0.4 μM mtCsA gave 50% inhibition of PT pore opening (Figures 4B and 4D, closed symbols). In the same system, CsA inhibited extramitochondrial CyP-A with a concentration profile similar to that of the PT pore (Figure 4B); this was expected given that CyP-D and CyP-A have similar binding affinities for CsA (Figure 1) and, being uncharged, CsA should equilibrate to the same free concentrations on either side of the inner membrane. In contrast, mtCsA inhibited PT pore formation considerably better than it inhibited CyP-A (Figure 4D), even though it binds to CyP-A and CyP-D with similar affinities (Figure 1). This indicates that mtCsA was accumulated in the mitochondrial matrix, where CyP-D is located, in preference to the external medium, containing CyP-A. SMBz-CsA gave similar inhibition profiles for the PT pore and CyP-A, indicating a similar free-SMBz-CsA concentration across the inner membrane (Figure 4F).
Figure 4. mtCsA preferentially inhibits intramitochondrial CyP-D, rather than extramitochondrial CyP-A, in a mixed in vitro system.

(A, C and E) Isolated liver mitochondria were incubated with added recombinant CyP-A. After preincubation with CsA, mtCsA or SMBz-CsA, as indicated, PT pore opening was initiated by infusion of Ca2+ and monitored by recording the decrease in A540 due to mitochondrial swelling. (B, D and F) The degree of PT pore opening was compared by using the absorbance decreases attained at the time indicated by the broken lines in (A, C and E) and replotted as percentage activity (black symbols). In parallel incubations, mitochondria were sedimented immediately after Ca2+ addition and CyP-A activity in the supernatant was determined (white symbols).
Evaluating the CyP-D selectivity of mtCsA in intact cells
Selectivity of mtCsA for CyP-D in intact cells was investigated using rat B50 neuroblastoma cells and a clone in which CyP-D is overexpressed by approx. 10-fold (CyP-D+). CyP-D overexpression did not affect CyP-A (Figure 5F). As shown previously [14], CyP-D+ cells maintain a relatively low ΔϕM, indicative of transient PT pore opening. As the lowering of ΔϕM is caused by excessive CyP-D, restoration of ΔϕM to wild-type values provides an unequivocal measure of CyP-D inhibition. Changes in ΔϕM were monitored from the uptake of TMRE, a fluorescent, lipophilic cation accumulated by mitochondria according to the magnitude of the potential. Figure 5(A) shows typical images of TMRE accumulated within the mitochondria of these cells. Mitochondria of the CyP-D+ clone accumulated considerably less TMRE than wild-type cells, but the difference was negated by CsA, which promoted uptake by the CyP-D+ cells. Maximal restoration of TMRE uptake by CyP-D+ cells was obtained with approx. 0.8 μM CsA (Figure 5C), 2.4 μM mtCsA (Figure 5D) and 2 μM SMBz-CsA (results not shown). The same concentrations did not affect TMRE uptake by wild-type cells [Figure 5B; also confirmed for SMBz-CsA (results not shown)]. It may be concluded that approx. 0.8 μM CsA, 2 μM SMBz-CsA and 2.4 μM mtCsA are sufficient to inhibit CyP-D in B50 cells.
Figure 5. mtCsA preferentially inhibits CyP-D, rather than CyP-A, in B50 neuronal cells.

(A) Wild-type B50 cells (WT), or a CyP-D+ clone overexpressing CyP-D (CLONE) were incubated with 50 nM TMRE for 30 min (panels 1 and 3) after which 1 μM CsA was added and the cells incubated for a further 30 min (panels 2 and 4). (B–D) Wild-type (WT) or CyP-D+ cells were incubated in the presence or absence of the indicated concentrations of CsA and mtCsA. TMRE uptake was measured by whole cell fluorescence determined at intervals. The results are the means±S.E.M. for four experiments. (E) CyP-D+ cells pretreated with CyP-A antisense oligodeoxynucleotide (AS; 42 h), 0.8 μM CsA, 2 μM SMBz-CsA or 2.4 μM mtCsA (for 10 min) were incubated with or without 100 μM nitroprusside (for 40 min) and extracts were assayed for caspase-3. *P<0.05, comparing test nitroprusside data with control (no addition) nitroprusside data over four experiments. (F) Immunoblots showing that CyP-D overexpression (CyP-D+) does not change CyP-A, and that CyP-A knockdown (AS CyP-A) does not change CyP-D.
To investigate whether mtCsA inhibited CyP-D selectively, i.e. without appreciable inhibition of CyP-A, we needed a marker of CyP-A activity. Previously, we reported that nitroprusside-induced caspase activation in B50 cells is largely prevented by CsA and by antisense suppression of CyP-A. This indicates that CsA inhibits caspase activation in this model by inhibiting CyP-A [23]. Although the specific site of action of CyP-A is unresolved, this system offers a measure of CyP-A activity. Antisense treatment decreased CyP-A expression by >85% without affecting CyP-D (Figure 5F), and antisense treatment, CsA and SMBz-CsA all reduced nitroprusside-induced activation of caspase-3 (Figure 5E). Unlike CsA and SMBz-CsA, however, 2.4 μM mtCsA (which was sufficient to inhibit mitochondrial CyP-D; Figure 4D), had no significant effect on caspase activation, indicating that it was not accumulated in the cytosol, where CyP-A is located.
I/R and glutamate-induced injury in hippocampal neurons
I/R was mimicked by incubating hippocampal neurons under OGD conditions for 30 min, after which glucose and oxygen were restored. To estimate the time period of OGD needed to remove oxygen sufficiently for impairment of mitochondrial electron transport, we followed TMRE loss from mitochondria of preloaded cells as an index of ΔϕM dissipation. TMRE was lost after approx. 5 min OGD, indicating respiratory inhibition at this time (results not shown). At the outset of each experiment, a group of hippocampal neurons were distinguished from underlying glial cells and the same neurons were imaged at intervals thereafter. The susceptibility of neuronal cells (but not glial cells) to OGD-induced necrosis increased with the number of days in culture, and results were obtained after culture for 24–28 days.
Following OGD, approx. 60% of neurons became necrotic within 90 min (Figure 6A), but mortality was approximately halved in the presence of mtCsA. Maximal protection was given with >0.8 μM mtCsA (Figure 6B). CsA was less protective (Figure 6B). There was a relatively small protection with 0.1 μM CsA, but this was reversed at higher CsA concentrations, suggesting the existence of secondary CsA targets outside mitochondria that counteracted the protection. Thus restricting the action of CsA to mitochondria using mtCsA improves its protective capacity against cell necrosis brought about by a period of OGD, indicating that CyP-D and the PT pore are major contributors to this form of injury.
Figure 6. Mitochondrial targeting enhances protection of hippocampal neurons by CsA following transient oxygen and glucose deprivation.

(A) Hippocampal neurons were preincubated under OGD for 30 min, and then returned to glucose-replete normoxia (OGD). The experiment was repeated with mtCsA included in both anoxic and normoxic media (OGD+mtCsA). The lower curve is without an OGD step (No ODG). Necrotic cells were quantified from nuclear staining with ethidium homodimer. (B) Cells were incubated with CsA or mtCsA and necrosis determined after 30 min OGD and 90 min glucose-replete normoxia. Results are means±S.E.M. for five cell preparations with >20 cells per preparation.
In vivo, a major component of I/R-induced neuronal injury is believed to be initiated by prolonged exposure to extracellular glutamate and overactivation of ionotropic glutamate receptors. To investigate PT pore involvement in glutamate toxicity, neurons were exposed to added glutamate under normoxic conditions in the presence of glucose. Following a short lag-period, this led to progressive necrosis (Figure 7A). Surprisingly, the relative protective capacities of CsA and mtCsA were now reversed with respect to OGD. Whereas CsA yielded good protection, mtCsA was ineffective (Figure 7B). In a further series of experiments (Figure 8A), mtCsA did reduce necrosis to a small extent (24%, but not significant) but, again, CsA was far more effective with 88% protection. Thus restricting the action of CsA to mitochondria with mtCsA largely removes protection by CsA, indicating that CsA attenuates glutamate toxicity by interacting with extramitochondrial CyPs, rather than intramitochondrial CyP-D.
Figure 7. CsA, but not mtCsA, strongly reduces glutamate toxicity in hippocampal neurons.

(A) Hippocampal neurons were incubated in Locke's medium with (●) and without (○) 1 mM glutamate at 37 °C and cell viability was determined from nuclear staining with ethidium homodimer. Results from a single representative preparation. (B) Neurons were incubated in Locke's medium with CsA (○) or mtCsA (●). After 10 min, 1 mM glutamate was added and cell necrosis determined 3 hours later. Results are means±S.E.M. of five cell preparations with >20 cells per preparation.
As expected, glutamate-induced necrosis was prevented by glutamate antagonists, i.e. by MK-801, an NMDA (N-methyl-D-aspartate) receptor antagonist, and by NBQX, an AMPA (amino-3-hydroxy-5-methyl-4-isoxazole proprionic acid) receptor antagonist (Figure 8A). However, glutamate antagonists had relatively little effect on OGD-induced necrosis (Figure 8B), indicating negligible contribution of glutamate toxicity under this protocol (any glutamate released from the cells during anoxia was presumably quickly diluted in the incubation medium). Thus the involvement of extramitochondrial CyPs in CsA protection correlates with glutamate toxicity, whereas their involvement in CsA counterprotection correlates with OGD-induced necrosis. To investigate whether these effects, both protective (glutamate) and counterprotective (OGD), might involve inhibition of calcineurin by the CsA–CyP-A complex, we used FK506 (an immunosuppressant macrolide) which, as the FK506–FKBP (FK506-binding protein) complex, also inhibits calcineurin. As shown in Figure 8(B), FK506 partially reversed mtCsA-protection against OGD-induced necrosis, indicating that calcineurin inhibition is counterprotective. In contrast, FK506 partially prevented glutamate-induced necrosis. Thus calcineurin appears to have opposing roles in OGD-induced and glutamate-induced cell death.
Calcineurin dephosphorylates, and thereby activates, neuronal NO synthase, and this activation is believed to contribute to glutamate toxicity [27]. However, the NO synthase inhibitor L-NAME did not yield significant protection. Indeed, it is notable that both L-NAME and FK506 were less effective than CsA (Figure 8A). To test whether the acute effectiveness of CsA against glutamate toxicity reflected a dual action in inhibiting both calcineurin and CyP-D (and the PT pore), we applied FK506 and mtCsA together; however this was no more effective than FK506 alone. This confirms that the inhibition of glutamate excitotoxicity by CsA is not due to inhibition of CyP-D.
The greater effectiveness of CsA compared with mtCsA plus FK506 in inhibiting glutamate-induced necrosis (Figure 8A) suggests that CsA-mediated inhibition of extramitochondrial CyPs (PPIase activity) may also be protective. This was further investigated using SMBz-CsA, which inhibits CyP PPIase activity (Figure 1), but not calcineurin when in complex with CyP-A (Figure 3). Similar to CsA, SMBz-CsA permeates into mitochondria and inhibits the PT pore (Figure 4E), but is not accumulated by mitochondria (Figure 4F) and it enters cells where it inhibits CyP-A (Figure 5E). As shown in Figure 9, SMBz-CsA was more cytoprotective than mtCsA, consistent with some protection due to inhibition of extramitochondrial CyPs. Figure 9 also shows that SMBz-CsA was a better cytoprotectant than CsA following OGD and that, unlike CsA (Figure 6), protection was not reversed with an increased SMBz-CsA concentration. This may reflect the lack of calcineurin inhibition by SMBz-CsA, as calcineurin inhibition appears to be counterprotective in OGD-induced necrosis (Figure 8B, mtCsA compared with mtCsA plus FK506). However, SMBz-CsA yielded less cytoprotection than mtCsA, suggesting that inhibition of extramitochondrial CyPs might also be counterprotective in OGD-induced necrosis (Figure 9). This is consistent with the fact that FK506 does not fully reverse cytoprotection by mtCsA (Figure 8B). In summary, the involvement of CyPs and calcineurin appears to differ between the two forms of injury; CyP-D inhibition is more protective after OGD than after glutamate, whereas inhibition of calcineurin and extramitochondrial CyP(s) is more protective after glutamate than after OGD (when these interventions actually reverse protection). These differences point to different pathogenic mechanisms.
Figure 9. Cytoprotection by SMBz-CsA.

Glutamate-induced and OGD-induced necrosis in hippocampal neurons was determined as in Figure 8 in the presence of 0.3–2.0 μM CsA, SMBz-CsA and mtCsA, as indicated. In the absence of CsA and derivatives, necrosis was 34±5% (glutamate) and 64±6% (OGD). Results are means±S.E.M. of five cell preparations with >20 cells per preparation. *P<0.05 for SMBz-CsA compared with mtCsA.
DISCUSSION
CsA is a lipophilic, cyclic undecapeptide which half-inserts, edge-on, into a cleft on the CyP surface, occluding the active PPIase site and forming interactions with 15 amino acids [28]. The binding region in CyPs is highly conserved. In particular, apart from a single conservative substitution (lysine for arginine), the binding residues in CyP-A are all present in CyP-D, and a high resolution structure of CyP-D has confirmed that the same residues as in CyP-A make contact with CsA [29]. Although differences in adjacent amino acids might offer potential for selective inhibition, the CsA-binding affinities of CyP-D and CyP-A are about the same (Figure 1). It is therefore difficult to envisage CyP-D selectivity based on binding affinities alone.
To address this problem, CsA was conjugated to the lipophilic cation TPP+ in order to allow electrophoretic accumulation within mitochondria. The CyP-binding amino acids in CsA have been identified as residues 9–11 and 1–3 [28]. However, substitution at the sarcosine in position-3 does allow retention of the tight binding to CyPs [30], and the same position was utilized in the present study for conjugation to TPP+. In the ternary CyP–CsA–calcineurin complex, positions 3–7 of the CsA ring form contacts with calcineurin [26,28]. As expected the position-3 conjugates, SMBz-CsA and mtCsA, therefore were unable to bind to calcineurin (Figure 3). The position-3 addition also reduced the binding affinity to CyPs (Figure 1). This reduction was marginal for SMBz-CsA, presumably reflecting steric factors, but was marked in the case of mtCsA. A possible explanation lies in the fact that the predominant conformation of free CsA is quite different from that bound to CyPs [28] and modifications that stabilize the unbound conformations would effectively lower the binding affinity for CyPs. In addition, mtCsA would be more lipophilic than CsA itself given the nature of the linking region. The TPP+ moiety, however, would not increase lipophilicity (the octanol/buffered saline partition coefficient of triphenylmethylphosphonium is 0.35 [31]). In this connection, analyses of radiolabelled CsA binding to mitochondria reveal that there are components besides CyP-D that bind CsA [32]; these components bind CsA far more weakly than CyP-D, but are present in excess. For mtCsA, which bound with a 30-fold lower affinity to CyP-D compared with CsA, the matrix free-mtCsA concentration needed to block the PT will be 30-fold higher than for CsA, and unspecific binding to other components will be correspondingly greater. Thus the combination of decreased CyP-D-binding affinity, increased partitioning into membrane phospholipids, and unspecific binding may explain why more mtCsA than CsA was needed to inhibit the PT pore (Figures 4B and 4D). Nevertheless, mtCsA displayed a good selectivity for intramitochondrial CyP-D over extramitochondrial CyP-A when tested in a mixed in vitro system (Figure 4D) and in intact B50 cells (Figure 5). The latter made use of the fact that nitroprusside-induced apoptosis in B50 cells is promoted by cytosolic CyP-A [23], but not by CyP-D [14]. In addition, although apoptosis in this system involves the release of cytochrome c from mitochondria [M. Capano (University College London, U.K.) and M. Crompton, unpublished work], CyP-D and the PT pore do not appear to be involved in this process [33]. Thus the fact that mitochondrial targeting of CsA largely removes its capacity to inhibit apoptosis in this system (Figure 5E) is consistent with mitochondrial accumulation of the conjugate out of the cytosol.
CyPs have both positive and negative roles in cell viability; CyP-D is linked to the PT, CyP-A (cytosol) participates in the translocation of AIF (apoptosis inducing factor) to the nucleus [34], in caspase activation [23] and in protection against oxidative stress [35] and CyP-B (endoplasmic reticulum) suppresses apoptosis associated with oxidative stress and altered Ca2+ metabolism [36]. In addition, the CyP-A–CsA complex inhibits calcineurin, which dephosphorylates pro-apoptotic BAD (Bcl-2/Bcl-XL-antagonist, causing cell death) [37] and the L-type Ca2+ channel [38] in neurons. The effects on calcineurin are avoided with some CsA derivatives, e.g. NIM811 and Debio-025 [39], as with SMBz-CsA (Figure 3), but these derivatives remain general CyP inhibitors and their overall effects can be difficult to interpret. For example, SMBz-CsA protected against both glutamate- and OGD-induced necrosis (Figure 9). At face value, this could be taken to indicate similar roles of CyP-D and the PT pore in the two cases. However, use of the CyP-D-specific mtCsA reveals that this is not the case.
The extent to which the CyP-D-dependent PT pore is the root cause of ischaemic cell death was addressed in neuronal cells, where the issue appears complex. Many models of transient ischaemia show that protection of hippocampal and other neuronal cells by CsA is equalled by FK506, suggesting that CsA inhibition of calcineurin-mediated processes, rather than the PT, is critical (reviewed in [39]). In contrast, other studies point to a primary role of the PT [8,40,41]. Thus CsA inhibition of both calcineurin and the PT may yield cytoprotection in different proportions depending on experimental conditions. To address this issue, we have taken account of the fact that hippocampal and other neuronal cells are subjected to two forms of pathogenic insult during I/R. In common with other kinds of cell, the normally high cellular energy state (the phosphorylation potential) becomes compromised in ischaemia as glycolysis and the tricarboxylic acid cycle are inhibited due to lack of glucose and oxygen, leading to ‘energy failure’. In addition, and unique to neuronal tissue, active transport systems for glutamate uptake in astrocytes and neurons (normally responsible for termination of synaptic transmission) operate in reverse during energy failure leading to high extracellular glutamate and over-activation of neuronal glutamate receptors [42]. Both types of insult produce qualitatively similar changes in critical factors needed for PT pore formation. Energy failure in ischaemia leads to losses of ATP and total adenine nucleotides and to a progressive increase in resting cytosolic Ca2+, and reperfusion generates ROS (reactive oxygen species) (reviewed in [43]). Similarly, addition of high exogenous glutamate under aerobic conditions causes excessive Ca2+ influx into the cell, depletion of ATP and ADP, and oxidative stress [40,44]. Moreover, there is evidence that glutamate toxicity requires mitochondrial Ca2+ uptake and induces mitochondrial dysfunction (ΔϕM dissipation) [40,45], consistent with PT pore formation. Accordingly, it might be expected that the PT pore would contribute similarly to injury resulting from transient energy failure alone and from glutamate toxicity. Surprisingly, however, comparisons of the cytoprotective capacities of CsA and mtCsA reveal markedly differing contributions of the PT pore in the two forms of pathogenesis.
Although CsA-mediated inhibition of glutamate toxicity is frequently ascribed to inhibition of CyP-D and prevention of the PT, we obtained no indication for this, as the inhibition of glutamate-induced necrosis by CsA was largely abolished by mitochondrial targeting. This mirrors a previous report showing that genetic ablation of CyP-D had no effect on glutamate toxicity in hippocampal neurons [45]. It appears that the major site of CsA-mediated protection against glutamate toxicity is extramitochondrial. In agreement, and in line with other studies [27,40], we obtained evidence that calcineurin plays a major part in the pathogenesis of cell necrosis induced by glutamate; in particular, FK506 was protective (Figure 8A) and SMBz-CsA was less protective than CsA (Figure 9). This may involve calcineurin-mediated activation of neuronal NO synthase. Neuronal NO synthase is a Ca2+/calmodulin-dependent enzyme, and activation of the synthase is believed to exert a prominent role in glutamate toxicity, aided by being ‘scaffolded’ to the NMDA receptor mediating Ca2+ influx [46]. Our results are consistent with this. However, as CsA was a better inhibitor of glutamate-induced necrosis than either FK506 (and hence calcineurin inhibition) or L-NAME (and hence NO synthase inhibition), and the extra inhibition is not explained by CyP-D inhibition, it appears that inhibition of extramitochondrial CyPs may also play a role in CsA-mediated protection against glutamate-induced necrosis. Consistent with this, SMBz-CsA was a better cytoprotectant than mtCsA (Figure 9).
In contrast, the major site of CsA-mediated protection against OGD-induced necrosis appears to be mitochondrial. Thus necrosis resulting from energy failure alone was substantially prevented by mtCsA. CsA itself yielded relatively poor protection against OGD-induced necrosis, with a narrow concentration range of effectiveness, similar to that found in cardiomyocytes subjected to transient OGD [4] and in perfused heart [7]. Secondary, extramitochondrial targets of CsA presumably negate protection gained by PT inhibition as the CsA concentration is increased. These counterprotective targets may include calcineurin, as mtCsA-mediated protection was decreased by FK506 (Figure 8B), and SMBz-CsA was a better cytoprotectant than CsA (Figure 9). Other extramitochondrial counterprotective targets cannot be ruled out, as mtCsA protection was only partially reversed by FK506 (Figure 8B) and mtCsA was a better cytoprotectant than SMBz-CsA (Figure 9). The augmented protective capacity of CsA against OGD-induced necrosis obtained by mitochondrial targeting cannot be explained by increased inhibition of mitochondrial CyP-D, as CsA was tested at concentrations (1 μM) sufficient to inhibit CyP-D completely (in B50 cells, 0.8 μM total CsA completely reversed the PT activation by CyP-D overexpression; Figure 5C). Rather, enhanced protection by mtCsA must reflect the relatively low mtCsA concentration outside mitochondria so that non-mitochondrial, counterprotective effects of CsA are minimized. Overall, enhanced protection by mtCsA indicates that it protects against energy-failure-induced necrosis by inhibiting mitochondrial CyP-D and supports the applicability of the PT pore model of pathogenesis to this form of injury.
In conclusion, we have developed a novel, mitochondrially targeted CsA and shown that it selectively inhibits the mitochondrial CyP-D-dependent PT pore compared with extramitochondrial CyP-A in intact cells. Application of this targeted CsA to hippocampal neurons exposes a marked difference between the contribution of CyP-D to necrosis induced by glutamate alone (a low contribution) and that arising simply from energy failure (a high contribution). The latter protocol may be representative of I/R injury to cells in general when glutamate does not contribute. In these cases, as selective targeting of CsA to mitochondria produces better protection against transient OGD-induced cell necrosis than CsA itself, the therapeutic potential of CsA in limiting reperfusion injury, recently tested in pilot trials [20], may be improved by mitochondrial targeting.
AUTHOR CONTRIBUTION
Experimental work was carried out by Henry Dube (chemical syntheses) and Sylvanie Malouitre (biological assays), both of whom also provided intellectual input throughout the course of the study. The project was conceived and directed by David Selwood (chemical aspects) and Martin Crompton (biological aspects). All authors contributed to writing this article.
ACKNOWLEDGEMENTS
We thank the following colleagues at University College London: Dr Philip Thomas for significant help in the culture of hippocampal neurons; Michela Capano for help with the CyP-A studies; and Dr Mina Edwards for providing the B50 cell cultures.
FUNDING
This work was supported by the Wellcome Trust [grant number 077357].
References
- 1.Crompton M., Costi A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate, and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur. J. Biochem. 1988;178:489–501. doi: 10.1111/j.1432-1033.1988.tb14475.x. [DOI] [PubMed] [Google Scholar]
- 2.Crompton M., Andreeva L. On the involvement of a mitochondrial pore in reperfusion injury. Basic Res. Cardiol. 1993;88:513–523. doi: 10.1007/BF00795416. [DOI] [PubMed] [Google Scholar]
- 3.Crompton M., Ellinger H., Costi A. Inhibition by cyclosporin A of a Ca2+-dependent mitochondrial pore activated by inorganic phosphate and oxidative stress. Biochem. J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- 4.Nazareth W., Yafei N., Crompton M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin A. J. Mol. Cell. Cardiol. 1991;23:1351–1354. doi: 10.1016/0022-2828(91)90181-k. [DOI] [PubMed] [Google Scholar]
- 5.Duchen M. R., McGuinness O., Brown L. A., Crompton M. On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc. Res. 1993;27:1790–1794. doi: 10.1093/cvr/27.10.1790. [DOI] [PubMed] [Google Scholar]
- 6.Haworth R. A., Hunter P. R. The Ca2+-induced membrane transition in mitochondria III transitional Ca2+ release. Arch. Biochem. Biophys. 1979;195:468–477. doi: 10.1016/0003-9861(79)90373-4. [DOI] [PubMed] [Google Scholar]
- 7.Griffiths E. J., Halestrap A. P. Protection by cyclosporin A of ischaemia-reperfusion induced damage in isolated rat hearts. J. Mol. Cell. Cardiol. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 8.Uchino H., Minamikawa-Tachino R., Kristian T., Perkins G., Narazaki M., Siesjo B. K., Shibasaki F. Differential neuroprotection by cyclosporin A and FK506 following ischaemia correlates with differing abilities to inhibit calcineurin and the mitochondrial permeability transition. Neurobiol. Dis. 2002;10:219–233. doi: 10.1006/nbdi.2002.0514. [DOI] [PubMed] [Google Scholar]
- 9.Javadov S. A., Lim K. H. H., Kerr P. M., Suleiman M. S., Halestrap A. P. Protection of hearts from reperfusion injury by propofol is associated with inhibition of the permeability transition. Cardiovasc. Res. 2000;45:360–369. doi: 10.1016/s0008-6363(99)00365-x. [DOI] [PubMed] [Google Scholar]
- 10.Kim J. S., Jin Y. G., Lemasters J.J. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischaemia-reperfusion. Am. J. Physiol. 2006;290:H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 11.Baines C. P., Kaiser R. A., Purcell N. H., Blair N. S., Osinska H., Hambleton M. A., Brunskill E. W., Sayen R. M., Gottlieb R. A., Dorn G. W., et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 12.Schinzel A. C., Takeuchi O., Huang Z., Fisher J. K., Zhou Z., Rubens J., Hetz C., Danial N. N., Moskowitz M. A., Korsmeyer S. J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischaemia. Proc. Natl. Acad. Sci. U.S.A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson N., Khan A., Virji S., Ward J. M., Crompton M. Import and processing of heart mitochondrial cyclophilin D. Eur. J. Biochem. 1999;263:353–359. doi: 10.1046/j.1432-1327.1999.00490.x. [DOI] [PubMed] [Google Scholar]
- 14.Li Y., Johnson N., Capano M., Edwards M., Crompton M. Cyclophilin-D promotes the mitochondrial permeability transition but has opposite effects on apoptosis and necrosis. Biochem. J. 2004;383:101–109. doi: 10.1042/BJ20040669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basso E., Fante L., Fowlkes J., Petronilli V., Forte M. A., Bernardi P. Properties of the mitochondrial permeability transition pore in mitochondria devoid of cyclophilin D. J. Biol. Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 16.Crompton M., Virji S., Ward J. M. Cyclophilin D binds strongly to complexes of the voltage dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 1998;258:729–753. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- 17.Brustovetsky N., Tropschug M., Heimpel S., Heidkaemoer D., Klingenberg M. A large Ca2+-dependent channel formed by recombinant ADP/ATP carrier from Neurospora crassa resembles the mitochondrial permeability transition pore. Biochemistry. 2002;41:11804–11811. doi: 10.1021/bi0200110. [DOI] [PubMed] [Google Scholar]
- 18.Kokoszka J. E., Waymire K. G., Levy S. E., Sligh J. E., Cal J., Jones D. P., MacGregor G. R., Wallace D. C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leung A. W. C., Varayuwatana P., Halestrap A. P. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piot C., Croisille P., Staat P., Thibault M. D., Rioufol G., Mewton N., Elbelghiti R., Cung T. T., Bonnefoy E., Angoulvant D., et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. New England J. Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 21.Kofron J. L., Kuzmic P., Kishore V., Colon-Bonilla E., Rich D. H. Determination of kinetic constants of peptidylprolyl cis–trans-isomerases by an improved spectrophotometric assay. Biochemistry. 1991;30:6127–6134. doi: 10.1021/bi00239a007. [DOI] [PubMed] [Google Scholar]
- 22.Henderson P. J. F. A linear equation that describes the steady-state kinetics of enzymes and subcellular particles interacting with tightly bound inhibitors. Biochem. J. 1972;127:321–333. doi: 10.1042/bj1270321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capano M., Virji S., Crompton M. Cyclophilin-A is involved in excitotoxin-induced caspase activation in rat neuronal B50 cells. Biochem. J. 2002;363:29–36. doi: 10.1042/bj3630029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy M. P., Smith R. A. J. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Ann. Rev. Pharm. Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 25.Seebach D., Beck A. K., Bossler H. G., Gerber C., Ko S. Y., Murtiashaw C., Naef R., Shoda S., Thaler A., Krieger M., et al. Modification of cyclosporin A: generation of an enolate at the sarcosine residue and reactions with electrophiles. Helv. Chim. Acta. 1993;76:1564–1590. [Google Scholar]
- 26.Jin L., Harrison S. C. Crystal structure of human calcineurin complexed with cyclosporin A and human calcineurin. Proc. Natl. Acad. Sci. U.S.A. 2002;99:13522–13526. doi: 10.1073/pnas.212504399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morioka M., Hamada J. I., Ushio Y., Miyamoto E. Potential role of calcineurin for brain ischaemia and traumatic injury. Progr. Neurobiol. 1999;58:1–30. doi: 10.1016/s0301-0082(98)00073-2. [DOI] [PubMed] [Google Scholar]
- 28.Taylor P. T., Husi H., Kontopidis G., Walkinshaw M. D. Structures of cyclophilin-ligand complexes. Progr. Biophys. Molec. Biol. 1997;67:155–181. doi: 10.1016/s0079-6107(97)00014-x. [DOI] [PubMed] [Google Scholar]
- 29.Kajitani K., Fujihashi M., Kobayashi Y., Shimizu S., Tsujimoto Y., Miki K. The crystal structure of human cyclophilin D in complex with its inhibitor cyclosporin A at 0.96 angstrom resolution. Proteins. 2008;70:1635–1639. doi: 10.1002/prot.21855. [DOI] [PubMed] [Google Scholar]
- 30.Baumgrass R., Zhang Y., Erdmann F., Thiel A., Weiwad M., Radbruch A., Fischer G. Substitution in position 3 of cyclosporin A abolishes the cyclophilin-mediated gain of function mechanism but not immunosuppression. J. Biol. Chem. 2004;279:2470–2479. doi: 10.1074/jbc.M304754200. [DOI] [PubMed] [Google Scholar]
- 31.Asin-Cayuela A., Manas A. B., James A. M., Smith R. A. J., Murphy M. P. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004;571:9–16. doi: 10.1016/j.febslet.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 32.McGuinness O., Yafei N., Costi A., Crompton M. The presence of two classes of high affinity cyclosporin binding sites in mitochondria Evidence that the minor component is involved in the opening of an inner-membrane, Ca2+-dependent pore. Eur. J. Biochem. 1990;194:671–679. doi: 10.1111/j.1432-1033.1990.tb15667.x. [DOI] [PubMed] [Google Scholar]
- 33.Gillick K., Crompton M. Evaluating cytochrome c diffusion in the intermembrane spaces of mitochondria during cytochrome c release. J. Cell. Sci. 2008;121:618–626. doi: 10.1242/jcs.021303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu C., Wang X., Deinum J., Huang Z., Gao J., Modjtahedi N., Neagu M. R., Nilsson M., Eriksson P. S., Hagberg H., et al. Cyclophilin A participates in the nuclear translocation of apoptosis inducing factor in neurons after cerebral hypoxia-ischaemia. J. Exp. Med. 2007;204:1741–1748. doi: 10.1084/jem.20070193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doyle V., Virji S., Crompton M. Evidence that cyclophilin-A protects cells against oxidative stress. Biochem. J. 1999;341:127–132. [PMC free article] [PubMed] [Google Scholar]
- 36.Kim J., Choi T. G., Ding Y. Overexpressed cyclophilin-B suppresses apoptosis associated with ROS and Ca2+ homeostasis after endoplasmic reticulum stress. J. Cell. Sci. 2008;121:3636–3648. doi: 10.1242/jcs.028654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang L., Omori K., Suzukawa J., Inagaki C. Calcineurin-mediated BAD Ser155 dephosphorylation in ammonia-induced apoptosis of cultured rat hippocampal neurons. Neurosc. Lett. 2004;357:73–75. doi: 10.1016/j.neulet.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 38.Norris C. M., Blalock E. M., Chen K. C., Porter N. M., Landfield P. W. Calcineurin enhances L-type Ca2+ channel activity in hippocampal neurons: increased effect with age in culture. Neuroscience. 2002;110:213–225. doi: 10.1016/s0306-4522(01)00574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waldmeier P. C., Zimmerman K., Qian T., Tintelnot-Blomley M, Lemasters J. J. Cyclophilin D as a drug target. Curr. Med. Chem. 2003;10:1485–1506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]
- 40.Ruiz F., Alvarez G., Ramos M., Hernandez M., Bogonez E., Satrastegui J. Cyclosporin targets involved in protection against glutamate toxicity. Eur. J. Pharm. 2000;404:29–39. doi: 10.1016/s0014-2999(00)00584-7. [DOI] [PubMed] [Google Scholar]
- 41.Rytter A., Cardoso C. M. P., Johanssohn P., Cronberg T., Hanssohn M. J., Mattiasson G., Elmer E., Wieloch T. The temperature dependence and involvement of the mitochondrial permeability transition and caspase activation in damage to organotypic hippocampal slices following in vitro ischaemia. J. Neurochem. 2005;95:1108–1117. doi: 10.1111/j.1471-4159.2005.03420.x. [DOI] [PubMed] [Google Scholar]
- 42.Bonde C., Noraberg J., Noer H., Zimmer J. Ionotropic glutamate receptors and glutamate transporters are involved in necrotic neuronal cell death induced by oxygen and glucose deprivation of hippocampal slice cultures. Neuroscience. 2005;136:779–794. doi: 10.1016/j.neuroscience.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 43.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 44.Reynolds I. J., Hastings T. G. Glutamate induces the formation of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abramov A. Y., Duchen M. R. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate toxicity. Biochim. Biophys. Acta. 2008;1777:953–964. doi: 10.1016/j.bbabio.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 46.Ishii H., Shibuya K., Ohta H., Mukai H., Uchino S., Takata N., Rose J. A., Kawato S. Enhancement of nitric oxide production by association of nitric oxide synthase with N-methyl-D-aspartate receptors via postsynaptic density 95 in genetically engineered Chinese hamster ovary cells: real-time fluorescence imaging using nitric oxide sensitive dye. J. Neurochem. 2006;96:1531–1539. doi: 10.1111/j.1471-4159.2006.03656.x. [DOI] [PubMed] [Google Scholar]