

Abstract
Bioassay-guided fractionation of an ethanol extract of a Madagascar collection of Elaeodendron alluaudianum led to the isolation of two new cardenolide glycosides (1 and 2). The 1H and 13C NMR spectra of both compounds were fully assigned using a combination of 2D NMR experiments, including 1H-1H COSY, HSQC, HMBC, and ROESY sequences. Both compounds 1 and 2 were tested against the A2780 human ovarian cancer cell line and the U937 human histiocytic lymphoma cell line assays, and showed significant antiproliferative activity with IC50 values of 0.12 and 0.07 μM against the A2780 human ovarian cancer cell line, and 0.15 and 0.08 μM against the U937 human histiocytic lymphoma cell line, respectively.
1. Introduction
In our continuing search for biologically active natural products from tropical rainforests as part of an International Cooperative Biodiversity Groups (ICBG) program,2 we obtained an extract of the stems of a plant initially identified as a Hippocratea sp. from Madagascar. The extract had significant antiproliferative activity against the A2780 ovarian cancer cell line, and it was thus selected for bioassay-guided fractionation. While the work was in progress the plant was reidentified as Elaeodendron alluaudianum H. Perrier (Celastraceae). As noted previously,3 there are about forty species in the genus Elaeodendron from the Mexican coast, Bermuda, Africa, Madagascar (incl. the Mascarenes), India, Melanesia, and Australia.4 The plants in this genus are usually glabrous trees or shrubs,4 and flavonoids,5 terpenoids,6 and cardenolides7 have been isolated from them. The cytotoxicities and cardiac activities of cardenolides have been widely studied.8
The extract of E. alluaudianum had an IC50 value of 3.3 μg/mL against the A2780 human ovarian cancer cell line. The crude extract afforded two new cardenolide glycosides, designated elaeodendroside V (1) and W (2), after solvent partitioning and reversed-phase C18 HPLC. Herein we report the structural elucidation of the two new cardenolide glycosides and their bioactivities against the A2780 human ovarian cancer cell line and the U937 human histiocytic lymphoma cell line.
2. Results and Discussion
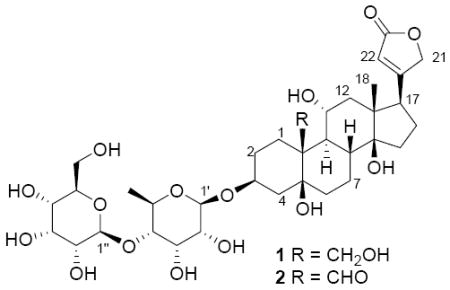
Elaeodendroside V (1) was obtained as a white amorphous solid. Its molecular formula was established as C35H54O16 on the basis of a protonated molecular ion peak at m/z 731.3496 in its HRFAB mass spectrum. Its 1H NMR spectrum in CD3OD showed characteristic signals of an α,β-unsaturated γ-lactone (δH 5.01, dd, J = 18.4, 1.6 Hz, H-21a; δH 4.92, dd, J = 18.4, 1.6 Hz, H-21b; and δH 5.91, s, H-22) (Table 1). Its 13C NMR spectrum contained 35 signals (Table 2), which were assigned as two methyls, eleven methylenes (including three oxymethylenes), sixteen methines (including twelve oxymethines and one olefinic carbon), and six quaternary carbons (including two oxyquaternary carbons, one olefinic carbon and one carbonyl carbon) based on 13C NMR (Table 2) and HSQC spectra.
Table 1.
1H NMR Data of Compounds 1 and 2c
| no. | 1a | 1b | 2b |
|---|---|---|---|
| 1 | 3.03 m | 2.24 m | 2.47 m |
| 2.85 td (13.8, 2.5) | 2.19 m | 2.24 m | |
| 2 | 2.45 m | 1.94 m | 1.92 m |
| 2.25 m | 1.82 m | 1.85 m | |
| 3 | 4.48 m | 4.15 m | 4.19 br s |
| 4 | 2.21 m | 2.10 dd (15.4, 3.0), 1.64 m | 2.09 m |
| 1.84 m | 1.72 m | ||
| 6 | 2.34 m | 1.87 m | 1.89 m |
| 1.67 br d | 1.47 m | 1.68 m | |
| 7 | 2.44 m | 1.99 m | 2.06 m |
| 1,49 m | 1.25 m | 1.28 m | |
| 8 | 2.40 m | 1.81 m | 2.00 m |
| 9 | 2.19 m | 1.78 m | 1.73 m |
| 11 | 4.52 m | 3.93 td (9.8, 4.2) | 3.94 dd (9.8, 4.7) |
| 12 | 1.97 m | 1.66 m | 1.67 m |
| 1.89 m | 1.54 dd (13.2, 9.8) | 1.49 dd (13.2, 10.8) | |
| 15 | 2.31 m | 2.17 m | 2.15 m |
| 1.97 m | 1.72 m | 1.68 m | |
| 16 | 2.09 m | 2.17 m | 2.14 m |
| 2.00 m | 1.91 m | 1.89 m | |
| 17 | 3.00 m | 2.93 t (7.2) | 2.94 t (7.2) |
| 18 | 1.13 s | 0.91 s | 0.90 s |
| 19 | 4.66 d (10.7) | 4.18 d (11.2) | 9.97 s |
| 4.38 m | 3.80 m | ||
| 21 | 5.28 br d (18.2) | 5.01 dd (18.4, 1.6), 4.92 dd (18.4, 1.6) | 5.00 br d (18.4) |
| 4.92 br d (18.4) | |||
| 5.03 m | |||
| 22 | 6.11 s | 5.91 s | 5.91 s |
| 1′ | 5.37 d (8.0) | 4.72 d (8.0) | 4.73 d (8.0) |
| 2′ | 3.90∼4.60d | 3.35 m | 3.35 m |
| 3′ | 3.90∼4.60d | 4.33 t (3.0) | 4.33 t (2.8) |
| 4′ | 3.90∼4.60d | 3.28 dd (9.6, 3.0) | 3.28 m |
| 5′ | 3.90∼4.60d | 3.85 m | 3.85 m |
| 6′ | 1.60 d (6.0) | 1.30 d (6.0) | 1.30 d (6.0) |
| 1″ | 5.49 d (7.7) | 4.73 d (7.6) | 4.73 d (8.0) |
| 2″ | 3.90∼4.60d | 3.33 m | 3.33 m |
| 3″ | 3.90∼4.60d | 4.05 t (2.9) | 4.05 t (2.8) |
| 4″ | 3.90∼4.60d | 3.53 dd (9.2, 2.9) | 3.52 dd (9.2, 2.9) |
| 5″ | 3.90∼4.60d | 3.67 m | 3.67 m |
| 6″ | 4.37 m | 3.84 m | 3.84 m |
| 4.30 m | 3.68 m | 3.69 m | |
| 5-OH | 5.88 s | ||
| 11-OH | 5.77 d (4.7) | ||
| 14-OH | 5.53 s |
in pyridine-d5.
in CD3OD.
δ (ppm) 500 MHz.
overlapped resonances not assigned.
Table 2.
13C NMR Data of Compounds 1-2c
 | |||
|---|---|---|---|
| no. | 1a | 1b | 2b |
| 1 | 22.7 | 22.4 | 21.3 |
| 2 | 27.6 | 27.7 | 27.1 |
| 3 | 74.8 | 76.2 | 75.5 |
| 4 | 35.3 | 35.9 | 35.7 |
| 5 | 76.3 | 77.5 | 75.5 |
| 6 | 37.3 | 37.0 | 38.7 |
| 7 | 25.0 | 25.1 | 25.5 |
| 8 | 40.8 | 41.1 | 42.2 |
| 9 | 45.1 | 45.2 | 46.1 |
| 10 | 45.4 | 45.7 | 56.7 |
| 11 | 68.7 | 69.1 | 68.5 |
| 12 | 50.7 | 50.6 | 50.1 |
| 13 | 51.0 | 51.8 | 51.0 |
| 14 | 85.0 | 85.9 | 85.4 |
| 15 | 33.5 | 33.5 | 33.1 |
| 16 | 28.0 | 28.0 | 28.0 |
| 17 | 51.4 | 51.1 | 51.5 |
| 18 | 18.2 | 17.8 | 17.6 |
| 19 | 65.6 | 65.9 | 211.1 |
| 20 | 174.8 | 177.2 | 177.2 |
| 21 | 74.0 | 75.5 | 75.5 |
| 22 | 118.1 | 118.1 | 118.1 |
| 23 | 175.8 | 177.7 | 177.5 |
| 1′ | 99.3 | 99.5 | 99.5 |
| 2′ | 65∼80d | 72.3 | 72.4 |
| 3′ | 65∼80d | 72.2 | 72.2 |
| 4′ | 83.8 | 84.0 | 84.0 |
| 5′ | 65∼80d | 69.9 | 69.9 |
| 6′ | 18.7 | 18.3 | 18.3 |
| 1″ | 104.3 | 103.7 | 103.7 |
| 2″ | 65∼80d | 72.4 | 72.4 |
| 3″ | 65∼80d | 73.3 | 73.3 |
| 4″ | 65∼80d | 68.6 | 68.6 |
| 5″ | 65∼80d | 75.4 | 75.4 |
| 6″ | 63.0 | 62.8 | 62.8 |
in pyridine-d5.
in CD3OD.
δ (ppm) 100 MHz.
resonances not assigned because of overlapping
1H NMR resonances in the HMBC spectrum.
The complete 1H and 13C NMR signal assignments and connectivity were determined from a combination of COSY, TOCSY, HSQC and HMBC data and comparison with the spectra of known cardenolides.9
COSY and TOCSY correlations established three spin systems, which were H2-1–H2-2–H-3–H2-4 in ring A, H2-6–H2-7–H-8–H–9–H-11–H2-12 in rings B and C, and H2-15–H2-16–H-17 in ring D (Figure 1). Further assembly of rings A-D and the α,β-unsaturated γ-lactone of the aglycone was determined on the basis of HMBC correlations. HMBC correlations of H2-19 to C-1, C-5 and C-9, H2-1 to C-9, and H2-6 to C-5 established the connectivity of rings A and B. Correlations of H2-12, H2-16, H-17 and H3-18 to C-13 and of H2-16 and H3-18 to C-14 indicated the connectivity of rings C and D. In the meantime, an HMBC correlation of H2-16 to C-20 suggested the location of the lactone ring at C-17 (Figure 1).
Figure 1.

Key COSY (bold) and HMBC (arrows) correlations of 1.
These correlations established the flat structure of the aglycone of 1. The structures of the sugar moieties of 1 were determined by 1D TOCSY, COSY and HMBC data. Two sugar units was clearly shown by COSY and TOCSY correlations of two spin systems, H-1′–H-2′ –H-3′ –H-4′ –H-5′ –H3-6′ and H-1″–H-2″ –H-3″ –H-4″ –H-5″ –H2-6″ (Figure 1). An HMBC correlation of H-3 to C-1′ indicated the sugars were connected to the aglycone at C-3. In the meantime, HMBC correlations of H-1″ to C-4′ and of H-4′ to C-1″ established that the two sugars were connected from C-1″ to C-4′ (Figure 1).
The relative configuration of the aglycone of 1 was established by analysis of its ROESY correlations and calculation of coupling constants (Figure 2, Table 1). The ROESY correlations of H3-18 to H-22, H2-21, H-8 and H-11, and of H2-19 to H-8 and H-11 indicated that the C-17 side chain, H-8, H-11 and the oxymethylene at C-10 were all β-oriented. Calculation of coupling constants of H-11 (td, J = 9.8, 4.2 Hz) suggested that the adjacent H-9 occupied the α-orientation. The above assignments indicated that the B/C ring junction was trans-fused. ROESY spectra of 1 obtained in pyridine-d5 showed a correlation of C-14-OH to H3-18 that suggested a cis-fused ring junction of rings C and D. Determination of the relative configuration of the remaining portions of 1 was carried out by comparison with literature data.9,8l These comparisons indicated that rings A and B were connected by cis-fused ring junctions and that the C-3 side chain (sugar moiety) existed in the β-orientation. The relative configuration of the sugar moiety was established mainly by calculation of coupling constants and was further proved by ROESY correlations (Table 1, JFigure 2). Coupling constants of H-1′ (d, = 8.0 Hz), H-3′ (t, J = 3.0 Hz) and H-4′ (dd, J = 9.6, 3.0 Hz) indicated that H-1′, H-2′, H-4′ and H-5′ were in the axial orientation, and that H-3′ was in the equatorial orientation. Coupling constants of H-1″ (d, J = 7.6 Hz), H-3″ (t, J = 2.9 Hz) and H-4″ (dd, J = 9.2, 2.9 Hz) suggested that H-1″, H-2″, H-4″ and H-5″ were in the axial orientation, and that H-3″ was in the equatorial orientation. Those assignments were proved by ROESY correlations of H-1′ to H-5′, H-1″ to H-5″ and H-2″ to H-4″. In addition, the structure of the sugar moiety of 1 was confirmed by comparison with the known compounds sarmentogenin-3β-O-[β-allosyl-(1→4)-β-6-deoxyalloside and securigenin-3β-O-[β-allosyl-(1→4)-β-6-deoxyalloside.8l The sugar moieties of 1 had identical 1H and 13C NMR data to those of the reported compounds. Therefore, the structure and configuration of 1 was determined as sarmentologenin-3β-O-[β-allosyl-(1→4)-β-6-deoxyalloside.
Figure 2.

Key ROESY correlations of 1
Elaeodendroside W (2) was obtained as a white amorphous solid. Its molecular formula was established as C35H52O16, which was two units less than that of 1, on the basis of a sodiated molecular ion peak at m/z 751.311 in its MALDI-TOF/TOF mass spectrum. The 1H NMR spectrum of 2 showed characteristic signals of a α,β-unsaturated γ-lactone (δH 5.00, br d, J = 18.4 Hz, H-21a, δH 4.92, br d, J = 18.4 Hz, H-21b, and δH 5.91, s, H-22) (Table 1). Comparison of the 1H NMR and 13C NMR spectra of compounds 1 and 2 showed that they were very similar, but that the oxymethylene resonances (δH 4.18, d, J = 11.2 Hz, H-19a, δH 3.80, m, H-19b and δC 65.9, C-19) that appeared in the spectra of 1 were absent in the spectra of 2, and that the aldehyde resonances (δH 9.97, s, H-19 and δC 211.1, C-19) that appeared in the spectra of 2 were absent in the spectra of 1 (Tables 1 and 2). Those data indicated that 2 had a similar structure to 1 except that 2 has an aldehyde instead of a hydroxymethyl group at C-19. Further comparison of the 1H and 13C NMR spectra of 2 and the known cardenolide glycoside, sarmentosigenin-3β-O-β-6-deoxyguloside, confirmed the assignments, since the 1H NMR and 13C NMR data of the aglycone of 2 were identical to the literature data for sarmentosigenin. The structure of 2 was further confirmed by analysis of 2D NMR spectra including COSY, HMQC, HMBC and ROESY spectra. Therefore, the structure and configuration of 2 was determined as sarmentosigenin-3β-O-[β-allosyl-(1→4)-β-6-deoxyalloside].
All of the isolated compounds were tested for antiproliferative activity against the A2780 human ovarian cancer cell line and the U937 human histiocytic lymphoma cell line. It was found that both 1 and 2 showed significant antiproliferative activity, with IC50 values of 0.12 and 0.07 μM against the A2780 human ovarian cancer cell line and 0.15 and 0.08 μM against the U937 human histiocytic lymphoma cell line, respectively. The antiproliferative activities of 1 and 2 do not appear to be correlated with the oxidation status of C-19 because their IC50 values were very close to each other. The known cardenolide glycoside, sarmentosigenin-3β-O-β-6-deoxyguloside, which possesses the same aglycone as 2 but contains a 6-deoxygulose, also showed significant cytotoxicity with an IC50 value of 0.074 μM against a KB cell line,8l while another cardenolide with an identical aglycone to 2 but a glycosylated with rhamnose instead of allose also showed significant cytotoxicity with an IC50 value of 0.049 μM (0.028 μg/mL) against the HSG cell line.10 Those data suggested that compounds with the same skeletons as 1 and 2 might show significant activities against cultured cancer cells. The cytotoxicity and antiproliferative activities of many structurally diverse cardenolide glycosides against cultured tumor cells have been widely investigated. Some recent reports are cited.8m,8n,11,12 This class of compounds has not however found any clinical applications as anticancer agents, in part because of unfavorable toxicity profiles.
3. Experimental Section
General Experimental Procedures
Optical rotations were recorded on a JASCO P-2000 polarimeter. IR and UV spectra were performed on MIDAC M-series FTIR and Shimadzu UV-1201 spectrophotometers, respectively. NMR spectra were obtained on JEOL Eclipse 500, Varian Inova 400, and Varian Unity 400 spectrometers. Mass spectra were obtained on a JEOL-JMS-HX-110 and an Applied Biosystems 4800 MALDI TOF/TOF instruments.
Chemical shifts are given in δ (ppm), and coupling constants (J) are reported in Hz. HPLC was performed Shimadzu LC-10A pumps coupled with a Varian Dynamax semipreparative C18 column (250 × 10 mm). Both HPLC instruments employed a Shimadzu SPD-M10A diode array detector.
Antiproliferative Bioassay
The A2780 ovarian cancer cell line assay was performed at Virginia Polytechnic Institute and State University as previously reported.13 The A2780 cell line is a drug-sensitive ovarian cancer cell line.14
The U937 human histiocytic lymphoma cell line assay was performed at Eisai Research Institute. The cells were cultured in 96-well plates in the absence or continuous presence of 0.005 to 10 ug/ml extract for 96 hours. Cell growth was assessed using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega) according to manufacturer's recommendations. Luminescence was read on the EnVision 2102 Multilabel Reader (Perkin-Elmer). IC50 values were determined as the concentration of an extract at which cell growth was inhibited by 50% compared to untreated cell population. Two independent repeating experiments were preformed.
Plant Material
Root, stem, and leaf samples of Elaeodendron alluaudianum H. Perrier (Celastraceae) were collected in the forest of Bemosa, a dense humid forest, in orthern Madagascar, at an elevation 200 m, at 13.14.17 S, 49.37.50 E, on November 2, 2005. The tree was 10 m high with diameter at breast height of 12 cm and white flowers. It was identified by R. H. Archer (South African National Biodiversity Institute); its assigned collector number is Randrianaivo et al. 1281.
Extraction and Isolation
The stems of the dried plant sample described above (270 g) were extracted with EtOH to give 5.78 g of extract designated MG 3593. A total of 1.63 g of extract was supplied to VPISU, and this had an IC50 value of 3.3 μg/mL against A2780 cells. A portion of this extract (96 mg) was suspended in 20 mL of 30% MeOH/CH2Cl2 and filtered. The filtrate was evaporated to afford 67 mg residue (IC50 0.14 μg/mL). The residue was suspended in aqueous MeOH (90% MeOH/H2O, 10 mL), and extracted with n-hexane (3 × 10 mL). The aqueous layer was then diluted to 60% MeOH (v/v) with H2O and extracted with CH2Cl2 (3 × 15 mL). The aqueous MeOH extract (44 mg) was found to be the most active (IC50 0.50 μg/mL) and a portion of this (39 mg) was loaded on a C18 SPE cartridge and eluted with solvent systems of 30% MeOH/H2O, 70% MeOH/H2O and MeOH to obtain 3 fractions (I-III). The most active fraction was Fraction I (IC50 0.14 μg/mL), and this was separated via semipreparative HPLC over a C18 column using MeOH-H2O (75:25) to afford 12 fractions (IV-XV). Fraction XII afforded elaeodendroside V (1, 5.3 mg, tR 35.7 min), and fraction XIV afforded elaeodendroside W (2, 2.4 mg, tR 44.1 min).
Elaeodendroside V (1) white amorphous solid; [α]D23 +1.6 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 215 (4.31) nm; IR: νmax 3382, 2944, 2828, 1732, 1028 cm-1; 1H and 13C NMR spectra, see Tables 1 and 2; HRFABMS m/z 731.3496 [M+1]+ (calcd for C35H55O16, 731.3490).
Elaeodendroside W (2): white amorphous solid; [α]D23 +1.6 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 214 (4.34) nm; IR: νmax 3382, 2945, 2833, 1731, 1026 cm-1; 1H and 13C NMR spectra, see Tables 1 and 2; MALDI-TOF/TOF-MS m/z 751.311 [M+Na]+ (calcd for C35H52O16Na, 751.315).
Acknowledgments
This project was supported by the Fogarty International Center, the National Cancer Institute, the National Science Foundation, the National Heart Lung and Blood Institute, the National Institute of Mental Health, the Office of Dietary Supplements, and the Office of the Director of NIH, under Cooperative Agreement U01 TW00313 with the International Cooperative Biodiversity Groups. This support is gratefully acknowledged. We also thank Mr. T. Glass and Mr. B. Bebout from the Chemistry Department at Virginia Polytechnic Institute and State University (Virginia Tech) and Dr. Keith Ray from the Virginia Tech Mass Spectrometry Incubator for obtaining NMR and HRMS spectra. We thank Dr. Robert H. Archer (South African National Biodiversity Institute, South Africa) for assistance with the plant identification.
References
- 1.Biodiversity Conservation and Drug Discovery in Madagascar, Part 34. For Part 33, see Cao S, Brodie PJ, Miller JS, Birkinshaw C, Rakotondrafara A, Andriantsiferana R, Rasamison VE, Kingston DGI. Antiproliferative Compounds of Helmiopsis sphaerocarpa from the Madagascar Rainforest. Nat Prod Res. doi: 10.1080/14786410802200479. submitted for publication
- 2.Hou Y, Cao S, Brodie PJ, Miller JS, Birkinshaw C, Ratovoson F, Andriantsiferana R, Rasamison VE, Kingston DGI. J Nat Prod. 2008;71:150–152. doi: 10.1021/np070437l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao S, Brodie PJ, Miller JS, Ratovoson F, Callmander M, Randrianasolo S, Rakotobe E, Rasamison VE, Kingston DGI. J Nat Prod. 2007;70:1064–1066. doi: 10.1021/np0701428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Archer RH, Van Wyk AE. S Afr J Bot. 1998;64:93–109. [Google Scholar]; (b) Islam MB, Simmons MP, Archer RH. Syst Bot. 2006;31:512–524. [Google Scholar]
- 5.(a) Weeratunga G, Bohlin L, Sandberg F, Kumar V. Acta Pharm Suecica. 1984;21:73–76. [PubMed] [Google Scholar]; (b) Weeratunga G, Bohlin L, Verpoorte R, Kumar V. Phytochemistry. 1985;24:2093–2095. [Google Scholar]
- 6.(a) Anjaneyulu ASR, Narayana Rao M. Phytochemistry. 1980;19:1163–1169. [Google Scholar]; (b) Weeratunga G, Kumar V, Sultanbawa MUS, Balasubramaniam S. J Chem Soc, Perkin Trans 1: Org Bioorg Chem. 1982:2457–2459. [Google Scholar]; (c) Weeratunga G, Kumar V, Sultanbawa MUS. Tetrahedron Lett. 1982;23:2031–2032. [Google Scholar]; (d) Weeratunga G, Kumar V, Sultanbawa MUS. Aust J Chem. 1983;36:1067–1072. [Google Scholar]; (e) Weeratunga G, Kumar V. Phytochemistry. 1985;24:2369–2372. [Google Scholar]; (f) Kubo I, Fukuhara K. J Nat Prod. 1990;53:968–971. [Google Scholar]; (g) Tsanuo MK, Hassanali A, Jondiko IJO, Torto B. Phytochemistry. 1993;34:665–667. [Google Scholar]
- 7.(a) Kupchan SM, Uchida I, Shimada K, Fei BY, Stevens DM, Sneden AT, Miller RW, Bryan RF. J Chem Soc Chem Comm. 1977:255–256. [Google Scholar]; (b) Shimada K, Nambara T, Uchida I, Kupchan SM. Heterocycles. 1979;12:1445–1448. [Google Scholar]; (c) Anjaneyulu ASR, Rao MN. Indian J Chem, Sect B. 1980;19B:944–949. [Google Scholar]; (d) Shimada K, Kyuno T, Nambara T, Uchida I. Heterocycles. 1981;15:355–360. [Google Scholar]; (e) Shimada K, Kyuno T, Nambara T, Uchida I. Chem Pharm Bull. 1982;30:4075–4081. doi: 10.1248/cpb.30.4075. [DOI] [PubMed] [Google Scholar]; (f) Shimada K, Kyuno T, Nambara T, Uchida I. Phytochemistry. 1985;24:1345–1350. [Google Scholar]; (g) Shimada K, Ishii N, Ohishi K, Ro JS, Nambara T. J Pharmacobio-Dyn. 1986;9:755–759. doi: 10.1248/bpb1978.9.755. [DOI] [PubMed] [Google Scholar]; (h) Shimada K, Masuda H, Ohtaki H, Kobayashi N, Nambara T. Heterocycles. 1990;30:441–450. [Google Scholar]; (i) Tsujino Y, Ogoche JIJ, Tazaki H, Fujimori T, Mori K. Phytochemistry. 1995;40:753–756. [Google Scholar]; (j) Kasai HF, Kawai K, Shimada K. Heterocycles. 2000;53:2689–2700. [Google Scholar]
- 8.(a) Hoffmann JJ, Cole JR. J Pharm Sci. 1977;66:1336–1338. doi: 10.1002/jps.2600660935. [DOI] [PubMed] [Google Scholar]; (b) Hembree JA, Chang CJ, McLaughlin JL, Peck G, Cassady JM. J Nat Prod. 1979;42:293–298. [Google Scholar]; (c) Koike K, Bevelle C, Talapatra SK, Cordell GA, Farnsworth NR. Chem Pharm Bull. 1980;28:401–405. doi: 10.1248/cpb.28.401. [DOI] [PubMed] [Google Scholar]; (d) Wagner H, Habermeier H, Schulten HR. Helv Chim Acta. 1984;67:54–64. [Google Scholar]; (e) Kamano Y, Sato N, Nakayoshi H, Pettit GR, Smith CR. Chem Pharm Bull. 1988;36:326–332. doi: 10.1248/cpb.36.326. [DOI] [PubMed] [Google Scholar]; (f) Kaneda N, Chai H, Pezzuto JM, Kinghorn AD, Farnsworth NR, Tuchinda P, Udchachon J, Santisuk T, Reutrakul V. Planta Med. 1992;58:429–431. doi: 10.1055/s-2006-961506. [DOI] [PubMed] [Google Scholar]; (g) Baek NI, Lee YH, Park JD, Kim SI, Ahn BZ. Planta Med. 1994;60:26–29. doi: 10.1055/s-2006-959401. [DOI] [PubMed] [Google Scholar]; (h) Decosterd L, Gustafson KR, Cardellina JH, II, Cragg GM, Boyd MR. Phytotherapy Res. 1994;8:74–77. [Google Scholar]; (i) Gil RR, Lin LZ, Chai HB, Pezzuto JM, Cordell GA. J Nat Prod. 1995;58:848–856. doi: 10.1021/np50120a005. [DOI] [PubMed] [Google Scholar]; (j) Kitanaka S, Takido M, Mizoue K, Nakaike S. Chem Pharm Bull. 1996;44:615–617. doi: 10.1248/cpb.44.615. [DOI] [PubMed] [Google Scholar]; (k) Kiuchi F, Fukao Y, Maruyama T, Obata T, Tanaka M, Sasaki T, Mikage M, Haque ME, Tsuda Y. Chem Pharm Bull. 1998;46:528–530. doi: 10.1248/cpb.46.528. [DOI] [PubMed] [Google Scholar]; (l) Ankli A, Heilmann J, Heinrich M, Sticher O. Phytochemistry. 2000;54:531–537. doi: 10.1016/s0031-9422(00)00144-8. [DOI] [PubMed] [Google Scholar]; (m) Laphookhieo S, Cheenpracha S, Karalai C, Chantrapromma S, Rat-a-pa Y, Ponglimanont C, Chantrapromma K. Phytochemistry. 2004;65:507–510. doi: 10.1016/j.phytochem.2003.10.019. [DOI] [PubMed] [Google Scholar]; (n) Cheenpracha S, Karalai C, Rat-a-Pa Y, Ponglimanont C, Chantrapromma K. Chem Pharm Bull. 2004;52:1023–1025. doi: 10.1248/cpb.52.1023. [DOI] [PubMed] [Google Scholar]; (o) Roy MC, Chang FR, Huang HC, Chiang MYN, Wu YC. J Nat Prod. 2005;68:1494–1499. doi: 10.1021/np0501740. [DOI] [PubMed] [Google Scholar]; (p) Lhinhatrakool T, Sutthivaiyakit S. J Nat Prod. 2006;69:1249–1251. doi: 10.1021/np060249f. [DOI] [PubMed] [Google Scholar]
- 9.Kawaguchi K, Asaka I, Hirotani M, Furuya T, Katsuki S. Phytochemistry. 1993;34:1317–1321. [Google Scholar]
- 10.Higano T, Kuroda M, Sakagami H, Mimaki Y. Chem Pharm Bull. 2007;55:337–339. doi: 10.1248/cpb.55.337. [DOI] [PubMed] [Google Scholar]
- 11.Cao S, Brodie PJ, Miller JS, Ratovoson F, Callmander MW, Randrianasolo S, Rakotobe E, Rasamison VE, Suh EM, TenDyke K, Kingston DGI. J Nat Prod. 2007;70:1064–1067. doi: 10.1021/np0701428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamed AI, Plaza A, Balestrieri ML, Mahalel UA, Springuel IV, Oleszek W, Pizza C, Piacente S. J Nat Prod. 2006;69:1319–1322. doi: 10.1021/np060228l. [DOI] [PubMed] [Google Scholar]
- 13.Cao S, Brodie PJ, Randrianaivo R, Ratovoson F, Callmander M, Andriantsiferana R, Rasamison VE, Kingston DGI. J Nat Prod. 2007;70:679–681. doi: 10.1021/np060627g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Louie KG, Behrens BC, Kinsella TJ, Hamilton TC, Grotzinger KR, McKoy WM, Winker MA, Ozols RF. Cancer Res. 1985;45:2110–2115. [PubMed] [Google Scholar]