

Abstract
Hyperphosphorylation of the microtubule associated protein tau is detected in the brains of individuals with a range of neurodegenerative diseases including Alzheimer's disease (AD). An imbalance in phosphorylation and/or dephosphorylation of tau at disease-related sites has been suggested to initiate the abnormal metabolism and toxicity of tau in disease pathogenesis. However, the mechanisms underlying abnormal phosphorylation of tau in AD are not fully understood. Here, we show that the DNA damage-activated Checkpoint kinase 2 (Chk2) is a novel tau kinase and enhances tau toxicity in a transgenic Drosophila model. Overexpression of Drosophila Chk2 increases tau phosphorylation at Ser262 and enhances tau-induced neurodegeneration in transgenic flies expressing human tau. The non-phosphorylatable Ser262Ala mutation abolishes Chk2-induced enhancement of tau toxicity, suggesting that the Ser262 phosphorylation site is involved in the enhancement of tau toxicity by Chk2. In vitro kinase assays revealed that human Chk2 and a closely related checkpoint kinase 1 (Chk1) directly phosphorylate human tau at Ser262. We also demonstrate that Drosophila Chk2 does not modulate the activity of the fly homolog of microtubule affinity regulating kinase, which has been shown to be a physiological tau Ser262 kinase. Since accumulation of DNA damage has been detected in the brains of AD patients, our results suggest that the DNA damage-activated kinases Chk1 and Chk2 may be involved in tau phosphorylation and toxicity in the pathogenesis of AD.
INTRODUCTION
Tau is a microtubule associated protein that is expressed in neurons and is predominantly localized in axons. The major function of tau is to regulate the assembly and stability of microtubules (1). Abnormally phosphorylated tau is found in the intracellular protein inclusions called neurofibrillary tangles (NFTs), which are associated with Alzheimer's disease (AD) and also with a range of neurodegenerative diseases called tauopathies (1–5). The occurrence of fibrillar tau inclusions in tauopathies, and the correlation of NFT density with cognitive decline in AD, suggests that the tau abnormality plays a key role in the observed clinical symptoms (6).
In vitro and in vivo studies have demonstrated that tau phosphorylation at some of the disease associated sites plays critical roles in the physiological and pathophysiological functions of tau. In vitro studies have revealed that phosphorylation at Ser262, Thr231 and Ser235 reduces tau binding to microtubules (7–9), and phosphorylation or pseudophosphorylation of a number of sites enhances tau fibril formation (6,10–12). In transgenic mice, overexpression of kinases that phosphorylate tau, including GSK-3β and cyclin-dependent kinase-5, modify tau phosphorylation, NFT formation and tau toxicity (13–19). These data suggest that an imbalance in phosphorylation and/or dephosphorylation of tau initiates the abnormal metabolism and toxicity of tau in disease pathogenesis (20).
Human tau toxicity has been modeled in transgenic Drosophila. Neuronal expression of human wild-type tau or tau with disease-associated mutations causes synaptic dysfunctions, memory defects and an age-dependent progressive neurodegeneration in the fly brain (21–26). Tau expression in fly eyes induces retinal toxicity in adult flies, which appears as a ‘rough’ eye phenotype, characterized by reduced external eye size, reduced thickness of the retina and a loss of the regular ommatidial organization (21,22). Accumulation of disease-associated conformational changes and phospho-epitopes in tau has been detected in the brain and eyes (21–23). NFT formation, a hallmark of both AD pathology and tauopathies, is not observed in fly neurons (23), indicating that tau toxicity is not conferred by large insoluble aggregates of tau in the Drosophila models. These results suggest that Drosophila models of tauopathies may recapitulate early, pre-tangle events in tau-associated neurodegeneration (27).
The transgenic Drosophila models have been used to address the role of distinct tau phosphorylation sites in controlling tau neurotoxicity (21–23,28–32). Studies of transgenic flies expressing tau mutants that contain phosphorylation-incompetent Ser/Thr kinase sites have revealed that mutating all 14 proline-directed kinase target sites (SP/TP sites), which are hyperphosphorylated in NFTs, significantly reduces tau toxicity. However, no single phosphorylation site among the SP/TP sites plays a dominant role in controlling tau toxicity (31).
Enhanced phosphorylation of tau at Ser262/356 is detected in pre-tangle neurons in AD (33). The introduction of alanine mutations at Ser262 and Ser356 dramatically reduces tau-induced neurodegeneration in fly eyes and brains (22), indicating that these sites play critical roles in tau toxicity. Ser262/356 can be phosphorylated by microtubule affinity regulating kinases (MARKs) (34), and in the fly tauopathy model, overexpression of the Drosophila homolog of MARK (dMARK, also called partitioning defective-1, PAR-1) increases tau phosphorylation at Ser262/356 and enhances tau toxicity (22,32). In contrast, overexpression of MARK rescues axonal transport defects and synaptic degeneration caused by overexpression of tau in hippocampal neurons (35,36). Whether other kinases can phosphorylate tau at Ser262/356 in vivo and control its toxicity is not clear.
Accumulation of DNA damage in neurons has been observed in normal aging and in AD (37–39). Key components of the DNA damage response are the central transducing kinases ataxia telangiectasia mutated (ATM) and ATR (ATM and Rad3-related), and the checkpoint effector kinases checkpoint kinase 1 (Chk1) and checkpoint kinase 2 (Chk2) (40,41). Chk1 activation is primarily downstream of ATR in response to detection of single-stranded DNA, although Chk2 is activated primarily by ATM in response to double strand breaks in DNA. Chk1 and Chk2 are Ser/Thr kinases and phosphorylate many proteins that influence the DNA damage response, including cell cycle checkpoint regulators, DNA repair and replication fork maintenance proteins, transcriptional regulators and apoptosis inducing proteins, as well as proteins that influence chromatin dynamics (40,41). In addition, recent studies have shown that Chk1 and Chk2 phosphorylate proteins involved in the circadian clock systems (42). Whether Chk1 and Chk2 phosphorylate tau in vitro and in vivo has not been previously determined.
In this study, we demonstrate that the DNA damage-activated checkpoint kinases Chk1 and Chk2 are novel tau Ser262 kinases, and overexpression of Chk2 enhances tau toxicity in vivo.
RESULTS
Overexpression of Drosophila Chk2 enhances human tau-induced retinal degeneration in flies
The activation of DNA repair is a tightly regulated process that is conserved across species (40). Chk2 is activated by DNA double strand break and plays a critical role in repair responses in mammals and in Drosophila (43–46). Drosophila Chk2, as well as human Chk2, mouse Chek2 and rat Chek2, contain a ‘Serine/threonine-protein kinase Chk2 domain’ (InterPro) (47), and belong to the ‘Chk2’ category by the ‘Panther classification system’ (48). Drosophila Chk2 protein is critical for radiation-induced apoptosis and cell cycle arrest (43) and the mitotic response to DNA damage and replication defects (46). These studies indicate that Drosophila Chk2 is also a functional homolog of mammalian Chk2. Using transgenic Drosophila expressing human wild-type 0N4R tau in fly eyes (23), we tested whether overexpression of Drosophila Chk2 increases human tau toxicity.
Human tau overexpression using the pan-retinal gmr-GAL4 driver caused eye degeneration, characterized by a rough surface and smaller eye size [(30) and Fig. 1B]. Overexpression of Drosophila Chk2 dramatically enhanced human tau toxicity in fly eyes (Fig. 1C). It has been reported that Chk2 overexpression itself does not cause apoptosis in fly eyes (44). We also found that the overexpression of Chk2 itself did not cause obvious eye degeneration (Fig. 1D), indicating that the observed enhancement of tau toxicity by Chk2 overexpression is not merely an additive effect.
Figure 1.

Enhancement of human tau-induced retinal degeneration by overexpression of Drosophila Chk2. (A) Eye size is normal in a control fly with the pan-retinal gmr-GAL4 driver only. (B) Expression of human tau in the eye reduces eye size. (C) Flies co-expressing tau and Chk2 have smaller eyes than flies expressing tau alone. (D) Expression of Chk2 alone does not change eye size. (E) Expression of expanded polyglutamine repeat peptides (108 polyQ repeat peptides) in the eye causes depigmentation. (F) Expression of Chk2 does not enhance eye phenotype induced by expanded polyglutamine repeat peptides. (G) Expression of two copies of Alzheimer's amyloid β 42 peptides with Arctic mutation (50) in the eye causes degeneration. (H) Expression of Chk2 does not enhance eye degeneration induced by Alzheimer's amyloid-β 42 peptides with Arctic mutation.
We also tested whether overexpression of Drosophila Chk2 increases eye degeneration caused by other neurodegenerative disease-associated peptides including expanded polyglutamine repeat peptides (108 polyQ repeat peptides with Myc/FLAG tag) or Alzheimer's amyloid-β 42 peptides (49,50). Overexpression of Drosophila Chk2 did not enhance neurodegeneration caused by these peptides in fly eyes (Fig. 1E–H), suggesting that the effect of Chk2 overexpression on tau toxicity is not due to non-specific enhancement of toxicity induced by aggregation-prone proteins.
Overexpression of Drosophila Chk2 increases phosphorylation of human tau at Ser262
Tau is abnormally phosphorylated in the brains of individuals with AD (2–5), and an imbalance in phosphorylation and/or dephosphorylation of tau at disease-related sites has been suggested to initiate the toxicity of tau. Among AD-related phosphorylation sites, tau phosphorylation at Ser262 has been reported to play a critical role in tau toxicity (22,32).
We examined whether overexpression of Drosophila Chk2 increases phosphorylation of tau at Ser262. Human tau and Drosophila Chk2 were expressed in fly eyes using the pan-retinal gmr-GAL4 driver. Using phospho-specific antibodies, we found that co-expression of Chk2 significantly increased tau phosphorylation at Ser262 (Fig. 2). We also tested whether Chk2 overexpression increases phosphorylation of tau at other AD-related sites including Ser202 and Thr231. In contrast to Ser262, Chk2 overexpression did not increase tau phosphorylation at these sites (Fig. 2). We verified the specificity of the phospho-tau-antibodies as shown in Supplementary Material, Fig. S1.
Figure 2.

Tau phosphorylation levels increase at Ser262, but not at Ser202 or Thr231, when Chk2 is co-expressed in fly eyes. Fly heads expressing human tau alone (tau) or with Drosophila Chk2 (tau + Chk2) driven by the pan-retinal gmr-GAL4 driver were subjected to western blotting with anti-tau (human tau) or anti-phospho-tau antibodies. The phosphorylation levels at each site in fly heads co-expressing tau and Chk2 are normalized to the tubulin level and shown as a ratio relative to that in fly heads expressing tau alone. Asterisks indicate significant differences from control (n = 4 or 5, P < 0.05, Student's t-test). Representative blots are shown.
The MARKs have been shown to be a tau Ser262 kinase (34). Tau phosphorylation at Ser262 by overexpression of dMARK increases phosphorylation levels of some of the tau SP/TP phosphorylation sites, including Ser202 (22). In contrast, Chk2 overexpression increased Ser262 phosphorylation without affecting Ser202, indicating that the increase in phosphorylation at Ser262 is not always correlated with that at Ser202.
We also tested whether a genetic reduction of Chk2 ameliorates tau-induced retinal degeneration. A heterozygous loss-of-function mutation of Chk2 (43) did not modify retinal degeneration induced by tau (Supplementary Material, Fig. S2). Additionally, the heterozygous loss-of-function mutation of Chk2 did not change tau Ser262 phosphorylation levels (Supplementary Material, Fig. S2). These results indicate that loss of one copy of Chk2 is not sufficient to reduce tau phosphorylation at Ser262 and toxicity.
The non-phosphorylatable Ser262Ala tau mutation abolishes Chk2-induced enhancement of human tau toxicity
Since phosphorylation of tau at Ser262 plays a critical role in tau toxicity, we hypothesized that Chk2 enhances tau toxicity via tau phosphorylation at Ser262. To test this hypothesis, we established transgenic fly lines that express human 0N4R tau with a S262A mutation (S262A tau) at levels similar to the expression of wild-type tau (Fig. 3A). Previous reports have shown that the double mutant S262A/S356A reduces tau toxicity (22,32). We found that the single S262A mutation was sufficient to reduce tau toxicity (Fig. 3B–D). Using S262A transgenic flies, we found that co-expression of Chk2 and S262A tau did not cause a reduction in external eye size (Fig. 3D and E), suggesting that the Ser262 phosphorylation site is critical for Chk2-induced enhancement of tau toxicity.
Figure 3.
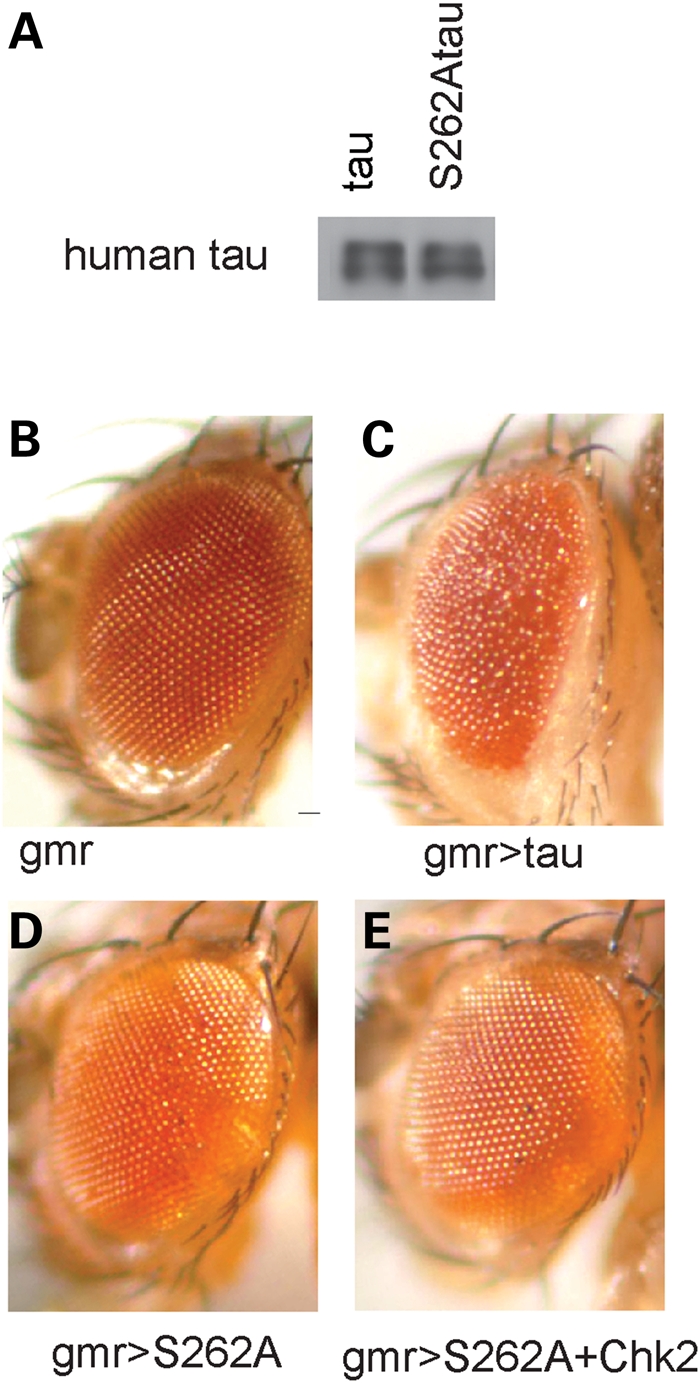
The S262A tau mutation greatly reduces tau toxicity and abolishes Chk2-induced enhancement of tau toxicity. (A) Establishment of transgenic flies expressing human 0N4Rtau with the S262A mutation in fly eyes with the pan-retinal gmr-GAL4 driver. Western blot of head lysates with anti-human tau show that human wild-type tau and human S262A tau are expressed at similar levels in the transgenic flies. (B–E) External eyes of flies carrying the gmr-GAL4 driver only (B), expressing wild-type human tau (C), expressing S262A tau (D) or flies co-expressing S262A tau and Drosophila Chk2 (E).
Human Chk1 and Chk2 directly phosphorylate recombinant human tau at Ser262 in vitro
Peptide library analyses have identified the Chk2 substrate target motif as R/F-R/Φ-L/I-K/R-R-X-X-S-F/I-F/I/R, although there are several exceptions to this consensus sequence (51,52). Although the sequence surrounding Ser262 (NVKSKIGS262TE) does not exactly match this consensus sequence, we tested whether recombinant active human Chk2 directly phosphorylates human tau at Ser262 in vitro. We found that recombinant active human Chk2 phosphorylated recombinant human tau at Ser262 (Fig. 4A and B). This phosphorylation was inhibited by a Chk2-specific inhibitor (Chk2-inhibitor II) in a dose-dependent manner (Fig. 4A and B).
Figure 4.

Checkpoint kinases directly phosphorylate human tau at Ser262 in vitro. Recombinant active human Chk2 (A and B) or recombinant active human Chk1 (C and D) were incubated with recombinant human tau in the presence or absence of inhibitors (Chk2 inhibitor II for Chk2 and UCN-01 for Chk1). Proteins were resolved by SDS-PAGE and visualized by western blotting using an anti-pSer262 antibody (A and C). The signals were quantified and the phosphorylation levels are shown as a ratio relative to that of recombinant tau incubated with recombinant kinase in the absence of inhibitor (B and D). Asterisks indicate significant differences (n = 4, P < 0.05, Student's t-test).
Since many substrates of the closely related kinase Chk1, which is activated by DNA single strand breaks, overlap with Chk2 substrates (40), we tested whether human recombinant Chk1 also phosphorylates human tau at Ser262 in vitro. We found that recombinant active human Chk1 directly phosphorylated tau at Ser262, and this phosphorylation was inhibited by a Chk1 inhibitor (UCN-01; Fig. 4C and D). These results demonstrate that DNA damage-activated checkpoint kinases, Chk1 and Chk2, are novel tau Ser262 kinases.
Chk2 overexpression does not activate Drosophila MARK
The MARKs have been shown to be a physiological tau Ser262 kinase (34,35). Studies in Drosophila have revealed that dMARK plays a critical role in tau toxicity via the Ser262 phosphorylation site in vivo (22,32). We tested whether dMARK was involved in increased phosphorylation of tau at Ser262 by overexpression of Chk2 in Drosophila.
dMARK activity depends on its phosphorylation at Thr408 (53). Overexpression of dMARK causes eye degeneration in flies, and co-expression of LKB1, an activator of dMARK, increases dMARK phosphorylation at Thr408 and enhances the dMARK-induced rough eye phenotype (53). To test whether dMARK phosphorylation at Thr408 was affected by Chk2 overexpression, a myc-tagged Drosophila dMARK was expressed alone or co-expressed with Chk2 in fly eyes using the pan-retinal gmr-GAL4 driver. Western blot with a phospho-specific antibody revealed that Chk2 co-expression did not increase dMARK phosphorylation at Thr408 (Fig. 5A). Furthermore, co-expression of Chk2 did not enhance the dMARK-induced eye degeneration (Fig. 5B). These results suggest that Chk2 does not modulate dMARK activity in fly eyes.
Figure 5.

Chk2 overexpression does not affect dMARK activity in fly eyes. A myc-tagged Drosophila dMARK was expressed alone or co-expressed with Drosophila Chk2 using the pan-retinal gmr-GAL4 driver. (A) Western blot with an anti-pT408 dMARK antibody (top) or an anti-myc antibody (bottom). The signals were quantified and the phosphorylation levels are shown as a ratio relative to that of flies expressing dMARK alone. No significant difference was detected (n = 4, P > 0.05, Student's t-test). Representative blots are shown. gmr: flies with the gmr-GAL4 driver only. The asterisk indicates a non-specific band. (B–D) External eyes from the gmr-GAL4 driver control (B), transgenic flies overexpressing dMARK alone (C) or flies co-expressing dMARK and Drosophila Chk2 (D). Chk2 overexpression did not enhance dMARK-induced eye degeneration.
DISCUSSION
Using a transgenic Drosophila model, we have shown that overexpression of Drosophila Chk2 increases human tau phosphorylation at Ser262 and enhances tau-induced neurodegeneration. We have also shown that the Ser262 phosphorylation site is involved in the enhancement of tau toxicity by Chk2. We further demonstrated that human Chk2 and the closely related DNA damage-activated kinase Chk1 directly phosphorylated human tau at Ser262 in vitro. Since accumulation of DNA damage has been detected in the brains of AD patients, our results suggest that checkpoint kinases may be involved in tau phosphorylation and toxicity in the pathogenesis of AD.
Chk2 enhanced tau-induced neurodegeneration, although Chk2 overexpression alone did not cause neurodegeneration (Fig. 1). These results indicate that Chk2-induced enhancement of tau toxicity is not merely an additive effect. Furthermore, Chk2 did not enhance eye degeneration caused by expanded polyglutamine repeat peptides or Alzheimer's amyloid β 42 peptides (Fig. 1), suggesting that the effect of Chk2 overexpression on tau toxicity is not due to non-specific enhancement of toxicity induced by aggregation-prone proteins. However, it has been shown that Chk2 phosphorylates p53 and enhances apoptosis during irradiation-induced or p53 overexpression-induced apoptosis (44). In addition, Chk2 phosphorylates Che-1, a RNA polymerase II binding protein that is involved in apoptosis and cell cycle control and also binds to tau (54–56). Thus, in addition to enhancement of tau toxicity through Ser262 phosphorylation, Chk2 overexpression may synergistically enhance tau-induced apoptosis downstream of tau phosphorylation.
Accumulation of oxidative damage in nucleic acids, as well as widespread single- and double-strand breaks, has been detected in neurons in the brains of patients with AD and with mild cognitive impairment, which possibly represents an early stage of AD (39,57–69). More DNA damage was found in the aging hippocampus, one of the vulnerable regions of the brain in AD, than in the aging cerebellum (70). In postmortem brains from patients with AD, the neurons that show NFT formation in AD are the same as those that show age-related accumulation of DNA damage (71). Increases in DNA repair gene expression have been reported in aged brains (37) and in brains from Down syndrome patients (72). Our results suggest that the increased activity of the DNA repair response in aging and under pathological conditions may initiate tau pathology and toxicity.
The accumulation of DNA damage in AD patients may be due to a decrease in the efficiency of DNA repair. Several groups have tested whether AD is accompanied by a deficiency in DNA repair enzymes. Some studies have confirmed a decrease in DNA repair efficiency (62,73–77), whereas other studies have not (78,79). In contrast, changes in DNA damage transducers, such as Chk1 and Chk2 in AD brains, have not been reported. It may be possible that a defect in the DNA repair machinery and failure to properly execute DNA repair may cause sustained activation of DNA damage sensors and transducers such as Chk1 and Chk2 in AD brains.
Oxidative stress induced by paraquat, a reactive oxygen species-generating chemical, has been reported to enhances tau toxicity by promoting tau-induced cell cycle re-entry in Drosophila models (80). Damage to nucleic acids caused by reactive oxygen species includes base modifications such as 8-hydroxydeoxyguanosine, single-strand breaks and double-strand breaks if single-strand breaks are in close proximity (81). Several studies indicate that the capacity for DNA repair is compromised in AD brains (62,73–77), and it is possible that reduced efficiency of DNA repair machinery in AD causes a failure of base-modification repair or single-strand break repair and leads to accumulation of double-strand breaks. It may be interesting to test whether Chk2 is involved in enhancement of tau toxicity induced by oxidative stress.
In summary, our results demonstrate that checkpoint kinases Chk1 and Chk2 are novel tau Ser262 kinases and overexpression of Chk2 enhances tau toxicity in vivo. Our results suggest that checkpoint kinases, whose inhibitors have entered early clinical development for cancer therapy (82), may be therapeutic targets for AD and tauopathies.
MATERIALS AND METHODS
Fly stocks and antibodies
The transgenic fly line carrying the human 0N4R tau was a kind gift from Dr Mel Feany (Harvard Medical School, MA, USA) (23). To establish transgenic fly lines carrying S262A mutant tau, human 0N4R tau [a kind gift from Dr Mike Hutton (Mayo Clinic Jacksonville)] was served as a template for the generation of alanine mutation. The codon for Ser262 was mutated to alanine using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA), and the resulting construct was subcloned into the pUAST Drosophila transformation vector and microinjected into fly embryos of the w1118 genotype. The authenticity of the construct was confirmed by sequencing, and lines with expression level equivalent to wild-type tau were determined by quantitative western blot analysis. The transgenic fly line carrying Alzheimer's amyloid-β 42 with Arctic mutation has been described previously (50). Other fly stocks were obtained from: Drs Wei Du (the University of Chicago, IL, USA) (Chk2[E51]), Bingwei Lu (Stanford University School of Medicine, CA, USA) (UAS-dPar1), Tak Mak (The Campbell Family Institute for Breast Cancer Research, Canada) (UAS-Chk2), Leslie Thompson (University of California, Irvine, CA, USA) (UAS-polyQ108) and the Bloomington Drosophila Stock Center (Indiana University, IN, USA) (gmr-GAL4). Crosses were maintained on standard cornmeal-based Drosophila medium at 25°C and analyzed at 1–3 days of age, except for the analysis of tau phosphorylation levels described in Figure 2. For this experiment, crosses were maintained at 18°C during developmental stages to reduce eye degeneration, since co-expression of tau and Chk2 overexpression causes severe reduction in tau protein levels presumably due to massive neurodegeneration when cultured at 25°C. After eclosion, adult flies were maintained at 25°C for 1 week to induce transgene expression and subjected to western blot analysis.
Western blotting
Antibodies were obtained from Drs Peter Davis (Albert Einstein College of Medicine, NY, USA) (CP13) and Bingwei Lu (Stanford University School of Medicine) (anti-pT408). Anti-myc (Invitrogen), Anti-tau (T46) (Zymed), anti-tau pS262 (Biosource and Calbiochem), anti-tau pT231 (AT180, Thermo Fisher Scientific, MA, USA) and Anti-tubulin (Sigma, St. Louis, MO, USA) was purchased. Fifteen fly heads for each genotype were collected at 1–3 day-after-eclosion and homogenized in SDS-Tris-Glycine sample buffer, separated by 6 or 10% Tris-Glycine gel and transferred to nitrocellulose membrane. The membranes were blocked with 5% milk (Nestle), blotted with the antibodies described above, incubated with appropriate secondary antibody and developed using ECL plus Western Blotting Detection Reagents (GE Healthcare). The signal intensity was quantified using ImageJ (NIH). Western blots were repeated a minimum of three times with different animals and representative blots are shown.
In vitro phosphorylation
Recombinant active human GST-tagged Chk1 (C0870, Sigma) and Chk2 (C0995, Sigma) were diluted to 1:2 or 1:5, respectively, mixed with 2 µg of recombinant human 0N4R tau (Sigma) in 5 mm MOPS, pH 7.2, 2.5 mm glycerol 2-phosphate, 5 mm MgCl2, 1 mm EGTA, 0.4 mm EDTA, 0.05 mm DTT and 5 µm ATP, with or without inhibitors [UCN-01 (Sigma) or Chk2 inhibitor II (Sigma)]. After incubation for 3 h at 30°C, samples were mixed with SDS-Tris-Glycine samples buffer and separated by 10% Tris-Glycine gel and transferred to nitrocellulose membrane. The membranes were blocked with 5% milk (Nestle), blotted with anti-pS262, incubated with anti-rabbit IgG-HRP and developed using ECL plus Western Blotting Detection Reagents (GE Healthcare). The signal intensity was quantified using ImageJ (NIH).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by start-up funds from the Farber Institute for Neurosciences, a pilot research grant from the Thomas Jefferson University, and grants from Gilbert Foundation/the American Federation for Aging Research, the Alzheimer's Association (NIRG-08-91985) and the National Institute of Health (R01AG032279-A1).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Peter Davis, Wei Du, Mel Feany, Mike Hutton, Bingwei Lu, Tak Mak, Leslie Thompson, Yi Zhong and the Bloomington stock center for fly stocks and antibodies. We thank Dr Mark Fortini for his insightful comments on the manuscript. We thank Dr Miki Fujioka for her help with establishment of transgenic fly strains. We thank Ms Linda Granger for technical helps.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Lee V.M., Goedert M., Trojanowski J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 2.Grundke-Iqbal I., Iqbal K., Quinlan M., Tung Y.C., Zaidi M.S., Wisniewski H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- 3.Grundke-Iqbal I., Iqbal K., Tung Y.C., Quinlan M., Wisniewski H.M., Binder L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kosik K.S., Joachim C.L., Selkoe D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1986;83:4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wood J.G., Mirra S.S., Pollock N.J., Binder L.I. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) Proc. Natl. Acad. Sci. USA. 1986;83:4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunden K.R., Trojanowski J.Q., Lee V.M. Advances in tau-focused drug discovery for Alzheimer's disease and related tauopathies. Nat. Rev. Drug Discov. 2009;8:783–793. doi: 10.1038/nrd2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biernat J., Gustke N., Drewes G., Mandelkow E.M., Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 8.Sengupta A., Kabat J., Novak M., Wu Q., Grundke-Iqbal I., Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch. Biochem. Biophys. 1998;357:299–309. doi: 10.1006/abbi.1998.0813. [DOI] [PubMed] [Google Scholar]
- 9.Cho J.H., Johnson G.V. Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J. Biol. Chem. 2003;278:187–193. doi: 10.1074/jbc.M206236200. [DOI] [PubMed] [Google Scholar]
- 10.Alonso A.C., Grundke-Iqbal I., Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 11.Necula M., Kuret J. Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro. J. Biol. Chem. 2004;279:49694–49703. doi: 10.1074/jbc.M405527200. [DOI] [PubMed] [Google Scholar]
- 12.Alonso Adel C., Mederlyova A., Novak M., Grundke-Iqbal I., Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J. Biol. Chem. 2004;279:34873–34881. doi: 10.1074/jbc.M405131200. [DOI] [PubMed] [Google Scholar]
- 13.Spittaels K., Van den Haute C., Van Dorpe J., Geerts H., Mercken M., Bruynseels K., Lasrado R., Vandezande K., Laenen I., Boon T., et al. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J. Biol. Chem. 2000;275:41340–41349. doi: 10.1074/jbc.M006219200. [DOI] [PubMed] [Google Scholar]
- 14.Engel T., Lucas J.J., Gomez-Ramos P., Moran M.A., Avila J., Hernandez F. Cooexpression of FTDP-17 tau and GSK-3beta in transgenic mice induce tau polymerization and neurodegeneration. Neurobiol. Aging. 2006;27:1258–1268. doi: 10.1016/j.neurobiolaging.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 15.Gomez-Sintes R., Hernandez F., Bortolozzi A., Artigas F., Avila J., Zaratin P., Gotteland J.P., Lucas J.J. Neuronal apoptosis and reversible motor deficit in dominant-negative GSK-3 conditional transgenic mice. EMBO J. 2007;26:2743–2754. doi: 10.1038/sj.emboj.7601725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noble W., Olm V., Takata K., Casey E., Mary O., Meyerson J., Gaynor K., LaFrancois J., Wang L., Kondo T., et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- 17.Plattner F., Angelo M., Giese K.P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 2006;281:25457–25465. doi: 10.1074/jbc.M603469200. [DOI] [PubMed] [Google Scholar]
- 18.Wen Y., Planel E., Herman M., Figueroa H.Y., Wang L., Liu L., Lau L.F., Yu W.H., Duff K.E. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J. Neurosci. 2008;28:2624–2632. doi: 10.1523/JNEUROSCI.5245-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gotz J., Gladbach A., Pennanen L., van Eersel J., Schild A., David D., Ittner L.M. Animal models reveal role for tau phosphorylation in human disease. Biochim. Biophys. Acta. 2009 doi: 10.1016/j.bbadis.2009.09.008. (in press). doi:10.1016/j.bbadis.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 20.Ballatore C., Lee V.M., Trojanowski J.Q. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 21.Jackson G.R., Wiedau-Pazos M., Sang T.-K., Wagle N., Brown C.A., Massachi S., Geschwind D.H. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 22.Nishimura I., Yang Y., Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–682. doi: 10.1016/s0092-8674(04)00170-9. [DOI] [PubMed] [Google Scholar]
- 23.Wittmann C.W., Wszolek M.F., Shulman J.M., Salvaterra P.M., Lewis J., Hutton M., Feany M.B. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 24.Mershin A., Pavlopoulos E., Fitch O., Braden B.C., Nanopoulos D.V., Skoulakis E.M. Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn. Mem. 2004;11:277–287. doi: 10.1101/lm.70804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chee F.C., Mudher A., Cuttle M.F., Newman T.A., MacKay D., Lovestone S., Shepherd D. Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol. Dis. 2005;20:918–928. doi: 10.1016/j.nbd.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 26.Mudher A., Shepherd D., Newman T.A., Mildren P., Jukes J.P., Squire A., Mears A., Drummond J.A., Berg S., MacKay D., et al. GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry. 2004;9:522–530. doi: 10.1038/sj.mp.4001483. [DOI] [PubMed] [Google Scholar]
- 27.Lee V.M., Kenyon T.K., Trojanowski J.Q. Transgenic animal models of tauopathies. Biochim. Biophys. Acta. 2005;1739:251–259. doi: 10.1016/j.bbadis.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 28.Shulman J.M., Feany M.B. Genetic modifiers of tauopathy in Drosophila. Genetics. 2003;165:1233–1242. doi: 10.1093/genetics/165.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fulga T.A., Elson-Schwab I., Khurana V., Steinhilb M.L., Spires T.L., Hyman B.T., Feany M.B. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- 30.Steinhilb M.L., Dias-Santagata D., Mulkearns E.E., Shulman J.M., Biernat J., Mandelkow E.M., Feany M.B. S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J. Neurosci. Res. 2007;85:1271–1278. doi: 10.1002/jnr.21232. [DOI] [PubMed] [Google Scholar]
- 31.Steinhilb M.L., Dias-Santagata D., Fulga T.A., Felch D.L., Feany M.B. Tau phosphorylation sites work in concert to promote neurotoxicity in vivo. Mol. Biol. Cell. 2007;18:5060–5068. doi: 10.1091/mbc.E07-04-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chatterjee S., Sang T.K., Lawless G.M., Jackson G.R. Dissociation of tau toxicity and phosphorylation: role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum. Mol. Genet. 2009;18:164–177. doi: 10.1093/hmg/ddn326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Augustinack J.C., Schneider A., Mandelkow E.M., Hyman B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- 34.Matenia D., Mandelkow E.M. The tau of MARK: a polarized view of the cytoskeleton. Trends. Biochem. Sci. 2009;34:332–342. doi: 10.1016/j.tibs.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 35.Mandelkow E.M., Thies E., Trinczek B., Biernat J., Mandelkow E. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J. Cell Biol. 2004;167:99–110. doi: 10.1083/jcb.200401085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thies E., Mandelkow E.M. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J. Neurosci. 2007;27:2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu T., Pan Y., Kao S.Y., Li C., Kohane I., Chan J., Yankner B.A. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 38.Lovell M.A., Markesbery W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer's disease. Nucleic Acids Res. 2007;35:7497–7504. doi: 10.1093/nar/gkm821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moreira P.I., Nunomura A., Nakamura M., Takeda A., Shenk J.C., Aliev G., Smith M.A., Perry G. Nucleic acid oxidation in Alzheimer disease. Free Radic. Biol. Med. 2008;44:1493–1505. doi: 10.1016/j.freeradbiomed.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Stracker T.H., Usui T., Petrini J.H. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst) 2009;8:1047–1054. doi: 10.1016/j.dnarep.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reinhardt H.C., Yaffe M.B. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2 and MK2. Curr. Opin. Cell Biol. 2009;21:245–255. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kondratov R.V., Antoch M.P. Circadian proteins in the regulation of cell cycle and genotoxic stress responses. Trends Cell Biol. 2007;17:311–317. doi: 10.1016/j.tcb.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 43.Xu J., Xin S., Du W. Drosophila Chk2 is required for DNA damage-mediated cell cycle arrest and apoptosis. FEBS Lett. 2001;508:394–398. doi: 10.1016/s0014-5793(01)03103-9. [DOI] [PubMed] [Google Scholar]
- 44.Peters M., DeLuca C., Hirao A., Stambolic V., Potter J., Zhou L., Liepa J., Snow B., Arya S., Wong J., et al. Chk2 regulates irradiation-induced, p53-mediated apoptosis in Drosophila. Proc. Natl. Acad. Sci. USA. 2002;99:11305–11310. doi: 10.1073/pnas.172382899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brodsky M.H., Weinert B.T., Tsang G., Rong Y.S., McGinnis N.M., Golic K.G., Rio D.C., Rubin G.M. Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol. Cell Biol. 2004;24:1219–1231. doi: 10.1128/MCB.24.3.1219-1231.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takada S., Kelkar A., Theurkauf W.E. Drosophila checkpoint kinase 2 couples centrosome function and spindle assembly to genomic integrity. Cell. 2003;113:87–99. doi: 10.1016/s0092-8674(03)00202-2. [DOI] [PubMed] [Google Scholar]
- 47.Hunter S., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bork P., Das U., Daugherty L., Duquenne L., et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas P.D., Campbell M.J., Kejariwal A., Mi H., Karlak B., Daverman R., Diemer K., Muruganujan A., Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steffan J.S., Bodai L., Pallos J., Poelman M., McCampbell A., Apostol B.L., Kazantsev A., Schmidt E., Zhu Y.Z., Greenwald M., et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 50.Iijima K., Chiang H.C., Hearn S.A., Hakker I., Gatt A., Shenton C., Granger L., Leung A., Iijima-Ando K., Zhong Y. Abeta42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS ONE. 2008;3:e1703. doi: 10.1371/journal.pone.0001703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O'Neill T., Giarratani L., Chen P., Iyer L., Lee C.H., Bobiak M., Kanai F., Zhou B.B., Chung J.H., Rathbun G.A. Determination of substrate motifs for human Chk1 and hCds1/Chk2 by the oriented peptide library approach. J. Biol. Chem. 2002;277:16102–16115. doi: 10.1074/jbc.M111705200. [DOI] [PubMed] [Google Scholar]
- 52.Seo G.J., Kim S.E., Lee Y.M., Lee J.W., Lee J.R., Hahn M.J., Kim S.T. Determination of substrate specificity and putative substrates of Chk2 kinase. Biochem. Biophys. Res. Commun. 2003;304:339–343. doi: 10.1016/s0006-291x(03)00589-8. [DOI] [PubMed] [Google Scholar]
- 53.Wang J.W., Imai Y., Lu B. Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. J. Neurosci. 2007;27:574–581. doi: 10.1523/JNEUROSCI.5094-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khurana V., Feany M.B. Connecting cell-cycle activation to neurodegeneration in Drosophila. Biochim. Biophys. Acta. 2007;1772:446–456. doi: 10.1016/j.bbadis.2006.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barbato C., Corbi N., Canu N., Fanciulli M., Serafino A., Ciotti M., Libri V., Bruno T., Amadoro G., De Angelis R., et al. Rb binding protein Che-1 interacts with Tau in cerebellar granule neurons. Modulation during neuronal apoptosis. Mol. Cell Neurosci. 2003;24:1038–1050. doi: 10.1016/j.mcn.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 56.Passananti C., Fanciulli M. The anti-apoptotic factor Che-1/AATF links transcriptional regulation, cell cycle control, and DNA damage response. Cell Div. 2007;2:21. doi: 10.1186/1747-1028-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smale G., Nichols N.R., Brady D.R., Finch C.E., Horton W.E., Jr Evidence for apoptotic cell death in Alzheimer's disease. Exp. Neurol. 1995;133:225–230. doi: 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- 58.Adamec E., Vonsattel J.P., Nixon R.A. DNA strand breaks in Alzheimer's disease. Brain Res. 1999;849:67–77. doi: 10.1016/s0006-8993(99)02004-1. [DOI] [PubMed] [Google Scholar]
- 59.Lassmann H., Bancher C., Breitschopf H., Wegiel J., Bobinski M., Jellinger K., Wisniewski H.M. Cell death in Alzheimer's disease evaluated by DNA fragmentation in situ. Acta Neuropathol. 1995;89:35–41. doi: 10.1007/BF00294257. [DOI] [PubMed] [Google Scholar]
- 60.Lucassen P.J., Chung W.C., Kamphorst W., Swaab D.F. DNA damage distribution in the human brain as shown by in situ end labeling; area-specific differences in aging and Alzheimer disease in the absence of apoptotic morphology. J. Neuropathol. Exp. Neurol. 1997;56:887–900. doi: 10.1097/00005072-199708000-00007. [DOI] [PubMed] [Google Scholar]
- 61.Davydov V., Hansen L.A., Shackelford D.A. Is DNA repair compromised in Alzheimer's disease? Neurobiol. Aging. 2003;24:953–968. doi: 10.1016/s0197-4580(02)00229-4. [DOI] [PubMed] [Google Scholar]
- 62.Shao C., Xiong S., Li G.M., Gu L., Mao G., Markesbery W.R., Lovell M.A. Altered 8-oxoguanine glycosylase in mild cognitive impairment and late-stage Alzheimer's disease brain. Free Radic. Biol. Med. 2008;45:813–819. doi: 10.1016/j.freeradbiomed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Markesbery W.R., Kryscio R.J., Lovell M.A., Morrow J.D. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann. Neurol. 2005;58:730–735. doi: 10.1002/ana.20629. [DOI] [PubMed] [Google Scholar]
- 64.Keller J.N., Schmitt F.A., Scheff S.W., Ding Q., Chen Q., Butterfield D.A., Markesbery W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 65.Ding Q., Markesbery W.R., Chen Q., Li F., Keller J.N. Ribosome dysfunction is an early event in Alzheimer's disease. J. Neurosci. 2005;25:9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Butterfield D.A., Poon H.F., St Clair D., Keller J.N., Pierce W.M., Klein J.B., Markesbery W.R. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol. Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 67.Wang J., Xiong S., Xie C., Markesbery W.R., Lovell M.A. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J. Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- 68.Su J.H., Anderson A.J., Cummings B.J., Cotman C.W. Immunohistochemical evidence for apoptosis in Alzheimer's disease. Neuroreport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- 69.Stadelmann C., Bruck W., Bancher C., Jellinger K., Lassmann H. Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis. J. Neuropathol. Exp. Neurol. 1998;57:456–464. doi: 10.1097/00005072-199805000-00009. [DOI] [PubMed] [Google Scholar]
- 70.Mandavilli B.S., Rao K.S. Neurons in the cerebral cortex are most susceptible to DNA-damage in aging rat brain. Biochem. Mol. Biol. Int. 1996;40:507–514. doi: 10.1080/15216549600201073. [DOI] [PubMed] [Google Scholar]
- 71.Ugolini G., Cattaneo A., Novak M. Co-localization of truncated tau and DNA fragmentation in Alzheimer's disease neurones. Neuroreport. 1997;8:3709–3712. doi: 10.1097/00001756-199712010-00010. [DOI] [PubMed] [Google Scholar]
- 72.Fang-Kircher S.G., Labudova O., Kitzmueller E., Rink H., Cairns N., Lubec G. Increased steady state mRNA levels of DNA-repair genes XRCC1, ERCC2 and ERCC3 in brain of patients with Down syndrome. Life Sci. 1999;64:1689–1699. doi: 10.1016/s0024-3205(99)00107-1. [DOI] [PubMed] [Google Scholar]
- 73.Parshad R.P., Sanford K.K., Price F.M., Melnick L.K., Nee L.E., Schapiro M.B., Tarone R.E., Robbins J.H. Fluorescent light-induced chromatid breaks distinguish Alzheimer disease cells from normal cells in tissue culture. Proc. Natl. Acad. Sci. USA. 1996;93:5146–5150. doi: 10.1073/pnas.93.10.5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jacobsen E., Beach T., Shen Y., Li R., Chang Y. Deficiency of the Mre11 DNA repair complex in Alzheimer's disease brains. Brain Res. Mol. Brain Res. 2004;128:1–7. doi: 10.1016/j.molbrainres.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 75.Robbins J.H., Brumback R.A., Polinsky R.J., Wirtschafter J.D., Tarone R.E., Scudiero D.A., Otsuka F. Hypersensitivity to DNA-damaging agents in abiotrophies: a new explanation for degeneration of neurons, photoreceptors, and muscle in Alzheimer, Parkinson and Huntington diseases, retinitis pigmentosa, and Duchenne muscular dystrophy. Basic Life Sci. 1985;35:315–344. doi: 10.1007/978-1-4899-2218-2_20. [DOI] [PubMed] [Google Scholar]
- 76.Krishna T.H., Mahipal S., Sudhakar A., Sugimoto H., Kalluri R., Rao K.S. Reduced DNA gap repair in aging rat neuronal extracts and its restoration by DNA polymerase beta and DNA-ligase. J. Neurochem. 2005;92:818–823. doi: 10.1111/j.1471-4159.2004.02923.x. [DOI] [PubMed] [Google Scholar]
- 77.Weissman L., Jo D.G., Sorensen M.M., de Souza-Pinto N.C., Markesbery W.R., Mattson M.P., Bohr V.A. Defective DNA base excision repair in brain from individuals with Alzheimer's disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35:5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Edwards J.A., Wang L.G., Setlow R.B., Kaminskas E. O6-methylguanine-DNA methyltransferase in lymphocytes of the elderly with and without Alzheimer's disease. Mutat Res. 1989;219:267–272. doi: 10.1016/0921-8734(89)90028-3. [DOI] [PubMed] [Google Scholar]
- 79.Kinsella T.J., Dobson P.P., Fornace A.J., Jr, Barrett S.F., Ganges M.B., Robbins J.H. Alzheimer's disease fibroblasts have normal repair of N-methyl-N’-nitro-N-nitrosoguanidine-induced DNA damage determined by the alkaline elution technique. Biochem. Biophys. Res. Commun. 1987;149:355–361. doi: 10.1016/0006-291x(87)90374-3. [DOI] [PubMed] [Google Scholar]
- 80.Dias-Santagata D., Fulga T.A., Duttaroy A., Feany M.B. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J. Clin. Invest. 2007;117:236–245. doi: 10.1172/JCI28769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weterings E., Chen D.J. The endless tale of non-homologous end-joining. Cell Res. 2008;18:114–124. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- 82.Antoni L., Sodha N., Collins I., Garrett M.D. CHK2 kinase: cancer susceptibility and cancer therapy—two sides of the same coin? Nat. Rev. Cancer. 2007;7:925–936. doi: 10.1038/nrc2251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.