

Abstract
Protein phosphorylation plays a central role in cellular regulation. Recent proteomics strategies for identifying phosphopeptides have been developed using the model organism Saccharomyces cerevisiae, and consequently, when combined with studies of individual gene products, the number of reported specific phosphorylation sites for this organism has expanded enormously. In order to systematically document and integrate these various data types, we have developed a database of experimentally verified in vivo phosphorylation sites curated from the S. cerevisiae primary literature. PhosphoGRID (www.phosphogrid.org) records the positions of over 5000 specific phosphorylated residues on 1495 gene products. Nearly 900 phosphorylated residues are reported from detailed studies of individual proteins; these in vivo phosphorylation sites are documented by a hierarchy of experimental evidence codes. Where available for specific sites, we have also noted the relevant protein kinases and/or phosphatases, the specific condition(s) under which phosphorylation occurs, and the effect(s) that phosphorylation has on protein function. The unique features of PhosphoGRID that assign both function and specific physiological conditions to each phosphorylated residue will provide a valuable benchmark for proteome-level studies and will facilitate bioinformatic analysis of cellular signal transduction networks.
Database URL: http://phosphogrid.org/
Background
Cellular responses to physiological signals, including cell growth, differentiation and death are mediated by post-translational protein modifications, most notably phosphorylation, which function to transmit signals to downstream effectors and target molecules (1,2). At least one half of all proteins in a typical eukaryotic cell are phosphorylated (3); site-specific phosphorylation on serine, threonine and tyrosine residues is thus the most abundant and well-characterized intracellular post-translational modification. The addition or removal of phosphate groups by protein kinases and phosphatases, respectively, can regulate protein interactions, activity and conformation (4). The budding yeast genome encodes 130 protein kinases and some 40 protein phosphatases (5,6), while the human genome encodes more than 500 protein kinases and over 100 protein phosphatases (7–9). The vast combinatorial possibilities afforded by the global kinase–phosphatase network presents an enormous challenge in deconvolving the information flow that underlies cellular behavior (10).
The development of high throughput strategies for detection and sequence determination of phosphopeptides offers the potential to exhaustively catalogue the phosphorylation status of the proteome under different conditions (11). However, the full biological significance of this information will only be realized through the identification of the enzymes that regulate each specific phosphorylation site, the conditions under which the phosphorylation occurs, and the functional consequences of the modification for protein function (12). Delineation of complete signaling networks and regulatory pathways will require a combination of approaches to assign these parameters, in combination with bioinformatics and modeling tools to organize and analyze the information.
Because of the powerful array of genetic, molecular biological, genomic and proteomic strategies developed for S. cerevisiae, this organism has become a model of choice for global characterization of cellular regulatory networks and for implementation of novel functional genomic methods. The scope of genomics and proteomics resources available for S. cerevisiae includes: protein interaction networks derived from two-hybrid and mass spectrometry data (13,14), genetic synthetic lethal interactions (15,16), subcellular compartmentalization (17), global gene expression patterns under a variety of conditions (18,19), global identification of protein-DNA interactions (20), and comparative fungal genomics (21,22). Combined with rapid progress in identification of phosphorylated residues, these resources should eventually enable comprehensive predication of phospho-regulatory networks (12).
In order to facilitate the analysis and prediction of protein kinase/phosphatase-substrate relationships and signaling networks, we have developed a database of experimentally verified in vivo protein phosphorylation sites for S. cerevisiae. The initial version of the database, designated PhosphoGRID, documents approximately 5000 individual phosphorylated residues on 1495 gene products annotated from the published literature. For each phosphorylated residue, where data is available, we record relevant protein kinases and phosphatases, specific conditions under which the modification occurs, and the effect on protein function. All entries in PhosphoGRID are linked to other existing online yeast resources, including the BioGRID interaction database (13) and the Saccharomyces Genome Database (SGD) (23). PhosphoGRID will also provide an important resource to benchmark mass spectrometry-based methods for the global assignment of phosphorylation sites (24–26).
Database construction and development
Rationale for yeast PhosphoGRID
Several online protein phosphorylation resources have been described previously, but most of these do not contain a significant focus on S. cerevisiae. NetPhos and Scansite are online search tools that enable prediction of phosphorylation sites based on consensus sequences defined in vitro (27,28). These web-based tools are useful in predicting candidate sites in cases where a kinase–protein substrate relationship has been established in vivo, but they suffer from over-prediction and therefore have limited usefulness for identifying phosphorylation sites with physiological relevance. Furthermore, because these prediction tools are largely devoted to metazoans, they are reported to be less reliable for prediction of potential sites in S. cerevisiae, which has at least 32 unique protein kinases (29). Consequently, a phosphorylation site prediction tool specific for Saccharomyces, NetPhosYeast (30), has recently been described. PhosphoSite is a curated web-based resource for physiologically relevant phosphorylations in mammals (31). A similar database, Phospho.ELM (formerly known as PhosphoBase), contains a collection of defined eukaryotic phosphorylation sites, but is not focused on any one species (32); less than 150 entries in Phospho.ELM represent sites from ‘other species’, including yeast. A number of phosphorylation site databases are focused on individual or a limited number of species, including for archea and prokaryotic organisms (33), Arabidopsis (34), and more recently PhosphoPep, which contains data from proteomics initiatives for model organisms including S. cerevisiae, Drosophila and C. elegans (35). Similarly, PHOSIDA contains data produced from mass spectrometry of phosphoproteomes from a variety of eukaryotic and prokaryotic species, but currently has no data from yeast (36,37). PhosphoGRID is thus the first online resource that currently focuses exclusively on experimentally defined phosphorylation sites in the budding yeast S. cerevisiae. PhosphoGRID documents sites from both mass spectrometry-based proteomics efforts and from focused studies on individual gene products; moreover, PhosphoGRID is the first resource to link each specific phosphorylation events with relevant physiological conditions, protein kinases and protein phosphatases.
PhosphoGRID design and architecture
PhosphoGRID is implemented on an open source software platform. The web interface was developed using PHP 5.2.3 (http://www.php.net) and is hosted on an Apache 2.0 web server (http://www.apache.org) running CentOS (http://www.centos.org). The package is designed to run on multiple platforms and has been tested successfully with older versions of PHP (4.x) and alternate web servers such as IIS and Apache 1.3. The PhosphoGRID web interface makes use of the Asynchronous JavaScript and XML (AJAX) package jQuery (http://www.jquery.com) to implement user interface transitions and effects such as highlighting of motifs within the protein sequences in search results. The open source database system MySQL 4.1 (http://www.mysql.com) is the primary database management system that drives both the web-based interface and storage of the phosphorylation site curation data for PhosphoGRID. The relational architecture of PhosphoGRID ensures data integrity and future expandability. In addition, PhosphoGRID makes use of custom-designed lookup tables that ensure rapid response on search queries. Maintenance, input, and manipulation of the database, which includes loading of new phosphorylation site data, is implemented via several Python (http://www.python.org) scripts and applications. These Python tools are designed to automate the procedure of updating and maintaining the database without requiring user intervention once the process has started.
Annotation of phosphorylation sites from the primary literature
Consistent annotation is essential in order to establish a non-redundant collection of phosphorylation sites on proteins and to ensure accuracy for search queries and curation efforts. PhosphoGRID utilizes annotation compiled from the Saccharomyces Genome Database including protein names, descriptions, aliases, sequences, Gene Ontology (GO) mappings, and external database identifiers (23). All ancillary information is compiled via an in-house annotation compilation system (ACS) written in Java SDK version 1.5 (java.sun.com). PhosphoGRID annotation tables are updated on a bi-monthly basis and seamlessly integrate with existing curation to ensure that searches always reflect current annotation.
Data contained within version 1.0 of PhosphoGRID is curated from all papers published prior to the end of 2008. We examined abstracts from approximately 1400 published manuscripts from PubMed with keywords relating to phosphorylation in S. cerevisiae (yeast, phosphorylation, residue, phosphorylation site, protein kinase), and/or that had been flagged with relevance to protein phosphorylation within the yeast BioGRID database (13). Abstracts from 514 of these papers indicated possible reference to specific phosphorylated residues, and these were examined in detail. Of this subset, 332 contained descriptions of specific phosphorylated residues. The vast majority of defined phosphorylation sites were derived from four large-scale mass spectrometry-based analyses of phosphopeptides (24,26,38,39). For each residue identified as a specific phosphorylation, we noted the evidence(s) for that phosphorylation, as well as whether a protein kinase or phosphatase, function, or specific condition was associated with the residue, and whether the phosphorylation had a defined effect on the protein activity. For each phosphorylation site listed in the dataset, we also verified that the residue number cited in the literature corresponds with the sequence in GenBank. We observed a substantial number of inaccurately stated residue positions that primarily arise because of a discrepancy in the actual translational start site, or because the open reading frames, as documented in SGD, generally do not reflect post-translational cleavage of the gene product. In such cases, the position of the phosphorylation site in PhosphoGRID was mapped to the corresponding residue in the ORF as documented in GenBank and a free text comment in a ‘Notes’ field was used to document the discrepancy. The main annotation categories in PhosphoGRID were assigned as follows:
Evidence for in vivo phosphorylation. We limited entries in the initial version of PhosphoGRID to residues for which there was published evidence for phosphorylation in vivo, as documented in data figures or tables. Some techniques for detection of phosphorylation sites are more definitive than others; for each residue in the dataset, we summarized the accumulated experimental evidence(s) for the in vivo modification (Table 1). Direct detection of phosphorylation sites in vivo can only be achieved through mass spectrometric or Edman degradation sequencing of phosphopeptides; this evidence is considered the most definitive. Immunoblot analysis with specific anti-phosphopeptide antibodies directed against specific sites on proteins of interest is also definitive, although reliability of this technique is highly dependent upon the quality of the available antibody. Phosphorylation at specific residues is often indirectly inferred from biochemical and genetic phenotypes produced by substitutions of hydroxyl amino acid residues; when rigorously controlled, this method is also very reliable. Finally, bioinformatic approaches can infer phosphorylation sites, either by identity with an ortholog from another species that is known to be phosphorylated at a specific site, or by matches to consensus sequences; however, in the absence of supporting evidence, these methods can only tentatively assign phosphorylation sites. A combination of methods is necessary to instill confidence that the phosphorylation actually exists in vivo, particularly in cases where detection is indirect. Accordingly, most phosphorylation sites documented in the dataset, primarily those with a defined function (see below), are supported by results from at least two and, in many cases, five or six different methodologies. Within the low throughput (LTP) dataset, ∼50% of the phosphorylations are documented by two or three different techniques, and 30% by four or more methods.
Function of the phosphorylation event. Mechanistic studies have demonstrated that most phosphorylation events characterized to date regulate inter- or intra-molecular interactions of proteins with binding partners, substrates or effectors (4). Specific phosphorylated residues may thus promote or inhibit interactions between proteins or protein domains, or regulate interactions with small molecule substrates or ligands (40,41). These interactions can control enzyme activity, subcellular localization and the assembly of signaling complexes and protein machines. For phosphorylation events that have a defined function, we have recorded both the mechanistic and more general consequences in a field termed ‘Function’ (summarized in Table 2). This flexible field enables documentation of all possible effects on protein function, even if the precise mechanism has not been defined. The directionality of these effects, i.e. activation or inhibition of protein function, is critical for network analysis and modeling. For example, protein phosphorylation may trigger substrate degradation by proteolytic enzymes, direct intracellular protein sorting or drive the assembly/disassembly of active complexes in signaling, transcription, translation, cell division and organelle biogenesis. These effects on protein function are typically a consequence of regulation of protein–protein interactions; for example, phosphorylation of the CDK inhibitor Sic1 by the cyclin-dependent kinase (CDK) Cdc28 causes Sic1 degradation by promoting its interaction with the WD40 domain on the Cdc4 subunit of the SCF ubiquitin protein ligase complex, thus targeting Sic1 for ubiquitination and subsequent degradation by the 26S proteasome (42,43). Other kinases appear to target Sic1 to modulate its degradation under different conditions (44–47). For each phosphorylated residue in the database shown to have a function, we record additional specific details on regulatory significance in a free text “Notes” field.
Specific conditions that modulate phosphorylation. Protein phosphorylation is the primary mechanism by which signals are transmitted within eukaryotic cells in response to specific physiological conditions (2). A major objective for the PhosphoGRID resource is to enable development of novel tools for predictive modeling of signaling network activity. We therefore document the specific condition(s) under which phosphorylated residues are observed in vivo (Table 3). We only note conditions for residues where there is clear evidence of differential phosphorylation; and consequently only 490 phosphorylations in version 1.0 of PhosphoGRID are associated with a specific condition. Approximately 140 of these were derived from two proteome-wide studies examining differential phosphorylation in response to pheromone (24,26). The ratio of the phosphorylated residue observed in the pheromone treated/untreated samples was noted in the free text notes field for each entry, where available. We expect that this aspect of PhosphoGRID will expand rapidly as more studies of this type are conducted to examine responses to additional conditions, including inhibition of TOR signaling by treatment with rapamycin (48), and DNA damage response (49). Importantly, these conditions will include different genetic contexts, such as the loss or gain of kinase or phosphatase function. It is important to emphasize that appearance of a phosphorylation site under a specific condition may occur through regulation of a protein kinase and/or protein phosphatase. For example, the Cdc14 phosphatase is activated at the end of mitosis, where it dephosphorylates and stabilizes Sic1, even in the face of high levels of CDK activity (50). Similarly, phosphatases downstream of TOR, including SIT4, PPH21 and PPH22 become activated in response to limiting nutrients to cause dephosphorylation of target proteins (51).
Enzymes regulating appearance of the phosphorylation site. Importantly, we document the specific protein kinases and phosphatases that have been shown to catalyze addition or removal of a specific phosphate, where this information is available. Furthermore, for both kinases and phosphatases, we indicate both the catalytic and regulatory subunits of the cognate enzyme. This information is particularly important for enzymes such as the CDKs, for which the regulatory cyclin subunit direct interactions and activity of the catalytic subunit towards specific protein substrates. For example, Cdc28 interacts with three G1 phase cyclins (Cln1, Cln2 and Cln3) and six S phase/M phase cyclins (Clb1–Clb6) to exert temporal control on phosphorylation of the manifold substrates required for cell cycle progression (52). Most eukaryotic species have a limited number of phosphoserine/threonine protein phosphatase catalytic subunits whose activity is directed to specific substrates by a myriad of different regulatory subunits (2). For example, Glc7p, the yeast homologue of human protein phosphatase type 1 (PP1), is involved in many different processes including glycogen metabolism, sporulation and mitosis, as mediated through its interactions with a variety of different regulatory subunits (e.g. Reg1p, Reg2p, Gip1p, Gip2p, Gac1p) that direct interactions with different physiological substrates (53). Evidence in support of catalytic and/or regulatory subunit function in a phosphorylation event typically includes a genetic requirement for in vivo phosphorylation, effects on substrate phosphorylation in vitro, or less definitively, inference from activity in other species or matches to a defined consensus sequence. In version 1.0 of PhosphoGRID, we document 59 different cognate protein kinases for 623 phosphorylated residues on 166 protein substrates (Table 4). Less information is available for protein phosphatases, and accordingly, we record 13 different phosphatase catalytic subunits that target 75 phosphorylated residues on 17 protein substrates. The annotation of specific subunit-dependent events will be important for analysis and predictive modeling of regulatory networks.
Table 1.
Summary of evidence codes for phosphorylation of specific amino acid residues in vivo
| Method for detecting phosphorylation | Residuesa | Proteinsb |
|---|---|---|
| Direct Detection | ||
| Mass spectrometry (sequencing/mass analysisc) | 4593 | 1280 |
| Edman degradation | 17 | 10 |
| Anti-phosphopeptide antibody | 77 | 27 |
| Indirect Detection—phenotype produced by a substitution | ||
| Shift in protein mobility in SDS–PAGE | 275 | 72 |
| Loss of 32P label from protein | 130 | 51 |
| Loss of phosphopeptide from fingerprint | 70 | 34 |
| Loss of isoelectric isoform | 38 | 13 |
| Loss of recognition by anti-pT/pS/pY antibody | 21 | 12 |
| Mutation of the residue affects activity | 390 | 124 |
| Phosphorylation of a peptide bearing the residue in vitro | 87 | 16 |
| Loss of phosphorylation of a protein in vitro | 213 | 76 |
| Identity to phosphorylation on ortholog from another species | 85 | 26 |
aNumber of individual phosphorylated residues in PhosphoGRID supported by the indicated evidence.
bNumber of proteins in PhosphoGRID where the indicated evidence supports existence of the phosphorylation.
cIncludes mass analysis without ambiguous hydroxylamino acids.
Table 2.
Defined functions for protein phosphorylation
| Effect of phosphorylation on protein function | Residuesa | Proteinsb |
|---|---|---|
| Specific effects on protein structure/function | ||
| Promotes a protein interaction | 184 | 35 |
| Inhibits a protein interaction | 93 | 18 |
| Modifies interaction with small molecule/ligand | 11 | 6 |
| Functional consequence of phosphorylation | ||
| Activates protein function | 209 | 88 |
| Inhibits protein function | 90 | 17 |
| Targets protein for degradation | 48 | 17 |
| Enhances protein stability | 19 | 4 |
| Modifies subcellular localization | 37 | 15 |
aNumber of individual phosphorylated residues in PhosphoGRID assigned the indicated function.
bNumber of proteins in PhosphoGRID bearing a phosphoresidue assigned the indicated function.
Table 3.
Specific conditions regulating protein phosphorylation
| Physiological conditiona | Residuesb | Proteinsc |
|---|---|---|
| Response to nutrients | ||
| Carbon source | 33 | 16 |
| Nitrogen | 40 | 8 |
| Phosphate | 13 | 2 |
| Response to stress conditions | ||
| Heat stress | 3 | 2 |
| Nutrient starvation | 25 | 10 |
| DNA damage | 70 | 18 |
| Oxidative stress | 1 | 1 |
| Cell wall/osmotic stress | 16 | 9 |
| Unfolded protein response | 1 | 1 |
| Cell cycle regulation | ||
| Pheromone response | 121 | 90 |
| Regulation during normal cell cycle | 222 | 43 |
aSpecific physiological condition under which phosphorylation is detected in vivo.
bNumber of individual phosphorylated residues in PhosphoGRID that are specifically detected under the indicated condition.
cNumber of proteins in PhosphoGRID that bear a phosphoresidue specifically detected under the indicated condition.
Table 4.
Documented substrate residues for yeast protein kinases
| Protein Kinase | Substrate residuesa |
|---|---|
| Bur1 | 20 |
| Cak1 | 6 |
| Cbk1 | 3 |
| Cdc5 | 56 |
| Cdc15 | 2 |
| Cdc28 (Cdk1) | 119 |
| Cla4 | 7 |
| Cdc7 | 7 |
| Chk1 | 9 |
| Cka1/Cka2b (Casein Kinase 2) | 36 |
| Ctk1 | 21 |
| Dun1 | 5 |
| Fus3 | 4 |
| Gcn2 | 2 |
| Hog1 | 12 |
| Hrr25 | 1 |
| Hsl1 | 1 |
| Ime2 | 11 |
| Ipl1 | 13 |
| Ire1 | 2 |
| Kin28 (Cdk7) | 22 |
| Mck1 | 2 |
| Mec1/Tel1b | 34 |
| Mps1 | 3 |
| Npr1 | 3 |
| Pbs2 | 2 |
| Pho85 | 41 |
| Pkc1 (Protein kinase C) | 13 |
| Pkh1/Pkh2 | 5 |
| Prk1 | 26 |
| Pks2 | 7 |
| Ptk2 | 1 |
| Rad53 | 11 |
| Rim11 | 3 |
| Sak1 | 1 |
| Sky1 | 1 |
| Slt2 | 4 |
| Snf1 (AMP-activated PK) | 9 |
| SSN3 (Cdk8) | 9 |
| Ste7 | 4 |
| Ste11 | 1 |
| Ste20 | 6 |
| Swe1 | 1 |
| Tor1 | 30 |
| Tpk1/Tpk2/Tpk3b (Protein kinase A) | 47 |
| Yak1 | 2 |
| Yck1/Yck2b | 11 |
| Ypk2 | 2 |
| Kinases with overlapping substratesc | |
| Bur1/Kin28 | 20 |
| Cla4/Cdc5/Cdc28 | 4 |
| Rim11/Mck1/Mrk1 | 3 |
aNumber of identified in vivo substrate residues for the indicated protein kinase.
bProtein kinases thought to have partial or complete redundancy.
cProtein kinases without genetic redundancy with overlapping in vivo substrate residues.
Dataset access
Phosphorylation information on any gene product of interest can be accessed through the search interface (Figure 1, top right). The search retrieval page display provides the protein amino acid sequence with all documented, experimentally verified phosphosites highlighted as red text (Figure 1). Upon mouse-over of each phosphosite, a pop-up window provides a summary of the phosphorylation site evidence, as well as the specific condition under which it occurs and functional consequence, where known. Consensus sequences for a limited number of protein kinases with well-defined specificity, which overlap verified phosphosites, are indicated in blue text on the amino acid sequence. This feature will be expanded in future updates as consensus sites for more yeast protein kinases are elaborated (30). Tables below the protein sequence provide details on each experimentally identified phosphosite, including experimental evidence, functional consequences (Figure 1, lower), and identity of the cognate protein kinases and/or phosphatases (Figure 2), and where relevant, specific regulatory subunits. For protein kinases and phosphatases themselves, and their corresponding regulatory subunits, an additional table displays sites of phosphorylation/dephosphorylation for known substrate proteins, and includes a summary of the evidence(s) for involvement in these reactions, as well as links to the corresponding substrate pages. An example of this feature is shown for the mating pheromone MAP kinase Fus3 (Figure 2). Each record also provides links to additional resources for each gene product provided at SGD and the NCBI protein database. Finally, for each site of phosphorylation and associated evidence codes, hyperlinks are provided to the original articles listed in PubMED from which the data was curated.
Figure 1.

Screen shot of PhosphoGRID webpage produced by a search for the mating pheromone MAPK Fus3. Phosphorylated amino acids are indicated on the protein sequence in red. Consensus sites for known protein kinases overlapping phosphosites are indicated in blue. Detailed information relating to each identified phosphosite is presented in table form below, with links to PubMED references for the evidence of phosphorylation, conditions under which phosphorylation occurs and effects on protein function.
Figure 2.

Screen shot of the Fus3 PhosphoGRID page. Details of relationships between phosphorylated residues and specific protein kinases and phosphatases are displayed in a second table for each gene product. Additionally, for protein kinases and phosphatases themselves, a summary of known substrate sites, with links to the relevant gene product is presented in an additional table (not shown).
All of the data within PhosphoGRID is freely downloadable in text file format through the ‘Downloads’ tab (Figure 1, top). Download data is refreshed regularly to correspond with new phosphorylation site entries as well as annotation updates via the ACS. In future updates, we will include support for additional download formats including PSI-MI2.5 (54), Osprey (55) and Cytoscape (56). In order to help maintain a current dataset, we have also implemented an online submission form, accessible through the ‘Contribute’ tab (Figure 1, top), through which users can contribute unpublished or newly published information. Contributions will be accepted for residues where evidence of in vivo phosphorylation is documented by one or more experimental evidence(s) as indicated in the ‘Experimental Evidence for Phosphorylation’ field. All PhosphoGRID corrections and clarifications can also be sent to admin@phosphogrid.org.
Overview of the PhosphoGRID dataset
Data in version 1.0 of PhosphoGRID was curated from S. cerevisiae publications up to 31 December 2008. The vast majority of phosphosites, greater than 4200, were generated from four seminal high throughput (HTP) proteomics studies based on mass spectrometric analysis of phosphopeptides derived from total cell protein (24,26,38,39). A total of 851 phosphorylated residues were identified by analysis of individual proteins and/or purified protein complexes in dedicated LTP studies. Surprisingly, the overlap between the HTP and LTP datasets is relatively modest as only 149 of the 851 sites in LTP data are found in HTP studies (Figure 3A). This limited concordance illustrates the difficulty in systematically mapping phosphorylation sites and suggests that existing phosphoproteome datasets are probably highly incomplete. Based on the overlap of phosphorylation sites identified in three large-scale studies (24,38,39), and overlap between sites identified in HTP versus LTP studies, we predict that the yeast proteome may contain on the order of 15 000 phosphorylated residues. Approximately 80% of the phosphorylated residues documented in PhosphoGRID occur on serines, with threonine and tyrosine representing 19 and 1.3% of phosphorylated residues, respectively (Figure 3B); these proportions are roughly similar for sites identified in HTP and LTP studies (not shown). Yeast do not have phosphotyrosine-specific protein kinases akin to those in metazoan cells (30), and so it is interesting that the relative proportion of phosphorylated tyrosine residues in vivo is similar to that observed in higher eukaryotes (57). This observation supports the view that some protein kinases have more relaxed hydroxyl amino acid specificity than is generally appreciated; indeed phosphorylation on tyrosine residues is frequently observed in vitro with various serine/threonine protein kinases (58).
Figure 3.
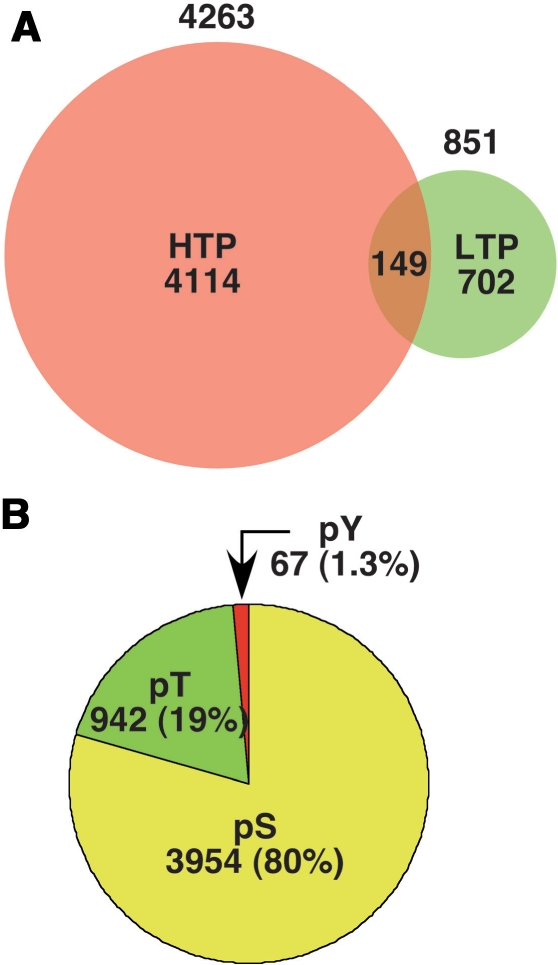
(A). The number of individual phosphorylation sites listed in PhosphoGRID version 1.0 identified by HTP mass spectrometry-based studies (red) and focused LTP studies on individual proteins (green). (B). The proportion of PhosphoGRID entries representing phosphoserine, phosphothreonine and phosphotyrosine residues.
To date, 1495 of the 5584 proteins encoded by the yeast genome appear to contain one or more phosphorylated residues (Figure 4). Given that the phosphoproteome is incompletely charted, it seems probable that most, if not all, yeast proteins will be phosphorylated under one or more conditions. Greater than one-third of phosphoproteins recorded in PhosphoGRID have a single identified phosphorylation site, while the remainder are multiply phosphorylated on anywhere between 2 and greater than 40 sites (Figure 4B). Proteins with large numbers of phosphorylated residues include Rpo21, Swe1, Cdh1, Net1 and Rad53, each having greater than 30 separate entries. Rpo21, also known as Rpb1, is the largest subunit of RNA Polymerase II that contains a C-terminal domain (CTD) consisting of 26 direct repeats of the heptapeptide YSPTSPS. Phosphorylation and dephosphorylation of serines 2, 5 and 7 on the heptapeptide repeat govern the transcription cycle through regulated assembly of various subcomplexes that modify polymerase function (59–62); combinations of CTD phosphorylation events might produce a CTD ‘code’ for transcription (63,64). We note, however, that evidence for phosphorylation of the CTD is limited to recognition by antibodies specific for Ser2, Ser5 and Ser7 phosphorylated heptapeptides, and there has been no direct demonstration of phosphorylation on individual repeats within the CTD, nor has the extent to which the CTD can be multiply phosphorylated in vivo been established.
Figure 4.

Distribution of multiply phosphorylated proteins in PhosphoGRID. The number of proteins with the indicated phosphorylated residues in PhosphoGRID are shown. The identity of gene products with 21 or greater identified phosphorylations are indicated to the right.
In considering proteins with numerous reported phosphorylation sites, it is apparent that there are biases in the identification of residues in HTP versus LTP approaches. For example, Net1, one of the most heavily phosphorylated proteins studied to date (Figure 4), has a total of 34 identified phosphosites; 9 of these are derived from proteomics efforts, and 25 from two studies that examined the role of phosphorylation in Net1 function (65,66); curiously though there are no sites in common between these studies (Table 5). Similarly, Pan1 has a total of 24 phosphosites, 8 from HTP studies and 16 from focused LTP studies (67,68), none of which are in common. Numerous additional similar anomalies exist (Table 5, and data not shown). For these heavily phosphorylated proteins, the differences may in part reflect the fact that most of the phosphorylations identified in focused studies occur under specific physiological conditions. For example, phosphorylation of Swe1, Net1 and Sic1 are primarily limited to specific phases of the cell cycle (42,66,69), and consequently these sites are likely to be underrepresented in samples from unsynchronized cells typically used for analysis in HTP studies. Similarly, most of the phosphorylation events characterized on Rad53 and Rad9 occur in response to DNA damage (70,71). Considering that 424 phosphorylation sites identified in LTP studies, nearly half of the total, are associated with a specific physiological condition (Table 3), the modest overlap with HTP may reflect the significant effects of environmental conditions. Apart from the major studies examining differential phosphorylation in pheromone-treated cells (24,26) (Table 3), there have not been other large-scale proteomics efforts examining phosphorylation under additional physiological conditions.
Table 5.
Summary of the most abundantly phosphorylated yeast proteins in PhosphoGRID Version 1.0
| Protein | Phos. residuesa | Proteomicsb | Overlapd | Other focusedc |
|---|---|---|---|---|
| SWE1 | 43 | 0 | 0 | 43 |
| RPO21 | 43 | 1 | 0 | 42 |
| CDH1 | 35 | 0 | 0 | 35 |
| NET1 | 34 | 9 | 0 | 25 |
| RAD53 | 32 | 2 | 2 | 32 |
| SEC16 | 29 | 29 | 0 | 0e |
| NPR1 | 27 | 7 | 6 | 26 |
| PAN1 | 24 | 8 | 0 | 16 |
| SPA2 | 24 | 24 | 0 | 0e |
| RAD9 | 22 | 1 | 1 | 22 |
| STE20 | 22 | 18 | 4 | 8 |
| SHS1 | 22 | 8 | 6 | 20 |
| EDE1 | 20 | 20 | 0 | 0e |
| SIC1 | 20 | 4 | 3 | 19 |
aTotal number of phosphorylated residues identified on the indicated protein.
bNumber of phosphoresidues identified in proteomics studies.
cNumber of phosphoresidues identified in focused studies on the indicated protein.
dNumber of phosphoresidues identified by both proteomics and focused studies.
eNo focused studies available.
As noted, an important feature of PhosphoGRID is that we have documented effects that each phosphorylation has on the target protein activity, where available. Currently, 490 phosphorylation sites are known to affect protein activity (Table 2). Encouragingly, approximately three-fourth of phosphorylation events identified in LTP studies are associated with phenotypic consequences, as revealed by mutational analysis (Figure 5, right); however, this strong correlation may result from study bias, in that only phosphorylation sites linked to a biological response are reported in the literature. Many phosphorylation events have a cumulative influence on protein activity such that phenotypes may be revealed only by combinatorial mutation of multiple phosphoacceptor sites. A well-characterized example is the finding that six Cdc28-dependent phosphorylation events on Sic1 are required for its recognition by Cdc4; this multisite dependence confers ultrasensitive or switch-like behavior on the degradation of Sic1 (42). The preponderance of multiply phosphorylated proteins in PhosphoGRID suggests that many phosphorylation-dependent responses may be imbued with similar qualities (72).
Figure 5.

Proportion of phosphorylation sites in PhosphoGRID with defined functions. Summaries of phosphorylation sites identified by HTP mass spectrometry-based studies (left chart) or focused LTP studies on individual proteins (right chart) are shown.
CONCLUSIONS AND PERSPECTIVE
PhosphoGRID is a repository for protein phosphorylation information in S. cerevisiae, particularly for data derived from LTP studies reported in the primary literature. As illustrated here, the LTP dataset provides a benchmark for HTP proteomic studies and will be an important resource for the construction of mathematical models of signaling networks. The initial release of PhosphoGRID contains all data published prior to 2009; we will build on this comprehensive dataset with regular curation updates, in conjunction with elaboration of the repertoire of search and display functions within the resource. Future PhosphoGRID releases will also have expanded capabilities, including documentation of in vitro phosphorylation of substrates by specific protein kinases, where specific residues have not been identified, demonstrated in both high throughput (58) and focused studies. In combination with expanded protein kinase consensus site prediction capability, this information will be important for bioinformatic analysis of signaling networks.
In order to provide an up-to-date and complete resource, we encourage community contributions of new data through the online data submission feature; in this latter regard, we also believe it will be important to report instances where phosphorylation site mutations do not yield an obvious phenotype, particularly as such data is rarely published. A long-term challenge for phosphoproteomics will be to fill in the enormous void in our understanding the functional consequences of the myriad of phosphorylation events in the cell; PhosphoGRID should help meet this challenge.
Funding
A Canada Research Chair in Functional Genomics and Bioinformatics, a Royal Society Wolfson Research Merit Award and the Scottish Universities Life Sciences Alliance through the Scottish Funding Council (to M.T.); Canadian Cancer Society Research Institute grant 0011258 (to I.S.), NIH National Center for Research Resources grant 1R01RR024031-01 (to M.T.) and Biotechnology and Biological Sciences Research Council grant BB/F010486/1 (to M.T.). Funding for open access charge: Canadian Cancer Society Research Institute.
Conflict of interest. None declared.
ACKNOWLEDGMENTS
The authors thank LeAnn Howe and Francis Ouellette for helpful discussions.
References
- 1.Hunter T. Signaling—2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 2.Cohen P. The origins of protein phosphorylation. Nat. Cell Biol. 2002;4:E127–E130. doi: 10.1038/ncb0502-e127. [DOI] [PubMed] [Google Scholar]
- 3.Zhou H, Watts JD, Aebersold R. A systematic approach to the analysis of protein phosphorylation. Nat. Biotechnol. 2001;19:375–378. doi: 10.1038/86777. [DOI] [PubMed] [Google Scholar]
- 4.Seet BT, Dikic I, Zhou MM, et al. Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol. 2006;7:473–483. doi: 10.1038/nrm1960. [DOI] [PubMed] [Google Scholar]
- 5.Hunter T, Plowman GD. The protein kinases of budding yeast: six score and more. Trends Biochem. Sci. 1997;22:18–22. doi: 10.1016/s0968-0004(96)10068-2. [DOI] [PubMed] [Google Scholar]
- 6.Jiang Y. Regulation of the cell cycle by protein phosphatase 2A in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2006;70:440–449. doi: 10.1128/MMBR.00049-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson SA, Hunter T. Kinomics: methods for deciphering the kinome. Nat. Methods. 2005;2:17–25. doi: 10.1038/nmeth731. [DOI] [PubMed] [Google Scholar]
- 8.Manning G, Whyte DB, Martinez R, et al. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 9.Mustelin T. A brief introduction to the protein phosphatase families. Methods Mol. Biol. 2007;365:9–22. doi: 10.1385/1-59745-267-X:9. [DOI] [PubMed] [Google Scholar]
- 10.Nurse P. Life, logic and information. Nature. 2008;454:424–426. doi: 10.1038/454424a. [DOI] [PubMed] [Google Scholar]
- 11.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 12.Linding R, Jensen LJ, Ostheimer GJ, et al. Systematic discovery of in vivo phosphorylation networks. Cell. 2007;129:1415–1426. doi: 10.1016/j.cell.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breitkreutz BJ, Stark C, Reguly T, et al. The BioGRID interaction database: 2008 update. Nucleic Acids Res. 2008;36:D637–D640. doi: 10.1093/nar/gkm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tarassov K, Messier V, Landry CR, et al. An in vivo map of the yeast protein interactome. Science. 2008;320:1465–1470. doi: 10.1126/science.1153878. [DOI] [PubMed] [Google Scholar]
- 15.Zhang LV, King OD, Wong SL, et al. Motifs, themes and thematic maps of an integrated Saccharomyces cerevisiae interaction network. J. Biol. 2005;4:6. doi: 10.1186/jbiol23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong SL, Zhang LV, Tong AH, et al. Combining biological networks to predict genetic interactions. Proc. Natl Acad. Sci. USA. 2004;101:15682–15687. doi: 10.1073/pnas.0406614101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huh WK, Falvo JV, Gerke LC, et al. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 18.Ghaemmaghami S, Huh WK, Bower K, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 19.Horak CE, Snyder M. Global analysis of gene expression in yeast. Funct. Integr. Genomics. 2002;2:171–180. doi: 10.1007/s10142-002-0065-3. [DOI] [PubMed] [Google Scholar]
- 20.Chua G, Robinson MD, Morris Q, et al. Transcriptional networks: reverse-engineering gene regulation on a global scale. Curr. Opin. Microbiol. 2004;7:638–646. doi: 10.1016/j.mib.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 21.Wolfe KH. Comparative genomics and genome evolution in yeasts. Philos. Trans. R. Soc. Lond. B, Biol. Sci. 2006;361:403–412. doi: 10.1098/rstb.2005.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galagan JE, Henn MR, Ma LJ, et al. Genomics of the fungal kingdom: insights into eukaryotic biology. Genome Res. 2005;15:1620–1631. doi: 10.1101/gr.3767105. [DOI] [PubMed] [Google Scholar]
- 23.Christie KR, Weng S, Balakrishnan R, et al. Saccharomyces Genome Database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res. 2004;32:D311–D314. doi: 10.1093/nar/gkh033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gruhler A, Olsen JV, Mohammed S, et al. Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol. Cell Proteomics. 2005;4:310–327. doi: 10.1074/mcp.M400219-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Bodenmiller B, Mueller LN, Mueller M, et al. Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat. Methods. 2007;4:231–237. doi: 10.1038/nmeth1005. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Gerber SA, Rudner AD, et al. Large-scale phosphorylation analysis of alpha-factor-arrested Saccharomyces cerevisiae. J. Proteome Res. 2007;6:1190–1197. doi: 10.1021/pr060559j. [DOI] [PubMed] [Google Scholar]
- 27.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blom N, Kreegipuu A, Brunak S. PhosphoBase: a database of phosphorylation sites. Nucleic Acids Res. 1998;26:382–386. doi: 10.1093/nar/26.1.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manning G, Plowman GD, Hunter T, et al. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002;27:514–520. doi: 10.1016/s0968-0004(02)02179-5. [DOI] [PubMed] [Google Scholar]
- 30.Ingrell CR, Miller ML, Jensen ON, et al. NetPhosYeast: prediction of protein phosphorylation sites in yeast. Bioinformatics. 2007;23:895–897. doi: 10.1093/bioinformatics/btm020. [DOI] [PubMed] [Google Scholar]
- 31.Hornbeck PV, Chabra I, Kornhauser JM, et al. PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–1561. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- 32.Diella F, Cameron S, Gemund C, et al. Phospho.ELM: a database of experimentally verified phosphorylation sites in eukaryotic proteins. BMC Bioinformatics. 2004;5:79. doi: 10.1186/1471-2105-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wurgler-Murphy SM, King DM, Kennelly PJ. The phosphorylation site database: a guide to the serine-, threonine-, and/or tyrosine-phosphorylated proteins in prokaryotic organisms. Proteomics. 2004;4:1562–1570. doi: 10.1002/pmic.200300711. [DOI] [PubMed] [Google Scholar]
- 34.Nuhse TS, Stensballe A, Jensen ON, et al. Phosphoproteomics of the Arabidopsis plasma membrane and a new phosphorylation site database. Plant Cell. 2004;16:2394–2405. doi: 10.1105/tpc.104.023150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bodenmiller B, Campbell D, Gerrits B, et al. PhosphoPep—a database of protein phosphorylation sites in model organisms. Nat. Biotechnol. 2008;26:1339–1340. doi: 10.1038/nbt1208-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gnad F, Ren S, Cox J, et al. PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 2007;8:R250. doi: 10.1186/gb-2007-8-11-r250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Macek B, Gnad F, Soufi B, et al. Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell Proteomics. 2008;7:299–307. doi: 10.1074/mcp.M700311-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Chi A, Huttenhower C, Geer LY, et al. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl Acad. Sci. USA. 2007;104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ficarro SB, McCleland ML, Stukenberg PT, et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 2002;20:301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 40.Salazar C, Hofer T. Multisite protein phosphorylation—from molecular mechanisms to kinetic models. FEBS J. 2009;276:3177–3198. doi: 10.1111/j.1742-4658.2009.07027.x. [DOI] [PubMed] [Google Scholar]
- 41.Narayanan A, Jacobson MP. Computational studies of protein regulation by post-translational phosphorylation. Curr. Opin. Struct. Biol. 2009;19:156–163. doi: 10.1016/j.sbi.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Orlicky S, Tang X, Willems A, et al. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell. 2003;112:243–256. doi: 10.1016/s0092-8674(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 43.Verma R, Annan RS, Huddleston MJ, et al. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science. 1997;278:455–460. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- 44.Escote X, Zapater M, Clotet J, et al. Hog1 mediates cell-cycle arrest in G1 phase by the dual targeting of Sic1. Nat. Cell Biol. 2004;6:997–1002. doi: 10.1038/ncb1174. [DOI] [PubMed] [Google Scholar]
- 45.Zinzalla V, Graziola M, Mastriani A, et al. Rapamycin-mediated G1 arrest involves regulation of the Cdk inhibitor Sic1 in Saccharomyces cerevisiae. Mol. Microbiol. 2007;63:1482–1494. doi: 10.1111/j.1365-2958.2007.05599.x. [DOI] [PubMed] [Google Scholar]
- 46.Sedgwick C, Rawluk M, Decesare J, et al. Saccharomyces cerevisiae Ime2 phosphorylates Sic1 at multiple PXS/T sites but is insufficient to trigger Sic1 degradation. Biochem J. 2006;399:151–160. doi: 10.1042/BJ20060363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wysocki R, Javaheri A, Kristjansdottir K, et al. CDK Pho85 targets CDK inhibitor Sic1 to relieve yeast G1 checkpoint arrest after DNA damage. Nat. Struct. Mol. Biol. 2006;13:908–914. doi: 10.1038/nsmb1139. [DOI] [PubMed] [Google Scholar]
- 48.Huber A, Bodenmiller B, Uotila A, et al. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009;23:1929–1943. doi: 10.1101/gad.532109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smolka MB, Albuquerque CP, Chen SH, et al. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl Acad. Sci. USA. 2007;104:10364–10369. doi: 10.1073/pnas.0701622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Visintin R, Craig K, Hwang ES, et al. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell. 1998;2:709–718. doi: 10.1016/s1097-2765(00)80286-5. [DOI] [PubMed] [Google Scholar]
- 51.Rohde J, Heitman J, Cardenas ME. The TOR kinases link nutrient sensing to cell growth. J. Biol. Chem. 2001;276:9583–9586. doi: 10.1074/jbc.R000034200. [DOI] [PubMed] [Google Scholar]
- 52.Andrews B, Measday V. The cyclin family of budding yeast: abundant use of a good idea. Trends Genet. 1998;14:66–72. doi: 10.1016/s0168-9525(97)01322-x. [DOI] [PubMed] [Google Scholar]
- 53.Ramaswamy NT, Dalley BK, Cannon JF. Analysis of protein interactions between protein phosphatase 1 and noncatalytic subunits using the yeast two-hybrid assay. Methods Mol. Biol. 1998;93:251–261. doi: 10.1385/0-89603-468-2:251. [DOI] [PubMed] [Google Scholar]
- 54.Hermjakob H, Montecchi-Palazzi L, Bader G, et al. The HUPO PSI's; molecular interaction format—a community standard for the representation of protein interaction data. Nat. Biotechnol. 2004;22:177–183. doi: 10.1038/nbt926. [DOI] [PubMed] [Google Scholar]
- 55.Breitkreutz BJ, Stark C, Tyers M. Osprey: a network visualization system. Genome Biol. 2003;4:R22. doi: 10.1186/gb-2003-4-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hunter T. Protein modification: phosphorylation on tyrosine residues. Curr. Opin. Cell Biol. 1989;1:1168–1181. doi: 10.1016/s0955-0674(89)80068-7. [DOI] [PubMed] [Google Scholar]
- 58.Ptacek J, Devgan G, Michaud G, et al. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–684. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- 59.Meinhart A, Kamenski T, Hoeppner S, et al. A structural perspective of CTD function. Genes Dev. 2005;19:1401–1415. doi: 10.1101/gad.1318105. [DOI] [PubMed] [Google Scholar]
- 60.Chapman RD, Heidemann M, Albert TK, et al. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science. 2007;318:1780–1782. doi: 10.1126/science.1145977. [DOI] [PubMed] [Google Scholar]
- 61.Egloff S, O'R;eilly D, Chapman RD, et al. Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science. 2007;318:1777–1779. doi: 10.1126/science.1145989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akhtar MS, Heidemann M, Tietjen JR, et al. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol. Cell. 2009;34:387–393. doi: 10.1016/j.molcel.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Egloff S, Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet. 2008;24:280–288. doi: 10.1016/j.tig.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 64.Buratowski S. The CTD code. Nat. Struct. Biol. 2003;10:679–680. doi: 10.1038/nsb0903-679. [DOI] [PubMed] [Google Scholar]
- 65.Loughrey Chen S, Huddleston MJ, Shou W, et al. Mass spectrometry-based methods for phosphorylation site mapping of hyperphosphorylated proteins applied to Net1, a regulator of exit from mitosis in yeast. Mol. Cell Proteomics. 2002;1:186–196. doi: 10.1074/mcp.m100032-mcp200. [DOI] [PubMed] [Google Scholar]
- 66.Shou W, Azzam R, Chen SL, et al. Cdc5 influences phosphorylation of Net1 and disassembly of the RENT complex. BMC Mol. Biol. 2002;3:3. doi: 10.1186/1471-2199-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zeng G, Cai M. Regulation of the actin cytoskeleton organization in yeast by a novel serine/threonine kinase Prk1p. J. Cell Biol. 1999;144:71–82. doi: 10.1083/jcb.144.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Toshima J, Toshima JY, Martin AC, et al. Phosphoregulation of Arp2/3-dependent actin assembly during receptor-mediated endocytosis. Nat. Cell Biol. 2005;7:246–254. doi: 10.1038/ncb1229. [DOI] [PubMed] [Google Scholar]
- 69.Harvey SL, Charlet A, Haas W, et al. Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell. 2005;122:407–420. doi: 10.1016/j.cell.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 70.Smolka MB, Albuquerque CP, Chen SH, et al. Dynamic changes in protein–protein interaction and protein phosphorylation probed with amine-reactive isotope tag. Mol. Cell Proteomics. 2005;4:1358–1369. doi: 10.1074/mcp.M500115-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Naiki T, Wakayama T, Nakada D, et al. Association of Rad9 with double-strand breaks through a Mec1-dependent mechanism. Mol. Cell. Biol. 2004;24:3277–3285. doi: 10.1128/MCB.24.8.3277-3285.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferrell J.E., Jr. Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem. Sci. 1996;21:460–466. doi: 10.1016/s0968-0004(96)20026-x. [DOI] [PubMed] [Google Scholar]