

Abstract
At least 10 enteroendocrine cell types have been identified, and the peptide hormones they secrete have diverse functions that include regulation of glucose homeostasis, food intake, and gastric emptying. Mice lacking individual enteroendocrine hormones, their receptors, or combinations of these have shed light on the role of these hormones in the regulation of energy homeostasis. However, because enteroendocrine hormones have partially overlapping functions, these loss-of-function studies produced only minor phenotypes, and none of the enteroendocrine hormones was shown to be essential for life. To examine the effect of loss of all enteroendocrine cells and hormones on energy homeostasis, we generated mice with intestinal-specific ablation of the proendocrine transcription factor neurogenin 3 (referred to herein as Ngn3Δint mice). Ngn3Δint mice were deficient for all enteroendocrine cells and hormones, and died with a high frequency during the first week of life. Mutant mice were growth retarded and had yellowish stool suggestive of steatorrhea. Subsequent analyses revealed that Ngn3Δint mice had impaired lipid absorption, reduced weight gain, and improved glucose homeostasis. Furthermore, intestinal epithelium of the mutant mice showed an enlarged proliferative crypt compartment and accelerated cell turnover but no changes to goblet and Paneth cell numbers. Enterocytes had shorter microvilli, but the expression of the main brush border enzymes was unaffected. Our data help unravel the role of enteroendocrine cells and hormones in lipid absorption and maintenance of the intestinal epithelium.
Introduction
Enteroendocrine cells are scattered individually in the lining of the gut epithelium and, despite the fact that their relative percentage is only around 1% within the gastrointestinal epithelium, quantitatively they constitute the major endocrine organ of the organism. However, in contrast to many other endocrine glands, enteroendocrine cells are embedded in a majority of nonendocrine cells, including the absorptive enterocytes, goblet, and Paneth cells. At least 10 different enteroendocrine cell types have been identified, and the various hormones produced by these endocrine cells — ghrelin, gastric inhibitory polypeptide (GIP), secretin, peptide YY (PYY), glucagon-like peptide-1 (GLP-1), GLP-2, neurotensin, serotonin, substance P, cholecystokinin (CCK), and motilin — control important physiological functions, such as glycemia, exocrine pancreatic secretion, growth and repair of the gut epithelium, motility of the gut wall, and gastric emptying (1–3). The incretin hormones GLP-1 and GIP, which are secreted by the L- and K-cells, respectively, are key in the regulation of glucose homeostasis by stimulating glucose-dependent insulin secretion in pancreatic β cells (1). Furthermore, results obtained by different groups suggest that GLP-1 also stimulates islet neogenesis and β cell proliferation (4–6), whereas GLP-2 promotes intestinal epithelial cell proliferation. Drucker and colleagues have shown that GLP-2 injection into mice resulted in elongated villi, mainly due to enhanced crypt cell proliferation and decreased enterocyte apoptosis (7). In addition, and as mentioned above, gut peptides have been shown to control gastric emptying, gastric acid secretion, and food intake (8). For instance, CCK, which when released from I-cells in the small intestine, stimulates gallbladder contraction, exocrine pancreatic secretion, and inhibition of gastric emptying and appetite (9, 10).
Neurogenin 3 (Ngn3) has been shown to be the key gene controlling the commitment of pluripotent progenitor cells toward the endocrine cell lineage in the developing pancreas (11) and intestine (12, 13). Ngn3-deficient mice do not develop any enteroendocrine and pancreatic endocrine cells, and mice show severe diabetes and die shortly after birth (11, 12). In the stomach, in which Ngn3 also marks all endocrine progenitor cells, endocrine differentiation does not entirely rely on Ngn3, as in the pancreas and intestine. In fact, serotonin- and ghrelin-expressing cells are still present in newborn Ngn3-knockout mice, whereas all other gastric endocrine cell types are lost (12, 14). Tracing studies performed in the adult intestine lineage demonstrated that enteroendocrine cells are constantly renewed throughout life from Ngn3-positive cells that are located in the proliferative compartment of the crypt (12, 15). Interestingly, these studies also showed that some Ngn3-expressing progenitors might differentiate into goblet and Paneth cells (15). NeuroD1, a direct target gene of Ngn3, has been shown to be important for the development of the enteroendocrine cell types CCK and S, expressing the hormones CCK and secretin, respectively (16). However, due to the early postnatal lethality of Ngn3-deficient mice, the role of this gene in the turnover and differentiation of enteroendocrine cell lineage in adult mice could not be studied so far.
We have generated mice carrying a floxed-Ngn3 allele conditionally deleted in the intestine to address its requirement for the development of enteroendocrine progenitors in the adult and to evaluate the consequence of their expected loss on glucose and intestinal cell homeostasis. Here we show that mice with a specific inactivation of Ngn3 in only the intestine do not develop any enteroendocrine cells and that mice die with a high frequency during their weaning period. Surviving mutant animals are smaller than wild-type littermates, show soft stool, impaired lipid absorption and glucose homeostasis, and an altered intestinal architecture. The significance of our findings, showing the importance of enteroendocrine cells/hormones for the regulation of energy homeostasis, is further supported by the recent identification of several patients carrying homozygote point mutations in Ngn3 (17). These patients show an almost complete lack of all enteroendocrine cells, which was classified as “enteroendocrine cell dysgenesis,” and are suffering, from the first days of life, from malabsorptive chronic diarrhea and the development of diabetes in late childhood (17, 18).
Results
Generation of intestinal-specific Ngn3-knockout mice.
To study the consequence of a complete and specific ablation of Ngn3 expression in the small and large intestine, we have generated mice carrying a floxed Ngn3 allele (Ngn3+/flox) (see Methods, Figure 1, and Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI40794DS1). Ngn3+/flox and Ngn3flox/flox mice developed normally, reached adulthood, were fertile, and showed normal glucose levels in the urine. In order to specifically ablate Ngn3 in the intestinal epithelium, we used transgenic mice expressing the Cre recombinase, under the control of a 9-kb regulatory region of the murine villin gene (vil-cre) (19). The 9-kb regulatory region of the villin gene has been shown to target stable and homogeneous expression of the Cre recombinase in small and large intestine along the crypt-villus axis, in the immature, undifferentiated cells of the crypt as well as in differentiated enterocytes (19). The following crosses were set up: Ngn3 heterozygous mice (Ngn3+/Neo) (11) or Ngn3+/flox mice carrying the vil-Cre transgene (Ngn3+/neo;vil-cre, Ngn3+/flox;vil-cre) were crossed with Ngn3+/flox or Ngn3flox/flox mice to obtain Ngn3neo/flox;vil-cre or Ngn3flox/flox;vil-cre mice, respectively. In the following, all experiments were done with mice coming from a pure CD1 background. As the efficiency of Ngn3 deletion in Ngn3neo/flox;vil-cre or Ngn3flox/flox;vil-cre mice was identical, judged by the complete absence of chromogranin A–positive cells at P3.5 or adult stages, they will in the following be referred to as Ngn3 knockout (Ngn3Δint) or mutant mice. As controls, Ngn3+/+ or Ngn3+/+;vil-cre littermates were used.
Figure 1. Generation of animals with a conditional Ngn3 allele.

(A) Schema depicting the Ngn3 locus and the targeting construct. (B) Targeted Ngn3 allele before and after the excision of the FRT flanked “PGK-Neo” selection cassette by the FLP recombinase. Stars in B indicate the position of the 5′- and 3′-external probes used for Southern blot analysis (see Supplemental Figure 1). (A and B) The black boxes indicate the Ngn3 coding sequence. The PGK-Neomycin selection cassette, the loxP, and FRT sites are indicated as well. X, XbaI; Sp, SpeI; E, EcoRI; Pm, PmeI; As, AscI.
Ngn3Δint mice exhibit severe growth retardation.
Ngn3Δint mice are born with the expected Mendelian frequency and are at P0.5 visually indistinguishable from control littermates. However, at this stage, Ngn3Δint mice already show, in average, a slightly lower body weight (control mice, 1.5 ± 0.17 g; mutant mice, 1.4 ± 0.14 g; P = 0.00809) and are from P3.5 on clearly smaller than control littermates (Figure 2A and Figure 3). In a CD1 background, 50% of the mutant mice died within the first 8 days of life after minimal weight gain. In order to rule out that the early death of some mutant mice is due to a problem in nursing, we dissected out the whole intestinal tract from dead and living mutant mice. The macroscopic analyses of the dissected whole intestine clearly showed the presence of milk in the stomach of surviving mutant mice (Figure 2C) and mutant mice found dead (Figure 2D). Importantly, surviving mice had soft stool, which did not cease with age. In the following, surviving male and female Ngn3Δint mice exhibited variant degrees of growth retardation, and even at adult stages, mutant mice stayed about 30% smaller than control littermates (Figure 3). However, although the intestinal tract of adult mutant mice seems visually to be shorter than the intestinal tract of control mice, its length is proportional to body weight (Supplemental Figure 2).
Figure 2. Fifty percent of mutant mice with an intestinal deletion of Ngn3 (Ngn3Δint) die within the first 8 days of life.

(A) Photography taken at P3.5 of a wild-type, mutant (MTa), and mutant mouse found dead (MTb) from the same litter. From P3.5 on, mutant animals start to be visibly smaller than control littermates. (B–D) Photography of the dissected intestinal tractus from the wild-type and mutant mice shown in A, taken with the same magnification (original magnification, ×0.8). The presence of milk in the stomach of mutant mice indicates their ability to suck milk.
Figure 3. Mutant mice gain less body weight than control littermates.

During the first 2 weeks of life, the weight of wild-type (control) and mutant mice was taken every day (left graph) and thereafter once a week for a period of 6 more weeks (right graph). Mutant mice do gain less weight than control mice and keep, at adult stages, about 30% lower body weight than control littermates. Control, n = 66; mutant, n = 35, for all time points measured; P < 0.008 (left graph). Male control, n = 8; male mutant, n = 5; female control, n = 5; female mutant, n = 4; *P < 0.05, #P < 0.01, †P < 0.001 (right graph).
Specific and efficient ablation of Ngn3 and of enteroendocrine cells in the intestine of Ngn3Δint mice.
To evaluate the ablation efficiency of Ngn3 in Ngn3Δint mice, total RNA from the intestine of E19.5 and adult mutant and wild-type mice was prepared, and the expression of Ngn3 was evaluated by quantitative RT-PCR (RT-QPCR). These analyses showed a 90%–100% reduction of Ngn3 mRNA expression (Supplemental Figure 3) all along the intestinal tract at embryonic and adult stages. Likewise, RT-QPCR and immunohistochemistry for chromogranin A showed an 95%–100% reduction of its mRNA (Supplemental Figure 3A) and a complete loss of chromogranin A+ cells (Supplemental Figure 4) in the intestine of E19.5 mutant embryos. The low amount of chromogranin A (Chga) mRNA detected in the intestine of some E19.5 mutant animals is most likely due to the contamination with pancreatic tissue, as we detected in 2 out of 6 animals analyzed significant levels of insulin-1 (Ins1) mRNA (data not shown), whereas the mRNA for Gip, an intestinal-specific endocrine hormone, is completely gone (Supplemental Figure 3A). Furthermore, immunohistochemistry analyses for Ngn3 showed a complete loss of Ngn3+ enteroendocrine progenitors along the proximal-distal axis of adult mutant intestine (Figure 4B). Taken together, these results show that already at embryonic stages Ngn3Δint mice showed a complete lack of all enteroendocrine cells, suggesting that the survival of some mutant mice is not due to a mosaic deletion of Ngn3 in these animals. We have previously shown that at embryonic stages all enteroendocrine cell development is Ngn3 dependent (12). However, since Ngn3 global knockout mice die shortly after birth, we could not analyze whether at adult stages enteroendocrine cell differentiation or the differentiation of the other intestinal cell types from intestinal stem cells is Ngn3 dependent or not. To evaluate the latter we performed immunohistochemistry analyses for chromogranin A, which marks all enteroendocrine cell types except CCK- and motilin-expressing cells, and for CCK/gastrin on tissue sections of Ngn3Δint and control mice at different adult stages. This analysis revealed a complete lack of chromogranin A– and CCK/gastrin-positive enteroendocrine cells all along the intestinal tract, demonstrating that enteroendocrine cell development is Ngn3 dependent at adult stages also (Figure 4, C–F). Importantly and as expected, glucagon- and insulin-expressing α- and β-cells, respectively, are detected in pancreatic islets (see below), confirming the specific intestinal deletion of Ngn3.
Figure 4. Conditional inactivation of Ngn3 in only the intestine results in a complete loss of all enteroendocrine cells all along the proximal-distal axis of the intestine.

Sections of adult duodenum, jejunum, ileum, and colon were examined for the presence of endocrine cells in control and mutant animals by immunofluorescence. Images presented are from the jejunum of control (A, C, E, G, and I) and mutant (B, D, F, H, and J) animals and are representative for the general loss of all enteroendocrine cells in mutant animals. Villin-Cre–mediated inactivation of Ngn3 results in a complete loss of all Ngn3+ enteroendocrine progenitors (B), which in wild-type animals are located in the intestinal crypt compartment (A, arrows). Likewise, mutant animals are also devoid of chromogranin A+ (D), Cck/gastrin+ (F), Glp1+ (H), and Gip+ (J) cells, normally located in the villi of wild-type mice (arrows in C, E, G, and I), respectively. The age of the animals analyzed is 10–12 weeks. Original magnification, ×10.
Intestinal ablation of Ngn3 leads to a perturbed intestinal morphology.
Histological analyses of the small intestine of adult Ngn3Δint and control mice revealed that in mutant Ngn3Δint mice villi are frequently blunted or club shaped and often show dilatation of up to 400 microns in diameter (Figure 5B). Immunohistochemistry staining for total laminin further demonstrates the frequent dilatation of mutant villi and the strong detachment of the epithelium from the basement membrane (Figure 5D). Published data showing a slight but significant increase of goblet cells in Ngn3 global knockout mice (12), which die shortly after birth, and cell lineage studies showing that Ngn3 progenitors can also contribute to some goblet and Paneth cells (15) prompted us to look at the distribution of these 2 cell types. Ngn3Δint mice showed no obvious change in the number and location of Paneth cells in the small intestine (Figure 5, E and F, and Supplemental Figure 5A). In contrast to the increased number of goblet cells found at early postnatal stages in Ngn3 global knockout mice, we did not observe in Ngn3Δint mice at adult stages a similar increase (Figure 5, G and H, and Supplemental Figure 5B). The histological analyses of the large intestine revealed a reduction in the length of the glands of up to 1.5 times compared with the wild-type mice (Figure 6, A, B, and E). In addition, goblet cells seemed to be larger and mostly devoid of mucus in mutant compared with control large intestine (Figure 6, C and D).
Figure 5. Intestinal ablation of Ngn3 leads to an altered morphology of the small intestine but normal Paneth and goblet cell differentiation.

Sections of adult wild-type and mutant duodenum, jejunum, and ileum were examined for their overall appearance (A–D) and the presence of Paneth (E and F) and goblet cells (G and H). Images presented are from the jejunum of control (A, C, E, and G) and mutant (B, D, F, and H) animals and are also representative for the phenotype observed in the duodenum and ileum of mutant animals. (A and B) H&E staining clearly shows the frequent blunt or club-shaped appearance of the villi and the disorganization of the crypt compartment of mutant animals compared with control small intestine. (C and D) Immunofluorescence analyses with an antibody recognizing all laminins, showing the frequent detachment of the intestinal epithelium from the lamina propria in mutant small intestine. (E and F) Immunohistochemistry with an anti-lysozyme antibody demonstrates normal appearance and location of Paneth cells (arrows in E and F) in mutant small intestine. (G and H) Likewise, periodic acid–Schiff staining shows that intestinal ablation of Ngn3 does not alter the location or number of goblet cells (arrows in G and H) in mutant animals. For Paneth and goblet cell counts, see Supplemental Figure 5. The age of the animals analyzed is 10–12 weeks.
Figure 6. The large intestine of mutant mice shows shorter glands.

Sections of adult wild-type (A and C) and mutant (B and D) large intestine were examined for their overall appearance (A and B) and the presence of goblet cells (C and D). (A and B) H&E staining clearly shows the reduction in the length of the glands in the large intestine of mutant mice (B, measurement in E) compared with control tissue. (C and D) Periodic acid–Schiff staining of goblet cells. (E) The colonic glands in mutant animals are on average 26% shorter than the colonic glands of control animals (n = 4; 50–60 glands were analyzed per genotype). **P < 0.01. The age of the animals analyzed is 10–12 weeks.
Ngn3-deficient small intestine showed an enlarged multifocal proliferating crypt compartment and an accelerated cell turnover. The most striking feature of Ngn3Δint intestine was the frequent disorganization of the crypt compartment. The transiently amplifying crypt compartment seemed to be larger and more abundant in Ngn3Δint mice than in control mice of the same litter and age (Figure 5, A and B, and Figure 7, E and F). To evaluate whether this is due to an increase in cell proliferation, we analyzed by immunohistochemistry the proliferation marker Ki67. This analyses clearly showed that the enlargement of the crypt compartment seen in mutant mice is due to an increase in the number of proliferating cells of up to 44% (±11%) compared with wild-type mice (Figure 7, A and B). However, the enlarged proliferative crypt compartments in mutant mice did not result in longer or more villi. In contrary, measurement of their length clearly showed that mutant villi are up to 40% shorter than the villi in control littermates (Figure 7G). We came up with 3 possibilities for why mutant mice have shorter villi despite the enlarged transiently proliferating crypt compartment: increased apoptosis, accelerated cell turn over, or both. Immunohistochemistry staining for the apoptotic cell marker caspase 3 revealed no difference between mutant and wild-type mice (data not shown). However, by performing a 24-hour BrdU chase, it became clear that mutant mice have an up to 1.6-fold accelerated cell turnover (Figure 7, C and D, and measurement in H), which most likely is the reason for the shorter villi seen in mutant mice. Importantly, at embryonic stage E19.5, the intervillus region seemed to be slightly de-organized but otherwise did not show an increase in proliferating Ki67+ cells (Supplemental Figure 4).
Figure 7. Altered cell homeostasis in Ngn3-deficient small intestine.

Sections of adult control (A, C, and E) and mutant (B, D, and F) intestine were examined for the status of the proliferative crypt compartment (A, B, E, and F), villus length (measurements in G), and cell turn over (C, D, and measurement in H). (A and B) Immunofluorescence staining for the proliferative cell marker Ki67 clearly demonstrates an up to 2-fold enlargement of the proliferative crypt compartment (dashed bars in A and B) in Ngn3-deficient intestine. Arrows point to chromogranin A+ cells. (G) Measurement of the villi length indicates an approximately 40% reduction in their length in mutant intestine. (C and D) Twenty-four hours before dissection, adult control and mutant mice were injected with BrdU, and BrdU-labeled cells were then visualized by immunofluorescence staining. Then the distance from the villus base to last labeled BrdU+ cell was measured (dashed bars in C and D), demonstrating a 1.6-fold accelerated cell turnover in Ngn3-deficient intestine. Arrows point to chromogranin A+ cells. (E and F) H&E staining showing the enlargement of the crypt compartment seen in Ngn3 mutant intestine. n = 3. The age of the animals analyzed is 10–12 weeks.
Strongly reduced intestinal absorptive surface area and impaired lipid absorption in Ngn3Δint mutant mice.
The growth retardation of mutant mice, their frequent death during the weaning period, the appearance of soft yellowish liquid stool, which suggested that they might have steatorrhea, and the perturbed intestinal morphology prompted us to further characterize the ultrastructure of the absorptive cells and look as well at the absorption of lipids. The presence of ample milk in the stomach of mutant mice (Figure 2C) already suggested that the frequent death and growth retardation of the surviving mutant mice results from malabsorption rather than malnutrition. The apical microvilli of the absorptive cells greatly enhance the absorptive surface area of the intestine. Surprisingly, electron microscopy analyses showed the microvilli on the absorptive cells of mutant mice to be sparser, approximately 60% shorter, but twice as large than in control littermates (Figure 8, I and J), resulting in an approximately 44% reduction of the brush border of the absorptive cells in the small intestine of mutant mice. As mentioned above, the yellowish stool during the weaning period of mutant mice, which at postweaning stages still is of a lighter brownish color compared with that of control mice, prompted us to look at the absorption of lipids by the enterocytes. Oil red O staining of the mutant gut revealed a clear reduction of the presence of lipids in the enterocytes and the lamina propria compared with control samples (Figure 9, A and B, and Supplemental Figure 6). In addition, electron microscopy analyses revealed a strong reduction in the number of chylomicrons, which transport dietary lipids from the intestine to other locations in the body, in the absorptive cells of mutant mice (Figure 9, C and D). These findings are also in agreement with the reduced levels of total cholesterol, HDL cholesterol, and triglycerides found in mutant mice (Figure 10A). The enteroendocrine hormones CCK and secretin are known to regulate the secretion of digestive enzymes from the pancreatic exocrine cells. As reduced levels of digestive enzymes, notably that of lipase, could have an effect on lipid absorption, we analyzed the levels of lipase in the blood of mutant and control mice. Control and mutant mice showed similar levels of lipase in the blood (Figure 10B), despite a complete loss of CCK- and secretin-secreting CCK and S cells, respectively. In addition, control and mutant mouse pancreas showed no difference in quantity or distribution of zymogen granules in acinar cells (Figure 9, E and F). Taken together, these results suggest that the reduced numbers of Oil red O–positive lipid droplets and chylomicrons and the reduced serum levels of total cholesterol, HDL cholesterol, and triglycerides found in mutant mice is rather due to impaired lipid absorption than due to impaired lipid digestion.
Figure 8. Strong reduction of the intestinal absorptive surface area but normal expression of brush border enzymes and glucose transporters in Ngn3-deficient mice.

Sections of control (A, C, E, G, and I) and mutant (B, D, F, H, and J) intestine were examined for the status of the absorptive cell population. Analyses of the lactase activity (A and B) and immunofluorescence staining for sucrase-isomaltase (C and D), the active glucose transporter Glut2 (E and F), and the passive glucose transporter SGLT1 (G and H) did not show any difference between control and mutant tissue, respectively. (I and J) Ultrastructural analysis of the brush border of the absorptive cells demonstrates a strong reduction of the microvilli length in mutant mice. The dashed lines in A and B indicate the bottom of the crypt compartment. The age of the mice analyzed in A and B is P1.5 and in C–J is 10–12 weeks. Original magnification, ×20 (A–H); ×40,000 (I and J).
Figure 9. Impaired lipid absorption in Ngn3Δint mutant mice.
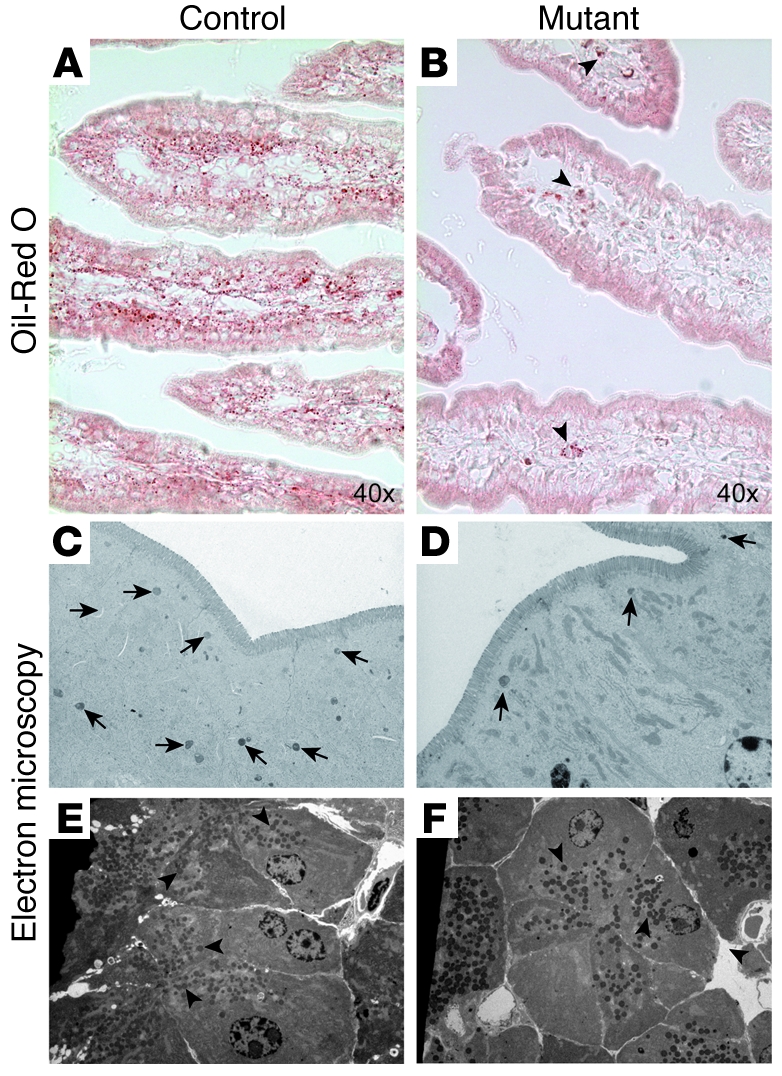
Oil red O, which stains neutral fats, was used to visualize lipid droplets in control (A) and mutant (B) tissue. Mutant small intestine clearly shows a strong reduction in the amount of lipid droplets (B) compared with control tissue (A). Arrowheads in B point to some lipid droplets found in mutant tissue. Ultrastructural analysis of the absorptive cells furthermore demonstrates a clear reduction in the number of neutral lipid-containing chylomicrons (arrows in C and D) in Ngn3–deficient intestine (D). (E and F) Ultrastructural analysis of exocrine pancreas at adult stage shows no difference in the number or quality of zymogen granules (arrowheads) in acinar cells in control (E) and mutant animals (F). The age of the mice analyzed is 10–12 weeks. Original magnification, ×10,000 (C and D); ×4,000 (E and F).
Figure 10. Reduced levels of cholesterol and triglyceride in the blood of Ngn3Δint mice.

(A) Twenty-seven–week-old Ngn3Δint mice have reduced cholesterol and triglyceride levels in the blood (n = 6; *P < 0.05, **P < 0.01). (B) Similar levels of lipase, a digestive enzyme mainly produced and secreted by the acinar cells in the pancreas, are found in the blood of 27-week-old control and mutant mice (n = 4–5).
Furthermore, we also analyzed by immunohistochemistry the expression of the brush border enzymes lactase and sucrase-isomaltase (Figure 8, A–D), respectively, and the active glucose transporter Glut2 (Figure 8, E and F) (20) and the passive glucose transporter SGLT1 (Figure 8, G and H) (20), which revealed no qualitative difference between mutant and wild-type mice. Likewise, RT-QPCR for Glut2 did not show any difference in the expression level between control and mutant intestinal tissue (data not shown), and the oral glucose tolerance tests (OGTTs) revealed that mutant animals take up glucose into the blood from the gastrointestinal tract with the same efficiency as wild-type littermates (Figure 11A, see time point 15 minutes).
Figure 11. Altered glucose homeostasis in Ngn3Δint mice.

Control (filled squares) and mutant mice (filled circles) were subjected to either an oral (A, OGTT) or intraperitoneal (B, IPGTT) glucose challenge or an ITT. Blood glucose levels were then measured at the indicated time points. (A) In the OGTT, mutant mice show a slightly improved glucose clearance from the blood. (B) In the IPGTT, at all time points measured, blood glucose levels of mutant mice do not rise to the same levels as in control mice. (C) The ITT clearly shows an improved insulin sensitivity of mutant compared with control mice. n = 6–7, for control and mutant mice. “0” indicates the blood glucose level before the glucose challenge. *P < 0.05, **P < 0.01, ***P < 0.001. The age of the mice analyzed is 7–10 weeks.
Altered glucose homeostasis in Ngn3Δint mice.
GLP-1 and GIP, 2 incretin hormones produced and secreted after food ingestion by intestinal L-cells and K-cells respectively, have been shown to potentiate glucose stimulated insulin secretion of pancreatic β cells (1). Compound knockout mice for GLP1 and GIP receptor display a greater glucose intolerance following oral glucose challenge (21). The absence of chromogranin A–positive cells in Ngn3Δint mice (Figure 4D) already suggest that mutant mice lack L and K cells secreting GLP1 and GIP, respectively. Indeed, immunohistochemistry and RT-QPCR analyses for GLP1 and GIP demonstrated their complete absence in mutant tissue (Figure 4, G–J, and Supplemental Figure 3). In addition, following a glucose challenge, we found in the blood serum of Ngn3Δint mice a complete lack of GIP (Figure 12). Unfortunately, our attempts to measure GLP1 in the blood serum failed, which is most likely is due to its known rapid degradation (22, 23). However, mutant mice showed also a complete lack of PYY (Figure 12), further supporting the ablation of L-cells. In the following, to evaluate the impact of a complete loss of all incretin hormones on disposal of a glucose load, we performed an OGTT (Figure 11A) and compared the results to an intraperitoneal glucose tolerance test (IPGTT) (Figure 11B). In the OGTT, mutant and control mice showed, after an overnight fasting period, the same fasting blood glucose concentration. However, although initially the glucose concentration in the blood of mutant and control mice rose to the same levels, mutant mice showed an improved glucose clearance from the blood (Figure 11A). Surprisingly, the IPGTT revealed an even more pronounced phenotype in our Ngn3Δint mice, where at all time points taken mutant mice had far lower glucose levels in the blood than control mice (Figure 11B). Already during all our dissections of adult Ngn3Δint mice, we observed a strong reduction in the extent of abdominal fat compared with control littermates of the same sex. Measurement of the body composition of 27-week-old mice clearly demonstrated that Ngn3Δint mice have strongly reduced body fat content, an improved lean mass and a better BMI than control littermates of the same sex (Figure 13, A–C). As reduced body fat results in improved insulin sensitivity and also an improved glucose uptake by the peripheral tissue, we subjected 14-week- and 27-week-old mutant and control mice to an insulin tolerance test (ITT). At both ages, analyzed Ngn3Δint mice have a slightly improved insulin sensitivity (Figure 11C and Figure 13D) compared with control littermates, which is also seen by their difference in average under the curve (AUC) between 0–45 minutes after the injection of insulin (Figure 13D, left inset). In addition, the ITT also revealed that Ngn3Δint mice had an approximately 30% lower fasting blood glucose level than control littermates (blood glucose levels at time 0 [T0], control mice, ~125 mg/dl, Ngn3Δint mice, ~88 mg/dl; Figure 13D, right inset) and a strongly blunted hypoglycemic response to the injected insulin (Figure 11C and Figure 13D). As our mutant mice showed improved insulin sensitivity, we could not exclude that in the IPGTT we have missed an early blood glucose peak. We therefore repeated the IPGTT and measured blood glucose levels, also at 5 minutes after glucose injection. This revealed that even in the early phase the blood glucose levels are much lower than in control mice (Supplemental Figure 7).
Figure 12. Reduced levels of several intestinal and pancreatic hormones in the blood of intestinal Ngn3-deficient mice.

After an oral glucose challenge, blood was taken from Ngn3Δint and control mice and analyzed. Ngn3Δint mice show a complete lack of the intestinal hormones GIP and PYY and reduced levels of ghrelin. Likewise, blood serum concentration levels of the pancreatic hormones insulin, amylin, and PP are also strongly reduced. The age of the mice analyzed is 9–10 weeks. n = 6, for mutant and control mice.
Figure 13. Improved BMI and insulin sensitivity in 27-week-old Ngn3Δint mice.

(A and B) The body composition of age- and sex-matched mutant and control mice was analyzed. Ngn3Δint mice have approximately 30% less body fat (A), are leaner (B), and show an improved BMI (C) compared with control mice. (D) Control (filled squares) and mutant (filled circles) mice were fasted and subjected to an ITT. Mutant mice show improved insulin sensitivity, seen also by the reduction in the average under the curve between 0–45 minutes after insulin injection (AUC, left inset). In addition, mutant mice show a clear difference in the fasting blood glucose level (right inset). n = 6–7. *P < 0.05, **P < 0.01.
Altered islet morphology in Ngn3Δint mice.
Results obtained by different groups suggest that GLP-1 not only potentiates glucose-stimulated insulin secretion but also stimulates islet neogenesis and β cell proliferation (4–6). In addition GLP–1 receptor–knockout (GLP-1R–knockout) mice exhibit an altered islet morphology with and shift from large islets to more medium or single islets (24). Moreover, these mice showed an increased proportion of islets with centrally located α-cells, which are normally located at their periphery. As our mutant mice showed no intestinal Glp1 expression (Figure 4 and Supplemental Figure 3B), we evaluated the distribution of islet sizes in Ngn3Δint mice according to the published criteria (24), which classified the islets as single (<300 μm2), small (300–5,000 μm2), medium (5,000–20,000 μm2), or large (>20,000 μm2). In each experimental group we analyzed 1,500–2,000 islets. This showed that, like in the GLP-1R–knockout mice (24), our Ngn3Δint mice, which lack all enteroendocrine cells, including the Glp1-secreting L-cells, show an shift from large to single islets (Figure 14, A and B). In addition, and like also seen in the GLP-1R–knockout mice (24), we also found an increasing number of medium and large islets with centrally located α-cells (Figure 14, C and D). These data confirm the published data (24) showing the importance of Glp1 for the organization of the adult endocrine islet cells. However, in contrast to Ngn3Δint mice GLP-1R–knockout mice exhibit mild fasting hyperglycemia and glucose intolerance after an oral glucose challenge (25).
Figure 14. Altered islet architecture in Ngn3Δint mice.

(A) Ngn3Δint mice show a decrease of large and an increase of single islets compared with control mice. (B) The contribution of islets of a particular size to the total islet volume is shifted from large to single and small islets in mutant mice compared with control mice. (C) Immunostaining for insulin (green) and glucagon (red) on a pancreatic section of control and Ngn3Δint mutant mice. In mutant mice, glucagon-positive cells appear in the periphery and ectopically in the center of the islets, whereas in control mice, they are normally exclusively located at the periphery. (D) Quantitative analyses of C. (A, B, and D) Control, black columns; mutant, white columns. Islet sizes were classified as follows: single, <300 μm; small, 300–5,000 μm; medium, 5,000–20,000 μm; and large, >20,000 μm; n = 5, all male; 1,500–2,000 islets counted per genotype. The age of the mice analyzed is 8–9 weeks. *P < 0.05, **P < 0.01, ***P < 0.001.
The intestinal food transit is accelerated in Ngn3Δint mice.
As mentioned in the beginning, Ngn3Δint mice fed with a low-fat standard diet showed all their life the appearance of soft stool. Likewise, so far, in all dissected Ngn3Δint mice, we hardly found “normal” dry excrement pellets in the colon. In severe cases, almost the whole intestinal tract was filled with liquid excrements, and these mice were in general in bad physical condition. Several hormones secreted by different enteroendocrine cells have been shown to regulate directly or indirectly the gastrointestinal motility (26). For example, ghrelin and motilin stimulate gastrointestinal motility, whereas GLP-1 and PYY inhibit it. To test whether in Ngn3Δint mice intestinal food transit is affected, mutant and control mice were fasted for a prolonged period and then given simultaneously access to artificially colored alimentation (27). The time of the appearance of colored stool was then measured and showed in average a 2.3-times faster intestinal food transit in Ngn3Δint mice compared with control littermates (Figure 15A). In addition, mutant mice excrete up to 2 times more stool than control littermates (Figure 15B). Importantly, the diarrhea seen in our mutant mice could also be the result of reduced expression of the water channels “Aquaporins” or deregulation of ionic transport. To address this point, we analyzed the expression of the Aquaporins 3 (Aqp3), 4 (Aqp4), and 8 (Aqp8), which are specifically expressed in the epithelium of the colon (28, 29), and performed and extensively analyzed the blood serum chemistry. These analyses showed no modulation of the expression of the Aqp3, Aqp4, and Aqp8 at embryonic stage E19.5 and only a very variable but weakly statistical significant increase of Aqp3 and Aqp4 in adult mutant mice (Supplemental Figure 8). In addition, blood chloride, sodium, calcium, potassium, magnesium, phosphorus, and iron levels were not altered and bicarbonate levels were only slightly increased in Ngn3Δint mice (Supplemental Figure 9).
Figure 15. Accelerated food transit and increased feces production in Ngn3Δint mice.

(A) Age- and sex-matched mutant and control mice were fasted overnight and given then simultaneously access to colored food. The time of the appearance of the first colored stool was then taken and normalized to their body weight. Mutant mice have a 2.3-fold accelerated intestinal food transit. (B) The feces of control and mutant mice were collected after 1 or 4 days, and their weight was taken. Mutant mice show an up to 2-fold increase in feces production compared with control mice. (A and B) n = 4–5. **P < 0.01, ***P < 0.001.
Discussion
We have generated mice with a conditional Ngn3 allele to specifically inactivate this gene in the intestine using the villin-Cre (19) mice, in order to address its requirement for the genesis and differentiation of enteroendocrine cells in the adult.
In the mouse embryo, the development of enteroendocrine cells relies on the proendocrine transcription factor neurogenin 3, which promotes the endocrine fate in pluripotent intestinal stem cells (12, 13, 15). Indeed, in Ngn3-deficient mice, neither enteroendocrine nor pancreatic endocrine cells develop, and the mice die postnatally due to severe diabetes (11, 12). Enteroendocrine cell differentiation starts mostly at late embryonic and postnatal stages, and Ngn3 expression persists in the adult intestine, in which enteroendocrine cells are constantly renewed throughout life. However, the perinatal death of Ngn3 global knockout mice precluded the analysis of the role of Ngn3 in intestinal cell differentiation, and thus it was not known whether Ngn3 has a similar proendocrine function in the adult mouse or other yet unknown functions.
As our data show, the villin-Cre transgene leads to a specific and complete inactivation of Ngn3 in the intestine and importantly, also to a complete lack of all differentiated enteroendocrine cell types. This result suggests that even at adult stages enteroendocrine cell differentiation is Ngn3 dependent. Surprisingly, histological analyses revealed a disorganized and enlarged crypt compartment in mutant Ngn3Δint animals. This finding was unexpected, since several enteroendocrine hormones have been shown to positively influence cell proliferation in the intestine, especially GLP2 but also PYY and gastrin (30), which suggested that the lack of one or several of these hormones might result in a reduction of the proliferative crypt. Our data showed that Ngn3Δint mice lack all enteroendocrine cells and, in particular, a complete absence of PYY and GLP2. However, in contrast to a possible reduction, Ngn3Δint mice clearly showed an enlargement of the proliferative crypt compartment, accompanied by an accelerated cell turnover and shorter villi. In addition, Ngn3Δint mice showed shorter and sometimes misshaped microvilli. These results are in part similar to the results published by Fre and coworkers, who used the villin promoter to target the expression of a constitutively active form of the mouse Notch 1 receptor in all the intestinal epithelium (31). These mice, which were referred to as Rosa-Notch/Cre+ mice, also show a strong reduction of Ngn3 expression and a marked reduction of enteroendocrine cells. However, the phenotype of Ngn3Δint mice is clearly different, in that, in mutant animals we do not see an increase in apoptosis or a change in the goblet or Paneth cell numbers or location. This difference most likely is due the fact that in Rosa-Notch/Cre+ mice, not only is Ngn3 expression downregulated but the expression of Math1 is also, which is essential for the development of all the secretory cell lineages. In contrast to the Notch 1 receptor gain-of-function results, Notch 1-2 compound knockout mice or gut-specific inactivation of Rbpj results in an almost complete loss of proliferating crypt progenitors (32, 33). In Ngn3Δint mice, in which we have conditionally inactivated Ngn3 in the intestine, the proliferative crypt compartment and cell turn over seemed to be deregulated, which is not due to increased inflammation, since we only find low numbers of lymphocytes and rare plasma cells. In addition, it is unlikely that this is due to the lack of endocrine progenitors at embryonic stages, since we do not see at E19.5 a difference in the proliferative status of the intervillus region in mutant animals. Taken together, our results suggest that at postnatal stages, enteroendocrine progenitors and/or differentiated enteroendocrine cells or their secreted hormones have a so far unknown role to our knowledge in maintaining the homeostasis of the intestinal crypt compartment.
As mentioned above, new born Ngn3 global knockout mice are diabetic showing high elevated urine glucose levels, which was suggested to be the most likely cause for their early death around day 3 (11). However, Ngn3 global knockout mice also do not develop any enteroendocrine cells (12), and although none of the so far published knockout mice for the diverse enteroendocrine hormones or their receptors, or even compound mutants (21, 34), show severe metabolic changes, one cannot exclude that first, the simultaneous loss of all enteroendocrine cells in the full Ngn3 knockout is contributing to their early lethality, and second, that the loss of only the enteroendocrine cells would lead to severe metabolic alterations and by itself be life threatening. As our data show, on a 100% CD1 background, approximately 50% of newborn mice with an intestinal Ngn3 ablation die within the first week of life. The capacity of mutant newborns to survive is not due to a mosaic deletion of Ngn3, since in all surviving mutant animals analyzed so far, embryonic, post-natal, or adult, we never found any chromogranin A– or hormone-positive enteroendocrine cell. The presence of milk in the stomach of dead mutant mice indicated that they do take up nutrition. We also separated mutant newborns and their mother from the rest of the litter to rule out that competition for parental resources is the reason for the mortality of Ngn3Δint mice. This step did not change the lethality ratio and suggests that surviving mutant mice reflect individual variability in sensitivity to the lack of enteroendocrine hormones. We can also rule out that Ngn3Δint mice, like Ngn3 global knockout mice, die from hyperglycemia, as from over 15 tested P3.5 Ngn3Δint mice, none showed elevated urine glucose levels. Overall, this high lethality ratio clearly shows the importance of enteroendocrine cells and their secreted hormones to sustain life.
The incretin hormones GLP-1 and GIP have been shown to potentiate glucose-stimulated insulin secretion (1), and compound knockout mice for GLP-1 and GIP receptor display a greater glucose intolerance after oral glucose challenge (21, 34). Surprisingly, Ngn3Δint mice, which completely lack GLP-1 and GIP, showed, in contrast to the GLP-1/GIP compound knockout mice, a slightly improved blood glucose clearance after an oral glucose challenge. In addition, in an IGTT, which does not stimulate intestinal GLP-1 and GIP secretion, mutant mice showed an even more blunted curve than control mice. Mutant mice have less body fat, a better lean mass, and showed slightly improved insulin sensitivity, which might contribute to the improved blood glucose clearance in the OGTT. As mutant mice showed very low accumulation of body fat, the improved insulin sensitivity might also enhance a direct uptake of the intraperitoneal injected glucose by the peripheral tissue. However, this might not fully explain the altered glucose homeostasis seen in our mutant mice, and we cannot exclude that the loss of several other enteroendocrine hormones contributes as well to the improved glucose clearance.
During the weaning period, mutant mice showed frequent yellowish stool, which suggested that they might have steatorrhea due to a problem with the absorption of lipids. In support of this hypothesis, surviving mutant mice did not gain weight at the same ratio as control littermates and even at adult stages still showed stool of a light brownish color, suggesting sustained lipid malabsorption. Accordingly, the strongly reduced Oil red O staining and the decreased number of chylomicrons in the intestine of mutant animals would suggest that lipids are not efficiently absorbed by the enterocytes. Lipid malabsorption is not due to an altered pancreatic exocrine function, which would result in an impaired enzymatic processing of lipids, as judged by the normal appearance and distribution of zymogen granules in acinar cells and the presence of similar blood lipase levels in mutant and control animals. Deficient lipid absorption is also not a consequence of an altered transport of lipids from the enterocytes to the lacteal vessels, which would result in an accumulation of chylomicrons and Oil red O staining in the enterocytes (35). The impaired lipid absorption might in part be due to the lack of GLP-2. Hsieh and coworkers (36) have recently shown that GLP-2 increases intestinal lipid absorption. However, the absorption of lipids, cholesterol and lipid soluble vitamins mostly depends on the function of bile acids (37, 38). In fact, bile acids owing to their amphipathic nature are essential for the solubilization of dietary lipids and their subsequent absorption in the digestive tract (37). In addition, there is increasing evidence that bile acids also affect energy and glucose homeostasis (39, 40). Considering the reduced body fat, the impaired lipid absorption and glucose homeostasis seen in our Ngn3Δint mice suggest that one or several enteroendocrine hormones may partially regulate bile acid homeostasis. A lack of enteroendocrine hormones could therefore lead to a deregulated bile acid homeostasis. In addition, the accelerated food transit and the reduced intestinal absorptive surface area of Ngn3Δint mice might contribute to their impaired weight gain. Taken together, our data suggest that the early lethality and the impaired weight gain of surviving Ngn3Δint mice are mostly due to malabsorption rather than malnutrition and clearly show that enteroendocrine cells and their secreted hormones are necessary to sustain life. Importantly, recently several patients with mutations in Ngn3 have been identified, suffering from birth onwards from unremitting diarrhea and the profound malabsorption of all nutrients, except water (17, 18), and the pathologic term given for the first case was enteroendocrine cell dysgenesis. Immunohistochemical analyses of the small intestine revealed few or a complete absence of enteroendocrine cells but normal numbers of Paneth, goblet, and absorptive cells, reinforcing the importance of enteroendocrine cells and/or hormones for nutrient absorption. Importantly and in contrast to our mice, which have only an intestinal deletion of Ngn3, Ngn3 function in these patients is affected globally, and besides impaired enteroendocrine cell function, they also develop diabetes by the age of 8 years. However, it is not known whether these patients, by the age of 8 years, still have some remaining endocrine cells left in the pancreas, like in the intestines of patients 2 and 3 (18), or whether they are completely lost. It is thus not clear whether the pathophysiology of patients with point mutations in Ngn3 results solely from enteroendocrine cell dysgenesis or also from the simultaneous altered Ngn3 function in the pancreas and/or hypothalamus. The malabsorption of all nutrients in the human patients versus specific lipid malabsorption in our mutant mice supports the latter hypothesis.
Taken together, from our data 2 important conclusions can be drawn. First, our results clearly show the importance of enteroendocrine cells/hormones in the regulation of energy homeostasis and that their loss is life threatening. They also point to the importance of enteroendocrine cells/hormones for lipid absorption by enterocytes. Second, enteroendocrine progenitor cells and/or one or several of the hormones secreted by differentiated enteroendocrine cells contribute, directly or indirectly, to the mechanisms regulating the homeostasis of the intestinal crypt compartment. The data presented here might also help to shed some further light on the pathophysiology of Ngn3 deficiency in humans and the contribution of enteroendocrine cell dysgenesis in malabsorption.
Methods
Generation of animals with a conditional Ngn3 allele.
Homologous recombination in ES cells and generation of chimeric mice were performed at the Institut Clinique de la Souris (ICS) (Mouse Clinical Institute), Illkirch, France. All mice were housed in an animal facility licensed by the French Ministry of Agriculture (agreement no. B67-218-5), and all animal experiments were supervised by G. Gradwohl (agreement no. 67-59 to G. Gradwohl, approved by the Direction des Services Vétérinaires, Strasbourg, France), in compliance with the European legislation on care and use of laboratory animals. The final targeting vector was linearized with an external unique SacII restriction site and electroporated into the ES cell line P1, derived from mouse strain 129 S2/Sv Pas ES, and geneticin selection was performed (catalog 10131-027; Gibco) at 250 μg/ml for 10 days. Homologous recombination events were detected in resistant colonies by Southern blotting using a 1.2-kb 3′-external probe generated from a KpnI–NotI Ngn3 genomic subclone. Independently targeted ES cell clones (n = 28) were obtained from 200 clones analyzed and further examined with a 0.48-kb 5′-external probe generated from an Ngn3 genomic clone by PCR, using the primers 5′CCCCTCTTCTCCCTTTGTTC and 3′ACACATGGATTTGGCACTGA. Two correct targeted independent ES cell clones (K23/67 and K23/104) were used to generate chimeras by standard procedures. Germline transmission was obtained by crossing the chimeras with C57BL/6J females, and the colony was amplified by crossing heterozygous animals with CD1 mice. Heterozygous females were crossed with CMV-Flip males (41) to excise the Neomycin selection cassette. When successful excision of Neomycin selection cassette occurred, a 550-bp PCR product was amplified, using the primers 5′CTCGAGGGATCCTCTAGTCGAGGAATTTGA and 3′CGGCAGATTTGAATGAGGGC from genomic tail DNA of the offspring.
Genotyping of Ngn3Δint mice.
Genomic tail DNA was analyzed by PCR, using the following primer pairs to distinguish Ngn3Δint mice from control mice: (a) Ngn3+/flox, 5′ primer CTCGAGGGATCCTCTAGTCGAGGAATTTGA and 3′ primer CGGCAGATTTGAATGAGGGC, resulting in an amplification of a 550-nt DNA fragment; (b) Ngn3+/+ or Ngn3flox/flox, 5′ primer TCTCGCCTCTTCTGGCTTTC and 3′ primer CGGCAGATTTGAATGAGGGC, resulting in an amplification of a 234-nt DNA fragment in the case of Ngn3+/+ and an 627-nt DNA fragment in the case of Ngn3flox/flox; (c) Cre recombinase, 5′ primer GCATTACCGGTCGATGCAACGAGTGATGAG and 3′ primer GAGTGAACGAACCTGGTCGAAATCAGTGCG; (d) Ngn3+/neo, 5′ primer GCAGCGCATCGCCTTCTATC and 3′ primer CGGCAGATTTGAATGAGGGC.
Glucose tolerance test.
For the OGTT, after a 16-hour overnight fast, age- and sex-matched 8- to 9-week-old mice received glucose by intragastric gavage (2 g/kg body weight of 15% d-glucose). For the IPGTT, after an 8-hour fasting period, age- and sex-matched 8- to 9-week-old mice received glucose by intraperitoneal injection (2 g/kg body weight of 15% d-glucose). For the OGTT and the IPGTT, circulating blood glucose was measured in tail blood at 0, 5, 15, 30, 45, 60, 90, and 120 minutes using a Glucofix Sensor from A. Menarini Diagnostics.
ITT.
Six-hour fasted mice of the same sex and age were given an intraperitoneal injection of human insulin (0.5–1 IU/kg depending on the animal; Umuline, Lilly). Circulating blood glucose was measured in tail blood at 0, 15, 30, 45, 60, and 90 minutes using a Glucofix Sensor from A. Menarini Diagnostics.
RT-QPCR analyses.
To isolate tissue of adult mouse, small intestine epithelium was prepared by 4 rounds of incubation at 37°C for 20 minutes in a solution containing PBS without Ca2+ and Mg2+ and 30 mM EDTA, followed by vigorous shaking. Total RNA was then isolated by TRIzol Reagent (Invitrogen). One μg of total RNA was used for cDNA synthesis, using the SuperScript II Reverse Transcriptase (Invitrogen). Quantitative PCR was performed using mouse-specific TaqMan primers and probes recognizing Ngn3 (Mm00437606_s1), chromogranin A (Mm00514341_m1), Insulin-1 (Mm01259683_g1), Cck (Mm00446170_m1), Sct (Mm00441235_g1), Gip (Mm00433601_m1), Glucagon/Glp1 (Mm00801712_m1; Gluc, Glp-1, and Glp-2 are encoded by the same mRNA), tryptophan hydroxylase 1 (Tph1) (Mm00493794_m1), Aqp3 (Mm01208559_m1), Aqp4 (Mm00802131_m1), Aqp8 (Mm00431846_m1), and β2 microglobulin (B2m) (Mm00437762_m1), with TaqMan Light Cycler 480 Probes Master Mix (Roche) on Light Cycler 480 (Roche). Gene expression results were normalized to B2m expression levels.
Immunohistochemistry.
Tissues of adult mice were fixed in 4% paraformaldehyde overnight at 4°C and embedded in paraffin or in Shandon Cryomatrix (Thermo Scientific), in the case of the anti-lysozyme antibody, and 6-μm sections were used. Slides were hydrated, treated in blocking buffer (5% goat serum in PBS, 0.1% Triton X-100 [PBST]) for 30 minutes at room temperature, and incubated with primary antibodies in blocking buffer overnight at 4°C. After washing for 5 minutes 3 times in PBST, the secondary antibodies were added for 1 hour at room temperature in blocking buffer. In the immunofluorescence assays, after washing for 5 minutes 3 times in PBST, nuclei were stained for 5 minutes with Dapi at 1:10,000 in PBS, washed, and mounted in Aqua-poly/mount (Polysciences). For immunohistochemistry analysis, endogenous peroxidase activity was blocked by incubation in 0.5% H2O2 diluted in methanol. After washing in PBST, the signal was revealed using the Vectastain Elite ABC Kit (Vector Laboratories) and the DAB chromogen (DakoCytomation), supplemented with 0.02% H2O2. Slides were dehydrated and mounted in Eukitt (Euromedex). For BrdU detection assays, BrdU was injected into pregnant females at 50 mg/kg body weight, 24 hours prior to dissection. If required, antigen retrieval was performed by incubating slides in 10 mM sodium citrate and microwaving for 5 minutes at 1,000 W and 15 minutes at 300 W. Slides were then allowed to cool down to room temperature for 30 minutes.
The first antibodies used were as follows: Ngn3, the guinea pig anti-NGN3 was used at 1:1,000 and was generated by the Sander’s lab against the same GST-NGN3 fusion protein as described previously (42); chromogranin A, rabbit anti-chromogranin A at 1:500 (Diasorin); CCK/gastrin, rabbit anti-CCK/gastrin at 1:750 (12); insulin, mouse anti-insulin at 1:1,000 (Sigma-Aldrich); glucagon, mouse anti-glucagon at 1:2,000 (Sigma-Aldrich); guinea pig anti-glucagon at 1:1,000 (Linco); global-laminin, rabbit anti-laminin recognizing the 3 constituent chains, α1, β1, and γ1 at 1:1,000 (43, 44); lysozyme, rabbit anti-lysozyme AB at 1:200 (Dako); Ki67, mouse anti-Ki67 at 1:100 (Novo Castra); BrDu, mouse anti-BrdU at 1:00 (Boehringer Mannheim); Glut2, rabbit anti-Glut2 at 1:750 (Chemicon); SGLT1, rabbit anti-SGLT1 at 1:1,000 (Abcam); GLP1, rabbit anti-GLP1 (7-36) at 1:500 (Phoenix); GIP, rabbit anti-GIP at 1:500 (Phoenix), and goat anti-rat sucrase-isomaltase (gift of Jacques Riby, University of California, Berkeley, California, USA).
The secondary antibodies used were as follows: Alexa Fluor 488 anti-mouse at 1:1,000 (Molecular Probes); Alexa Fluor 568 anti-rabbit at 1:1,000 (Molecular Probes); Cy3 anti-rabbit and anti-guinea pig at 1:1,000 (Jackson ImmunoResearch Laboratories Inc.); Alexa Fluor 488 anti-goat at 1:500; biotin-coupled anti-rabbit at 1:200 (Vector Laboratories).
Goblet cells were stained for mucin using the PAS reaction.
Lactase activity.
Detection of lactase activity was performed as previously described (45). Briefly, detection of lactase activity was performed for 30 minutes at room temperature in 0.1 M maleate buffer, pH 6.5, containing 1 mM p-chloromercuribenzoate, 3.5 mM potassium ferrocyanide, 3.5 mM potassium ferricyanide, and 1 mM 5-bromo-4-chloro-3-indolyl-b-fucopyranoside (X-Fuc, Sigma-Aldrich). Slides were rinsed in PBS, dehydrated, and mounted in phenylenediamine with Eukitt (Euromedex).
Intestinal food transit.
Intestinal transit was determined by assessing the appearance in stools of an artificial colored alimentation. Eight-week-old mice from the same sex were fasted for 36 hours and given simultaneously access to powdered chow mixed with a green food dye, and the time of the first colored stools that appeared was noted (27).
Blood analyses.
All described blood analyses were done on adult mice from the same sex and were performed by the ICS ( http://www.ics-mci.fr).
BMI, body fat, and lean mass.
Analyses were performed on 27-week-old mice from the same sex by the ICS ( http://www.ics-mci.fr).
Statistics.
Values are presented as mean ± SD. P values were determined using the 2-tailed Student’s t test with unequal variance. P values of less than 0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank Stéphanie Muller for her expert advice and Caitlin Collin for technical assistance and careful reading of the manuscript. We also thank Nadia Messaddeq for her excellent electron microscopy analyses. We are grateful to the ICS transgenic facility for generation of the Ngn3 mice. This work was supported by INSERM, CNRS, and by a grant from the Agence National de la Recherche (ANR2008 GENOPAT to G. Gradwohl). The G. Gradwohl laboratory is a member of the JDRF Center for BetaCellTherapy in Diabetes, supported by the European Union, and the Beta Cell Biology Consortium, supported by NIDDK. A. Beucher is a recipient of a Fellowship from the Fondation pour la Recherche Médicale.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2010;120(5):1708–1721. doi:10.1172/JCI40794.
References
- 1.Drucker DJ. The role of gut hormones in glucose homeostasis. J Clin Invest. 2007;117(1):24–32. doi: 10.1172/JCI30076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy KG, Bloom SR. Gut hormones and the regulation of energy homeostasis. Nature. 2006;444(7121):854–859. doi: 10.1038/nature05484. [DOI] [PubMed] [Google Scholar]
- 3.Rindi G, Leiter AB, Kopin AS, Bordi C, Solcia E. The “normal” endocrine cell of the gut: changing concepts and new evidences. Ann N Y Acad Sci. 2004;1014:1–12. doi: 10.1196/annals.1294.001. [DOI] [PubMed] [Google Scholar]
- 4.Stoffers DA. The development of beta-cell mass: recent progress and potential role of GLP-1. Horm Metab Res. 2004;36(11–12):811–821. doi: 10.1055/s-2004-826168. [DOI] [PubMed] [Google Scholar]
- 5.Stoffers DA, et al. Insulinotropic glucagon-like peptide 1 agonists stimulate expression of homeodomain protein IDX-1 and increase islet size in mouse pancreas. Diabetes. 2000;49(5):741–748. doi: 10.2337/diabetes.49.5.741. [DOI] [PubMed] [Google Scholar]
- 6.Wang H, et al. Suppression of Pdx-1 perturbs proinsulin processing, insulin secretion and GLP-1 signalling in INS-1 cells. Diabetologia. 2005;48(4):720–731. doi: 10.1007/s00125-005-1692-8. [DOI] [PubMed] [Google Scholar]
- 7.Drucker DJ, Erlich P, Asa SL, Brubaker PL. Induction of intestinal epithelial proliferation by glucagon-like peptide 2. Proc Natl Acad Sci U S A. 1996;93(15):7911–7916. doi: 10.1073/pnas.93.15.7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drucker DJ. Glucagon-like peptides: regulators of cell proliferation, differentiation, and apoptosis. Mol Endocrinol. 2003;17(2):161–171. doi: 10.1210/me.2002-0306. [DOI] [PubMed] [Google Scholar]
- 9.Beglinger C, Degen L. Fat in the intestine as a regulator of appetite--role of CCK. Physiol Behav. 2004;83(4):617–621. doi: 10.1016/j.physbeh.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 10.Rehfeld JF. Clinical endocrinology and metabolism. Cholecystokinin. Best Pract Res Clin Endocrinol Metab. 2004;18(4):569–586. doi: 10.1016/j.beem.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97(4):1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenny M, et al. Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. EMBO J. 2002;21(23):6338–6347. doi: 10.1093/emboj/cdf649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez-Diaz L, et al. Intestinal Neurogenin 3 directs differentiation of a bipotential secretory progenitor to endocrine cell rather than goblet cell fate. Dev Biol. 2007;309(2):298–305. doi: 10.1016/j.ydbio.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee CS, Perreault N, Brestelli JE, Kaestner KH. Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev. 2002;16(12):1488–1497. doi: 10.1101/gad.985002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schonhoff SE, Giel-Moloney M, Leiter AB. Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev Biol. 2004;270(2):443–454. doi: 10.1016/j.ydbio.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 16.Naya FJ, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11(18):2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, et al. Mutant neurogenin-3 in congenital malabsorptive diarrhea. N Engl J Med. 2006;355(3):270–280. doi: 10.1056/NEJMoa054288. [DOI] [PubMed] [Google Scholar]
- 18.Cortina G, et al. Enteroendocrine cell dysgenesis and malabsorption, a histopathologic and immunohistochemical characterization. Hum Pathol. 2007;38(4):570–580. doi: 10.1016/j.humpath.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 19.el Marjou F, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39(3):186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 20.Drozdowski LA, Thomson AB. Intestinal sugar transport. World J Gastroenterol. 2006;12(11):1657–1670. doi: 10.3748/wjg.v12.i11.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Preitner F, et al. Gluco-incretins control insulin secretion at multiple levels as revealed in mice lacking GLP-1 and GIP receptors. J Clin Invest. 2004;113(4):635–645. doi: 10.1172/JCI20518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drucker DJ. The biology of incretin hormones. Cell Metab. 2006;3(3):153–165. doi: 10.1016/j.cmet.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Hui H, Zhao X, Perfetti R. Structure and function studies of glucagon-like peptide-1 (GLP-1): the designing of a novel pharmacological agent for the treatment of diabetes. Diabetes Metab Res Rev. 2005;21(4):313–331. doi: 10.1002/dmrr.553. [DOI] [PubMed] [Google Scholar]
- 24.Ling Z, et al. Glucagon-like peptide 1 receptor signaling influences topography of islet cells in mice. Virchows Arch. 2001;438(4):382–387. doi: 10.1007/s004280000374. [DOI] [PubMed] [Google Scholar]
- 25.Scrocchi LA, et al. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat Med. 1996;2(11):1254–1258. doi: 10.1038/nm1196-1254. [DOI] [PubMed] [Google Scholar]
- 26.Sanger GJ, Lee K. Hormones of the gut-brain axis as targets for the treatment of upper gastrointestinal disorders. Nat Rev Drug Discov. 2008;7(3):241–254. doi: 10.1038/nrd2444. [DOI] [PubMed] [Google Scholar]
- 27.Rahuel C, et al. Genetic inactivation of the laminin alpha5 chain receptor Lu/BCAM leads to kidney and intestinal abnormalities in the mouse. Am J Physiol Renal Physiol. 2008;294(2):F393–F406. doi: 10.1152/ajprenal.00315.2007. [DOI] [PubMed] [Google Scholar]
- 28.Ma T, Verkman AS. Aquaporin water channels in gastrointestinal physiology. J Physiol. 1999;517(Pt 2):317–326. doi: 10.1111/j.1469-7793.1999.0317t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuzaki T, Tajika Y, Ablimit A, Aoki T, Hagiwara H, Takata K. Aquaporins in the digestive system. Med Electron Microsc. 2004;37(2):71–80. doi: 10.1007/s00795-004-0246-3. [DOI] [PubMed] [Google Scholar]
- 30.Drozdowski L, Thomson AB. Intestinal hormones and growth factors: effects on the small intestine. World J Gastroenterol. 2009;15(4):385–406. doi: 10.3748/wjg.15.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435(7044):964–968. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- 32.Riccio O, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 2008;9(4):377–383. doi: 10.1038/embor.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Es JH, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435(7044):959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 34.Hansotia T, et al. Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of DPP-IV inhibitors. Diabetes. 2004;53(5):1326–1335. doi: 10.2337/diabetes.53.5.1326. [DOI] [PubMed] [Google Scholar]
- 35.Van Dyck F, et al. Loss of the PlagL2 transcription factor affects lacteal uptake of chylomicrons. Cell Metab. 2007;6(5):406–413. doi: 10.1016/j.cmet.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh J, et al. Glucagon-like peptide-2 increases intestinal lipid absorption and chylomicron production via CD36. Gastroenterology. 2009;137(3):997–1005, 1005.e1–1005.e4. doi: 10.1053/j.gastro.2009.05.051. [DOI] [PubMed] [Google Scholar]
- 37.Alrefai WA, Gill RK. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm Res. 2007;24(10):1803–1823. doi: 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- 38.Iqbal J, Hussain MM. Intestinal lipid absorption. Am J Physiol Endocrinol Metab. 2009;296(6):E1183–E1194. doi: 10.1152/ajpendo.90899.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Houten SM, Watanabe M, Auwerx J. Endocrine functions of bile acids. EMBO J. 2006;25(7):1419–1425. doi: 10.1038/sj.emboj.7601049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7(8):678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez CI, et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25(2):139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- 42.Henseleit KD, Nelson SB, Kuhlbrodt K, Hennings JC, Ericson J, Sander M. NKX6 transcription factor activity is required for alpha- and beta-cell development in the pancreas. Development. 2005;132(13):3139–3149. doi: 10.1242/dev.01875. [DOI] [PubMed] [Google Scholar]
- 43.Simo P, et al. Changes in the expression of laminin during intestinal development. Development. 1991;112(2):477–487. doi: 10.1242/dev.112.2.477. [DOI] [PubMed] [Google Scholar]
- 44.De Arcangelis A, Neuville P, Boukamel R, Lefebvre O, Kedinger M, Simon-Assmann P. Inhibition of laminin alpha 1-chain expression leads to alteration of basement membrane assembly and cell differentiation. J Cell Biol. 1996;133(2):417–430. doi: 10.1083/jcb.133.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jost B, Duluc I, Vilotte JL, Freund JN. Lactase is unchanged in suckling mice fed with lactose-free milk. Gastroenterol Clin Biol. 1998;22(11):863–867. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.