

Abstract
Here we report a versatile method by which the interaction between a protein and a small molecule, and the disruption of that interaction by competition with other small molecules, can be monitored electrochemically directly in complex sample matrices.
Methods for the detection and quantification of protein–small molecule interactions have proven useful for a wide range of purposes. Examples include drug screening, in which enormous libraries of small molecules are tested for their ability to bind or otherwise inhibit a specific protein, and competition assays to detect small molecule targets via their ability to displace a labeled (e.g., fluorescent) variant bound to a protein-based specificity agent (e.g., an antibody).1
Recent efforts to develop fieldable competition assays for the detection of nitroaromatic explosives, such as TNT, illustrate the need for the convenient (e.g., rapid, robust, portable) detection of protein–small molecule interactions. Current EPA-accepted methods for the detection of these materials in environmental samples, where they represent an important class of pollutants,2 require the chromatographic analysis of organic solvent extracts (e.g., EPA method 83303), which, while sensitive (reported detection limits to ∼0.2 nM4), are cumbersome batch processes ill-suited for field use. In response, a number of alternative methods have been reported in recent years in which TNT is detected indirectly via its ability to compete with (i.e. competitively inhibit) the binding of an anti-TNT antibody to a labeled (e.g., fluorescent) or surface-attached TNT analog. Unfortunately, however, fluorescence-based assays,4–7 while sensitive and convenient, generally fail in samples that are optically dense or display significant autofluorescence.8 Platforms based on surface-attached TNT analogs, such as those based on surface plasmon resonance (SPR),9–12 are likewise sensitive and convenient but typically require complex, multi-step procedures in order to avoid false positives that arise from the non-specific binding of the interferants inevitably present in realistic samples.10 Thus, although some progress has been made with regard to field-portable immunosensors for the detection of TNT, these devices inevitably require lengthy sample preparation to minimize variability.13,14
Motivated by the growing need for convenient, rapid methods to monitor protein–small molecule interactions for analytical purposes, including competition assays and the screening of combinatorial libraries, we report here a bio-electrochemical approach that is not only user-friendly but also selective enough to deploy directly in complex sample matrices such as crude soil extracts and seawater. Our approach is based on the previously described “E-DNA scaffold” platform, a reagentless, highly selective bio-electrochemical method for the detection of protein–small molecule interactions.15 The first generation E-DNA scaffold sensors comprised a double-stranded DNA element in which one strand (the anchoring strand) is tethered to an interrogating electrode and modified with a redox reporter (analogous to Fig. 1).15 The complementary strand (the recognition strand) was modified with a small molecule that binds to the desired target. Upon target binding, the efficiency with which the redox tag can approach the electrode is reduced, impeding electron transfer and generating a readily measurable change in faradaic current. Earlier applications of E-DNA scaffold sensors were focused on the use of a small molecule recognition element for the detection of specific protein targets as the binding of such macromolecules significantly reduces the collision rate of the attached redox probe.
Fig. 1.
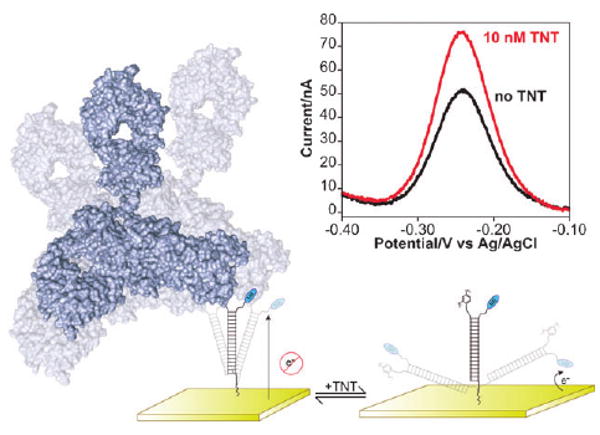
Here we demonstrate a method for monitoring protein–small molecule interactions. Our approach employs a DNA probe, one end of which is chemi-adsorbed to an interrogating electrode and the other end of which is modified with a recognition element and with a redox reporter. The binding of a protein to the recognition element reduces the efficiency with which the redox reporter approaches the electrode, minimizing the observed faradaic current. The addition of a small molecule that disrupts protein binding reverses this effect. (Inset) Shown are representative square wave voltammograms illustrating the competitive detection of TNT via this approach.
Here we explore alternative geometries for the E-DNA scaffold sensing architecture and employ the approach to monitor the binding of a family of small molecules, the nitroaromatic explosives, to an anti-TNT antibody. Specifically, we have fabricated a set of sensors employing the small molecule recognition element dinitrophenyl (DNP). In the presence of anti-TNT antibodies (e.g., clone TNT A/1.1.1 described in ref. 16) the signaling current of the sensor is suppressed. When challenged with TNT or other cross-reactive molecules,10,16 the antibody releases from the probe, producing a readily measurable increase in signaling current (Fig. 1), presumably due to an associated increase in probe dynamics (see Fig. S1, ESI†).
In an effort to optimize sensor performance, we have explored both double- and single-stranded probes. The double-stranded architecture, which is analogous to the first reported sensors in this class,15 utilizes a 27-base anchoring strand containing a 5′ thiol and a 3′ redox tag (methylene blue). A DNP-containing recognition strand complementary to (and centered on) this anchoring strand places the DNP distal to the electrode (Fig. 1). We find that, as with earlier sensors,15 the signal gain of this architecture is a function of the length (and thus rigidity) of the double-stranded region of the probe and the density with which the probe DNAs are packed onto the sensor surface.15 Varying the length of the recognition strand from 15 to 25 bases (centered on a 27-base anchoring strand) we find that optimal signal change is obtained with a 25-base recognition strand (strand DNP25; Fig. S2, ESI†) with no significant change in target affinity (Fig. S2, ESI†). This contrasts with the ∼19-base optima observed for other sensors in this class,15 indicating that the optimal recognition strand length likely depends on the size and geometry of the target. The gain of the sensor also increases strongly with decreasing probe density, a trend that continues to the lowest densities that produce a measurable faradaic current (Fig. S3, ESI†), an effect that has been observed previously.15 As the signaling mechanism relies on a binding-induced change in dynamics, higher surface coverage likely results in smaller signal changes due to reduced collision dynamics in the target-free state.
In an effort to better understand the “fabrication-space” of these sensors, we have explored 17-base, single-stranded probe DNAs containing an anchoring thiol on their 5′ termini and a redox tag and DNP recognition element on their opposite termini (Fig. S4, ESI†). We did so motivated by the argument that the dynamics of single-stranded probes will also be modulated by the binding of an antibody. We have explored two single-stranded probes differing only in the precise placement of the redox tag and the recognition element: for probe sequence TL1 the redox tag is located on the 3′ terminus and the DNP receptor is on a thymine 3 bases away, and for probe sequence TL2 these are reversed. Unfortunately, however, neither of these single-stranded probes respond robustly to the presence of anti-TNT antibodies. That is, while the double-stranded scaffold exhibits a 42% reduction in signal (using recognition strand DNP25) at 30 nM antibody, the two single-stranded scaffolds exhibit only 14% and 4% signal suppression under these same conditions (TL1 and TL2 respectively, Fig. S4, ESI†). The poor signaling of the single-stranded probes presumably occurs because the dynamics of these highly flexible, single-stranded probes do not change significantly upon target binding, minimizing measured signal change [see, by analogy, ref. 17]. The slightly greater suppression observed with probe TL1 may be the result of steric interactions between the antibody and the redox tag; for this construct the antibody binds between the redox tag and electrode surface. Given the generally poor performance of our single-stranded probes, our follow-on studies employed only double-stranded probes.
Optimal signaling upon TNT detection is achieved at an intermediate concentration of the antibody. If the antibody concentration is too low, some probes are unoccupied even in the absence of the small molecule target, thus increasing the background current observed in the absence of TNT. Alternatively, the sensor's detection limit will also be degraded if the concentration of anti-TNT antibody is too great as excess antibody will sequester some of the free TNT without contributing to the observed signal change. We accordingly employed our antibody at the 4 nM affinity of DNP25 in order to balance these competing effects.
Our electrochemical assay readily and conveniently detects the interaction between TNT and its antibody, and thus, in turn, it can also sense TNT itself at low nanomolar (parts-per-trillion) concentrations. For example, the IC50 (the concentration at which the signal change is half that at saturation) of the optimized sensor is just 3 nM, corresponding to 700 ppt (Fig. 2, top). The approach is also selective: the observed TNT IC50 is not changed when the sensor is challenged with complex samples such as seawater and soil extracts (Fig. 2, top, inset), and sensor performance remains excellent when challenged with crude cell lysates, further supporting its potential utility in, for example, the screening of candidate drug libraries (Fig. S5, ESI†). In terms of convenience, the sensor is rapid and reusable. For example, at 50 nM TNT the sensor exhibits single exponential relaxation kinetics with a time constant of ∼2 min (Fig. 2, bottom). Finally, the sensor is readily reusable: the addition of saturating TNT (to release the antibody) followed by rinsing with buffer and adding fresh antibody regenerates the sensor, allowing it to be reused multiple times without exhibiting significant degradation (Fig. S6, ESI†).
Fig. 2.
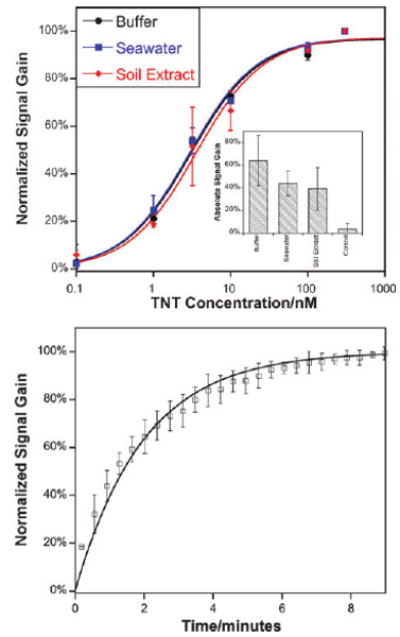
(Top) As shown, the E-DNA scaffold readily detects protein–small molecule interactions even in complex samples such as seawater and crude soil extracts. The concentrations of half maximal inhibition (IC50) observed in these complex sample matrices are within error of the 3.1 ± 0.5 nM observed in buffer. Data represent titrations normalized to the same final signal gain. The inset shows the average absolute signal gain for each condition.15 (Bottom) The assay is rapid; shown are normalized sensor responses of three independently fabricated sensors upon the addition of 50 nM TNT. The error bars in these panels and the following figure represent the standard deviation of three electrodes and are dominated by inter-electrode variability.
The specificity of the assay (the ability to discriminate between TNT and its close analogs) is high (Fig. 3), and is comparable to that of the antibody itself.16 For example, whereas the IC50 of TNT is just 3 nM, the IC50 of the close analog 2,6-DNT is some three orders of magnitude higher and that of dinitrophenol another order of magnitude higher still, comparable to prior studies.16 Likewise, no binding is observed for 4-(dimethylamino)phenylacetic acid, a small aromatic compound of similar size lacking nitro groups.
Fig. 3.
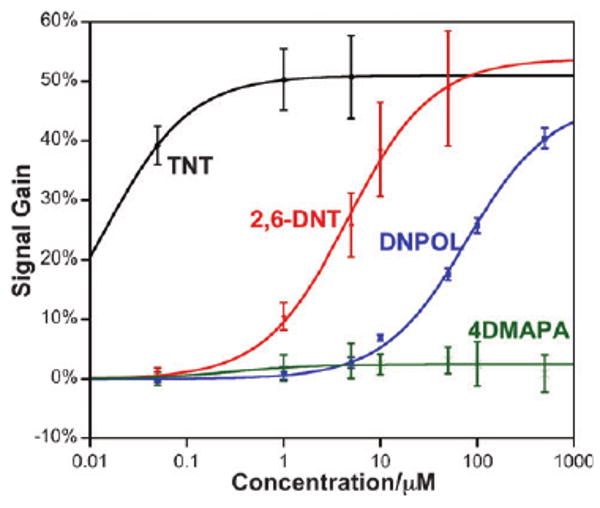
Our approach is as specific as the protein–small molecule interaction upon which it is based. For example, as expected,16 the IC50 for 2,6-DNT is orders of magnitude poorer than that of TNT, and the IC50 for dinitrophenol (DNPOL) is another order of magnitude poorer still. No detectable response is observed for the non-nitroaromatic control compound 4-(dimethylamino)phenylacetic acid (4DMAPA) even at the highest concentrations we have employed.
Here we have demonstrated an electrochemical assay by which small molecule–protein interactions are rapidly and conveniently monitored even in crude, highly complex sample matrices. The approach appears to be versatile, requiring only the ability to conjugate an appropriate small molecule recognition element to DNA in a manner that does not significantly disrupt protein binding. Moreover, as an electrochemical method our approach is easily scalable and parallelizable18 for applications such as microtiter plate-based assays.19 These attributes suggest our approach is well suited for both massively parallel drug screening in the laboratory and the detection of specific small molecules in the field.
Supplementary Material
Acknowledgments
This work was supported by the NIH through grant GM062958-01 and by the Institute for Collaborative Biotechnologies through grant DAAD19-03-D-0004 from the US Army Research Office. K. J. C. is supported by funds from the California HIV/AIDS Research Program of the University of California, Grant Number D07-SB-417.
Footnotes
Electronic supplementary information (ESI) available: Detailed experimental methods as well as additional experimental data. See DOI: 10.1039/b911558g
Notes and references
- 1.Silverman L, Campbell R, Broach JR. Curr Opin Chem Biol. 1998;2:397–403. doi: 10.1016/s1367-5931(98)80015-x. [DOI] [PubMed] [Google Scholar]
- 2.Spain JC. Annu Rev Microbiol. 1995;49:523–555. doi: 10.1146/annurev.mi.49.100195.002515. [DOI] [PubMed] [Google Scholar]
- 3.U. S. EPA. Washington, DC: 2006. [Google Scholar]
- 4.Narang U, Gauger PR, Ligler FS. Anal Chem. 1997;69:1961–1964. [Google Scholar]
- 5.Anderson GP, Moreira SC, Charles PT, Medintz IL, Goldman ER, Zeinali M, Taitt CR. Anal Chem. 2006;78:2279–2285. doi: 10.1021/ac051995c. [DOI] [PubMed] [Google Scholar]
- 6.Bromage ES, Lackie T, Unger MA, Ye J, Kaattari SL. Biosens Bioelectron. 2007;22:2532–2538. doi: 10.1016/j.bios.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 7.Goldman ER, Medintz IL, Whitley JL, Hayhurst A, Clapp AR, Uyeda HT, Deschamps JR, Lassman ME, Mattoussi H. J Am Chem Soc. 2005;127:6744–6751. doi: 10.1021/ja043677l. [DOI] [PubMed] [Google Scholar]
- 8.Owicki JC. J Biomol Screening. 2000;5:297–306. doi: 10.1177/108705710000500501. [DOI] [PubMed] [Google Scholar]
- 9.Kawaguchi T, Shankaran DR, Kim SJ, Gobi KV, Matsumoto K, Toko K, Miura N. Talanta. 2007;72:554–560. doi: 10.1016/j.talanta.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 10.Mizuta Y, Onodera T, Singh P, Matsumoto K, Miura N, Toko K. Biosens Bioelectron. 2008;24:191–197. doi: 10.1016/j.bios.2008.03.042. [DOI] [PubMed] [Google Scholar]
- 11.Shankaran DR, Kawaguchi T, Kim SJ, Matsumoto K, Toko K, Miura N. Anal Bioanal Chem. 2006;386:1313–1320. doi: 10.1007/s00216-006-0699-4. [DOI] [PubMed] [Google Scholar]
- 12.Shankaran DR, Matsumoto K, Toko K, Miura N. Sens Actuators, B. 2006;114:71–79. [Google Scholar]
- 13.Ciumasu IM, Kramer PM, Weber CM, Kolb G, Tiemann D, Windisch S, Frese I, Kettrup AA. Biosens Bioelectron. 2005;21:354–364. doi: 10.1016/j.bios.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 14.Gauger PR, Holt DB, Patterson CH, Charles PT, Shriver-Lake L, Kusterbeck AW. J Hazard Mater. 2001;83:51–63. doi: 10.1016/s0304-3894(00)00327-7. [DOI] [PubMed] [Google Scholar]
- 15.Cash KJ, Ricci F, Plaxco KW. J Am Chem Soc. 2009;131:6955–6957. doi: 10.1021/ja9011595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeck A, Weller MG, Niessner R. Fresenius' J Anal Chem. 1999;364:113–120. [Google Scholar]
- 17.Ricci F, Lai RY, Plaxco KW. Chem Commun. 2007:3768–3770. doi: 10.1039/b708882e. [DOI] [PubMed] [Google Scholar]
- 18.Swensen JS, Xiao Y, Ferguson BS, Lubin AA, Lai RY, Heeger AJ, Plaxco KW, Soh HT. J Am Chem Soc. 2009;131:4262–4266. doi: 10.1021/ja806531z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piermarini S, Micheli L, Ammida NHS, Palleschi G, Moscone D. Biosens Bioelectron. 2007;22:1434–1440. doi: 10.1016/j.bios.2006.06.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.