

Abstract
We describe in detail a new electrochemical DNA (E-DNA) sensing platform based on target-induced conformation changes in an electrode-bound DNA pseudoknot. The pseudoknot, a DNA structure containing two stem-loops in which the first stem’s loop forms part of the second stem, is modified with a methylene blue redox tag at its 3′ terminus and covalently attached to a gold electrode via the 5′ terminus. In the absence of a target, the structure of the pseudoknot probe minimizes collisions between the redox tag and the electrode, thus reducing faradaic current. Target binding disrupts the pseudoknot structure, liberating a flexible, single-stranded element that can strike the electrode and efficiently transfer electrons. In this article we report further characterization and optimization of this new E-DNA architecture. We find that optimal signaling is obtained at an intermediate probe density (~1.8 × 1013 molecules/cm2 apparent density), which presumably represents a balance between steric and electrostatic blocking at high probe densities and increased background currents arising from transfer from the pseudoknot probe at lower densities. We also find that optimal 3′ stem length, which appears to be 7 base pairs, represents a balance between pseudoknot structural stability and target affinity. Finally, a 3′ loop comprised of poly(A) exhibits better mismatch discrimination than the equivalent poly(T) loop, but at the cost of decreased gain. Optimization over this parameter space significantly improves the signaling of the pseudoknot-based E-DNA architecture, leading to the ability to sensitively and specifically detect DNA targets even when challenged in complex, multicomponent samples such as blood serum.
The rapid, sequence-specific detection of nucleic acids would be of value in the detection of pathogens across a range of clinical, environmental, and food safety applications.1,2 Consequently, a large number of electronic DNA detection methods have been described to date.3–8 However, the large majority of these technologies require the addition of exogenous, label-containing secondary probes and/or complicated, multicomponent deposition/amplification, and washing steps.3–8 In response to these relatively cumbersome methods, electrochemical DNA (E-DNA) sensors have been developed that operate by detecting hybridization-induced conformational changes in a redox-modified, electrode-bound DNA probe.9–14 These E-DNA sensors require no addition of reagents or target labeling, and detection is a rapid single-step process. Additionally, as the signaling mechanism is linked to a specific conformational change, these sensors are capable of functioning in complex, multicomponent samples such as blood serum.10
Both signal-on and signal-off E-DNA architectures have been reported. In a “signal-off” sensor target binding limits collisions between the redox tag and the electrode thereby reducing the signaling current.9–12 Examples include sensors based on electrode-bound stem loops9,10 and linear, single-stranded DNA probes.11,12 In the case of “signal-on” sensors the presence of target DNA leads to an increase in faradaic current. Examples include architectures in which target binding induces the probe to form a hairpin structure, forcing the redox tag into close proximity with the electrode,13 and a strand-displacement approach in which target binding liberates a flexible, single-stranded signaling element that improves electron transfer efficiency and increases redox current.14
Both signal-on and signal-off E-DNA sensors produce a signal indicating the presence of target DNA without the addition of exogenous reagents. Each approach, however, has its own strengths and limitations. For example, the signal-off E-DNA is very stable because it only consists of a single, continuous DNA probe.9–11 As a result they are readily reusable, sequence-specific, and selective enough to perform measurements even when placed directly in blood serum, soil, foodstuffs, or other complex materials.10 As signal-off sensors, however, their gain may be limited because targets can suppress no more than 100% of the original current. In contrast, the previously reported signal-on architectures consisting of a redox modified DNA sequence have the potential for greatly improved sensitivity because the gain increases without limit under ideal conditions but at a cost of greater complexity and, often, poorer stability and reusability. The first reported signal-on E-DNA sensor, for example, requires a complicated, difficult synthesis step of DNA-polymer-DNA triblock signaling probe.13 Likewise, we have reported an E-DNA design that uses a strand-displacement mechanism, exhibits exceptional gain, and is comprised of a readily synthesized, double-stranded DNA probe.14 But because this sensor contains a non-covalent signal-generating strand it does not perform well in complex samples matrixes (where the two probe strands dissociate) and is not readily reusable.
In response to the above concerns, we have recently developed an E-DNA sensor based on the target-induced resolution of an electrode-bound DNA pseudoknot15 (Figure 1). Being fully covalent and comprised of a single, readily synthesized DNA probe stand, the new signal-on architecture exhibits the stability, reusability, and simplicity of synthesis of earlier signal-off sensor designs. Here we report the further characterization and optimization of this new electrochemical sensing approach, and compare the specificity of different pseoduknot structure-based sensors against mismatch targets in simple buffer and in a complex matrix of blood serum.
Figure 1.

Signal generation in the pseudoknot electrochemical DNA (E-DNA) sensor occurs via a binding induced conformation change in a redox-tagged, electrode-bound probe DNA. (Left) In the absence of a target the probe’s pseudoknot structure holds the methylene blue tag away from the interrogating electrode, limiting electron transfer. (Right) Upon addition of a complementary target the pseudoknot unfolds, allowing the redox tag to collide with the electrode which, in turn, increases electron transfer.
EXPERIMENTAL SECTION
Reagents
Tris-(2-carboxyethyl) phosphine hydrochloride (TCEP), 6-mercaptohexanol, and fetal calf serum were purchased from Sigma-Aldrich, Inc. (U.S.A.) and used as received without further purification. Thiolated, methylene blue (MB)-tagged DNA probes were synthesized and purified by Biosearch Technologies, Inc. (Novato, CA). The sequences of the modified oligomers employed are
(1) 5′-HS(CH2)6-GGCGAGGTA4CGACGGCCAGCCTCGCCG-A16GCCGTCG-(CH2)7-MB-3′
(2) 5′-HS(CH2)6-GCGAGGTA4CGACGGCCAGCCTCGCG-A16GCCGTC-(CH2)7-MB-3′
(3) 5′-HS(CH2)6GGCGAGGTA4CGACGGCCAGCCTCGCCG-A16GCCGTCGT-(CH2)7-MB-3′
(4) 5′-HS(CH2)6-GGCGAGGTA4CGACGGCCAGCCTCGCCG-T16GCCGTCG-(CH2)7-MB-3′
All target oligonucleotides were synthesized and purified by Integrated DNA Technologies Inc. (Coralville, IA). The sequences of these targets, with mismatch locations underlined, are
Perfectly matched DNA target (PM): 5′-GCTGGCCGTCGTTTTAC-3′
1-base mismatched DNA target (1MM): 5′-ACTGGCCGTCGTTTTAC-3′
2-base mismatched DNA target (2MM): 5′-ACTGGCCCTCGTTTTAC-3′
3-base mismatched DNA target (3MM): 5′-GCTGGAATTCGTTTTAC-3′
Electrode Cleaning and E-DNA Sensor Preparation
The sensors were fabricated on polycrystalline gold disk electrodes (1.6 mm diameter; BAS, West Lafayette, IN). The electrodes were cleaned according to a published procedure.16 After cleaning the electrodes were modified with the relevant probe DNA by immersion in a 0.1 μM solution of thiolated MB-labeled DNA oligomer in a high salt phosphate buffer (100 mM phosphate, 1.5 M NaCl, 1 mM Mg2+, pH = 7.2) for 16 h at room temperature. Prior to immobilization, the DNA oligomers were incubated for 1 h in 2 μM TCEP to reduce disulfide bound oligomers. Following probe immobilization, the electrode surface was rinsed with deionized water, then passivated by immersion in 1 mM 6-mercaptohexanol in high salt phosphate buffer for 4 h at room temperature. The electrodes were then rinsed with deionized water and stored in high salt phosphate buffer. The stability of probe-modified electrodes was monitored through multiple electrochemical interrogations (see below).
Electrochemical Measurements
Alternating-current voltammetry (ACV) was performed on a CHI 603 potentiostat (CH Instruments, Austin, TX) in a standard cell with a platinum wire counter electrode and a Ag/AgCl reference electrode for all measurements. These measurements were conducted either in the high salt phosphate buffer or in fetal calf serum diluted to 10%, 20%, 30%, and 50% by phosphate buffer with NaCl to control pH and give a final NaCl concentration of 1.5 M. This buffer was employed to improve both signal gain and sensor signal stability; studies at higher and lower ionic strength (data not shown) indicate that this ionic strength produces optimal performance. The ionic strength was controlled to avoid nonspecific signal changes of duplexes resulting from the changes of ionic strength between buffer and serum. The sensors were incubated in each sample for 1 h at room temperature before being monitored using ACV measurements with a step potential of 10 mV, amplitude of 25 mV, and a frequency of 100 Hz. E-DNA sensor regeneration was obtained via a simple 30 s deionized water rinse at room temperature. Surface density of DNA oligomers was determined using cyclic voltammetry17 and assuming perfect electron transfer efficiency. Given that this assumption is likely incorrect, the reported probe densities should be considered relative rather than absolute measurements.
RESULTS AND DISCUSSION
The foundation of our sensor is a single DNA strand that forms a pseudoknot containing two stem-loops in which the first loop forms one strand of the second stem.18 To convert the pseudoknot structure into a reagentless, electrochemical DNA sensor, its 5′ terminus is covalently attached to an interrogating electrode via gold-thiol chemistry, and its 3′ terminus is modified with the redox reporter methylene blue (MB)19 (Figure 1, left). In the absence of a target the pseudoknot structure presumably fixes the MB tag away from the electrode, reducing faradaic current. Hybridization with the target sequence liberates a single-stranded element, allowing the MB to strike the electrode and efficiently transfer electrons (Figure 1, right). Because they support a wide range of loop and stem lengths, pseudoknots represent a diverse group of DNA structures each, presumably, with differing signaling characteristics when employed in the E-DNA sensor. We report on efforts to optimize our probe pseudoknots to achieve optimal E-DNA performance.
Effect of 3′ Stem Length on Signaling
Stem stability, which is a function of stem length, greatly influences sensor performance. While a longer 3′ stem likely stabilizes the pseudoknot conformation, reducing background current and improving gain, it concomitantly reduces target affinity as a result of stem stability. To optimize 3′ stem length we have fabricated E-DNA sensors containing 6-, 7-, or 8-base pairs (bp) at the 3′ terminus, each containing the same 7-bp sequence as its 5′ stem (Figure 2, left). All three constructs include the same target recognition element contained within the 5′ terminal loop (5′ loop), and a poly(A) sequence in the 3′ terminal loop (3′ loop) which, when librated by target binding, allows the MB tag to have flexibility to collide with the electrode surface.
Figure 2.

E-DNA signaling is sensitive to the length (and thus stability) of the 3′ stem. (Left) Here we have characterized sensors employing 3′ stems of 6, 7, or 8 base pairs (the 5′ stem was held fixed at 7 bp). (Right) Among these constructs optimal signal gain (defined as the relative current change observed in the presence of a saturating, perfectly matched target) is observed with a 3′ stem length of 7 base pairs. Of note, the estimated surface coverage of DNA (1.2 × 1013 molecules/cm2) and the concentration of target DNA (100 nM) were held fixed for all constructs in Figures 2–4.
As expected, E-DNA signaling is sensitive to the stability of the 3′ stem upon target recognition. For example, a 7-bp 3′ stem pseudoknot (1) produces a 50% signal change when challenged with its 17-base, perfectly matched (PM) target (at 100 nM) (Figure 2, right). In contrast, under the same conditions, a 6-bp stem pseudoknot (2) produces only a 22% increase in current (Figure 2, right), presumably because the decreased stability of this pseudoknot produces a larger background current. Further, the 8-bp (3) stem pseudoknot only exhibits 18% signal gain (Figure 2, right), presumably because of the increased stability of this pseudoknot structure; the hybridization of a 17-base target would likely compete poorly with the free energy required to open the 15 total base pairs contained within this structure. Consistent with these mechanistic speculations, the estimated melting temperatures (Tm) of the three pseudoknots (from Mfold20) are 65.6, 75.0, and 80.9 °C for 6-bp, 7-bp, and 8-bp stems, respectively.
Effect of the 3′ Terminal Loop Sequence on Signaling
The signaling mechanism of the pseudoknot E-DNA sensor is a strong function of the flexibility of the 3′ terminal loop (3′ loop), which is likely dependent on the composition of the sequence. Specifically, Bonnet has found that poly(T)-containing molecular beacons are far more flexible than the equivalent constructs containing poly(A), which may be due to increased stacking interactions among adenines or the smaller size of thymines.21,22 Consistent with this, we find that the flexibility of this loop affects sensor performance: when challenged with a perfectly matched target, the signal gain resulting from the flexible poly(T) containing probe is twice that of the corresponding less flexible poly(A) containing probe (Figure 3).
Figure 3.

(Left) Composition of the 3′ loop affects E-DNA signaling. (Right) The gain of sensors employing a more flexible poly(T) loop is twice that of sensors employing a less flexible poly(A) loop (both constructs contain 7-bp stems).
Effect of Probe Density on Sensor Performance
DNA probe sensor surface density is an important parameter in optimizing the signal gain of E-DNA sensors.23 We varied the surface coverage of a pseudoknot probe (composed of the 3′ poly (T) loop with fixed 7-bp stems at each termini) from 6 × 1011 to 3.6 × 1013 molecules/cm2. The probe density increases monotonically with increasing probe concentration until an apparent density of 3.6 × 1013 molecules/cm2 is obtained. We find that apparent probe densities below 6 × 1011 molecules/cm2 fail to produce stable, active monolayers (data not shown). In contrast, apparent probe densities above 6 × 1011 molecules/cm2 give rise to well-defined, reproducible peak currents (all RSD < 5%).
Sensor gain is a complex function of probe packing density. Maximum gain (100%) is observed at intermediate packing densities (1.8 × 1013 molecules/cm2) and falls at lower and higher densities. For example, at higher probe densities (~3.0 × 1013 molecules/cm2) the gain is only two-thirds of the maximum signal gain (at 100 nM target). Similarly, at lower probe densities (below 2.4 × 1012 molecules/cm2), the gain also falls slightly. We presume that this behavior arises because of two competing effects: whereas at high probe density target accessibility may be limited because of the crowded, highly charged DNA monolayer,23 at low probe densities even the formed pseudoknot structure may be capable of colliding with the surface, increasing the background current and thereby decreasing signal despite increasing hybridization. Further insight can be gained through calculating the mean probe-to-probe spacing for the observed surface densities. For optimal surface coverage, spacing is 2.5 nm, whereas the highest and lowest surface densities used show spacing of 1.8 and 14 nm, respectively (based on the assumption of evenly distributed, close packed pseudoknots). An estimate for the pseudoknot diameter, based on the known structure of a RNA pseudoknot of similar length is 2 nm.24 This suggests that optimal surface coverage reflects a compromise: if the pseudoknots are close, interactions between them limit detrimental collisions with the surface in their native, unbound state, but close packing likewise leads to steric and/or electrostatic repulsion of the target.
Specificity, Selectivity, and Regenerability of Pseudoknot-Based E-DNA Sensors
We have previously noted that the specificity of the signal-on pseudoknot-based sensor is significantly better than that of the original, signal-off E-DNA sensor.15 To more fully characterize this specificity, we have measured the signal difference among perfectly matched, and 1bp-, 2bp-, or 3bp-mismatched DNA targets. These experiments were conducted with pseudoknot-based DNA sensors with two fixed 7-bp stems and employing either the poly (T) or the poly (A) 3′ loops. Despite having a lower overall signal gain, we find that the poly(A) pseudoknot-based sensors show better mismatch discrimination compared with the poly(T) sensors. Using the poly(A) pseudoknot the signal gain from a perfect match (PM) target is 49%. The same concentration of a 1bp-mismatch target (1MM) yields a signal gain of roughly a third of the PM target. When the poly(T) pseudoknot is used, the signal gain for a PM target is 100%, but the signal gain from 1MM is over half of the PM signal (Figure 4). The poly(T) system shows a higher signal gain but at the cost of decreased specificity. The reason for the specificity difference between the two constructs remains unclear.
Figure 4.
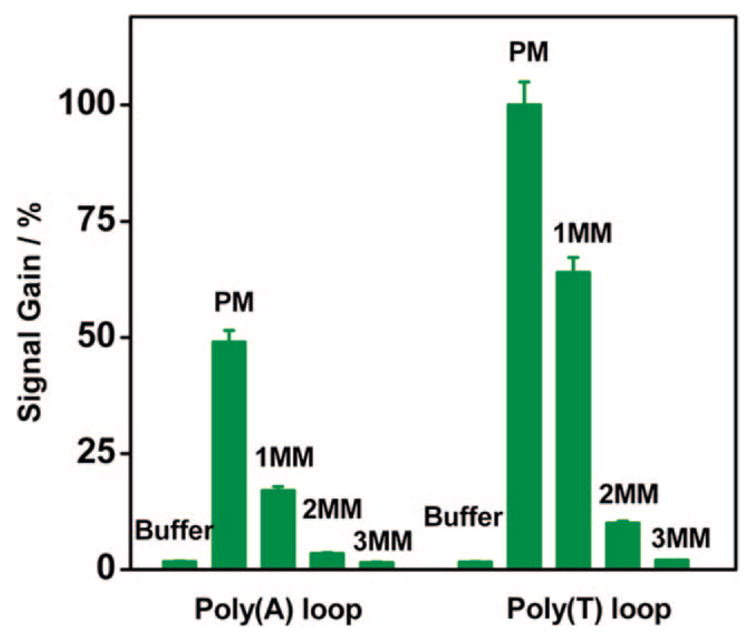
Despite decreased signal gain, the poly(A) loop construct exhibits better mismatch discrimination. The signal gain of the poly(A) sensor against a perfectly matched (PM) target is three times greater than that observed for a single mismatched target (1MM). In contrast, this ratio is only 1.6 for a poly(T) sensor, indicating a poorer level of discrimination. Neither structure responds significantly to a two or three mismatched target (2MM and 3MM respectively).
Signal generation in this new E-DNA sensor is based on a specific, binding-induced conformational change in the pseudoknot probe, and thus the sensor should be relatively impervious to false signals arising because of the nonspecific adsorption of interferants to the sensor surface. Consistent with this, the sensor responds selectively even when challenged with realistically complex samples. For example, target-free serum samples produce less than a 2% change in peak current relative to those observed in simple phosphate buffer, and the signal gain observed at 100 nM target is almost indistinguishable between these two very different sample matrixes (Figure 5). These results were consistent with previous studies showing that a similar DNA-based sensor can be stored in excess of 1 week in room temperature blood serum without exhibiting substantial degradation or loss of function.25 Likewise, the sensor’s specificity is unaffected by complex sample matrixes. For example, the 7-bp stem and 3′ poly(T) loop pseudoknot-based sensor readily distinguishes mismatched targets from the perfectly matched target in blood serum. We observe an average signal gain of 98% in serum doped with the perfectly matched target but only 65% for a 1-bp mismatched, 9% for a 2-bp mismatched, and less than 2% for 3-bp mismatched targets (at a concentration of 100 nM, Figure 5). Additionally, less than a 2% signal change is measured using a 3-bp mismatched DNA target in 20-fold excess over the amount of the perfectly matched target employed (data not shown). Of note, the effectively indistinguishable performance in buffer and serum occurs despite the presumably poor passivation properties of the hydroxyl-terminated monolayer covering our electrodes. Similar selectivity, however, has been observed in numerous other sensors in this class.10,11,15
Figure 5.
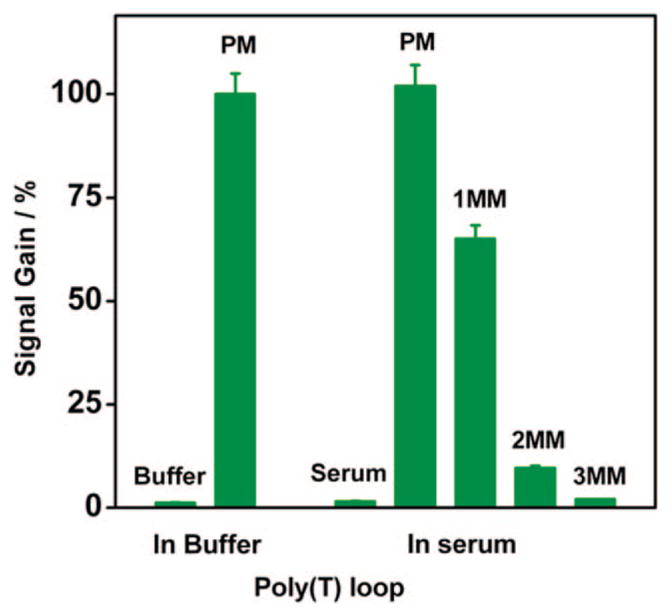
Selectivity and stability of the pseudoknot E-DNA sensor are excellent, allowing for the sensitive, specific detection of DNA directly in blood serum. Measurements taken in 50% blood serum (diluted with phosphate buffer) show the same response to those taken in pure buffer.
Because the MB-tagged pseudoknot is a single, fully covalent DNA strand strongly chemi-adsorbed to its interrogating gold electrode, our sensor is readily reusable. A low ionic strength wash (30 s in room temperature deionized water) is sufficient to produce effective recovery (96%) of the original sensor signal (for intermediate probe densities) even for sensors that have been challenged directly in 50% blood serum.15 This regeneration allows for E-DNA sensor reuse for more than six times with a mean recovery of >96% of the original signal before significant degradation is observed.15 In contrast, low and high-density sensors exhibit only 75% and 82% recovery, respectively, presumably because of the poorer organization or stability of these probe DNA monolayers (data not shown).
CONCLUSIONS
It has previously been shown that E-DNA-like sensing is supported by a wide range of binding-induced conformational changes, including hybridization-linked disruption of a DNA stem-loop,9 target-induced DNA strand displacement,14 and the binding-induced folding of DNA aptamers.26 This work demonstrates a sensitive electrochemical DNA detection platform that is based on a target-induced resolution of an electrode-bound DNA pseudoknot tertiary structure. We find that varying the length of the 3′ stem and the sequence composition of the 3′ loop optimize the signaling characteristics of this sensor, including both its gain and its specificity. For example, the 1:2 ratio of signal gain between the 3′ poly(A) loop and the 3′ poly(T) loop of pseudoknot sensors indicates that a more flexible 3′ loop results in a larger signal change upon target recognition. However, the poly(A) loop can give better discrimination ability against mismatched targets in comparison with the poly(T) loop.
This fully covalent E-DNA architecture couples stability, sensitivity, and reusability while simultaneously exhibiting excellent specificity for mismatched DNA targets directly in blood serum. Taken with the observation that pseudoknot structure might be broken by target binding induced-folding of aptamers, and given that any single-stranded nucleic acid sequence can be designed to form a pseudoknot structure, this sensor architecture may also prove to be a general platform for the detection of non-nucleic acid targets such as proteins or small molecules in complex samples.
Acknowledgments
This work was supported by the Institute for Collaborative Biotechnologies through Grant DAAD19-03-D-0004 from the U.S. Army Research Office, and by the National Institutes of Health (EB007689-02).
References
- 1.Patolsky F, Lichtenstein A, Willner I. J Am Chem Soc. 2001;123:5194–5205. doi: 10.1021/ja0036256. [DOI] [PubMed] [Google Scholar]
- 2.Wang J. Chem–Eur J. 1999;5:1681–1685. [Google Scholar]
- 3.Hwang S, Kim E, Kwak J. Anal Chem. 2005;77:579–584. doi: 10.1021/ac048778g. [DOI] [PubMed] [Google Scholar]
- 4.Park SJ, Taton TA, Mirkin CA. Science. 2002;295:1503–1506. doi: 10.1126/science.1067003. [DOI] [PubMed] [Google Scholar]
- 5.Gore MR, Szalai VA, Ropp PA, Yang IV, Silverman JS, Thorp HH. Anal Chem. 2003;75:6586–6592. doi: 10.1021/ac034918v. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Liu GD, Zhu QY. Anal Chem. 2003;75:6218–6222. doi: 10.1021/ac034730b. [DOI] [PubMed] [Google Scholar]
- 7.Kim E, Kim K, Yang H, Kim YT, Kwak J. Anal Chem. 2003;75:5665–5672. doi: 10.1021/ac034253x. [DOI] [PubMed] [Google Scholar]
- 8.Floch LF, Ho HA, Harding-Lepage P, Bedard M, Neagu-Plesu R, Leclerc M. Adv Mater. 2005;17:1251–1254. [Google Scholar]
- 9.Fan CH, Plaxco KW, Heeger AJ. Proc Natl Aad Sci USA. 2003;100:9134–9137. doi: 10.1073/pnas.1633515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lubin AA, Lai RY, Baker BR, Heeger AJ, Plaxco KW. Anal Chem. 2006;78:5671–5677. doi: 10.1021/ac0601819. [DOI] [PubMed] [Google Scholar]
- 11.Ricci F, Lai RY, Plaxco KW. Chem Commun. 2007:3768–3770. doi: 10.1039/b708882e. [DOI] [PubMed] [Google Scholar]
- 12.Anne A, Bouchardon A, Moiroux J. J Am Chem Soc. 2003;125:1112–1113. doi: 10.1021/ja028640k. [DOI] [PubMed] [Google Scholar]
- 13.Immoos CE, Lee SJ, Grinstaff MW. J Am Chem Soc. 2004;126:10814–10815. doi: 10.1021/ja046634d. [DOI] [PubMed] [Google Scholar]
- 14.Xiao Y, Lubin AA, Baker BR, Plaxco KW, Heeger AJ. Proc Natl Aad Sci USA. 2006;103:16677–17780. doi: 10.1073/pnas.0607693103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao Y, Qu XG, Plaxco KW, Heeger AJ. J Am Chem Soc. 2007;129:11896–11897. doi: 10.1021/ja074218y. [DOI] [PubMed] [Google Scholar]
- 16.Xiao Y, Lai RY, Plaxco KW. Nat Protoc. 2007;2:2875–2880. doi: 10.1038/nprot.2007.413. [DOI] [PubMed] [Google Scholar]
- 17.Willner I, Riklin A. Anal Chem. 1994;66:1535–1539. [Google Scholar]
- 18.Brierley I, Digard P, Inglis SC. Cell. 1998;57:537–547. doi: 10.1016/0092-8674(89)90124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levicky R, Herne TM, Tarlov MJ, Satija SK. J Am Chem Soc. 1998;120:9787–9792. [Google Scholar]
- 20.Zuker M. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawai T. Ann NY Acad Sci. 1998;852:230–242. [Google Scholar]
- 22.Bonnet G, Krichevsky O, Libchaber A. Proc Natl Aad Sci USA. 1998;95:8602–8606. doi: 10.1073/pnas.95.15.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricci F, Lai RY, Heeger AJ, Plaxco KW, Sumner JJ. Langmuir. 2007;23:6827–6834. doi: 10.1021/la700328r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Theimer CA, Blois CA, Feigon J. Mol Cell. 2005;17:671–682. doi: 10.1016/j.molcel.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 25.Lai RY, Seferos DS, Heeger AJ, Bazan GC, Plaxco KW. Langmuir. 2006;22:10796–10800. doi: 10.1021/la0611817. [DOI] [PubMed] [Google Scholar]
- 26.Xiao Y, Lubin AA, Heeger AJ, Plaxco KW. Angew Chem, Int Ed. 2005;44:5456–5459. doi: 10.1002/anie.200500989. [DOI] [PubMed] [Google Scholar]