

Abstract
Developmental and pathological death of neurons requires activation of a defined pathway of cell cycle proteins. However, it is unclear how this pathway is regulated and whether it is relevant in vivo. A screen for transcripts robustly induced in cultured neurons by DNA damage identified Sertad1, a Cdk4 (cyclin-dependent kinase 4) activator. Sertad1 is also induced in neurons by nerve growth factor (NGF) deprivation and Aβ (β-amyloid). RNA interference-mediated downregulation of Sertad1 protects neurons in all three death models. Studies of NGF withdrawal indicate that Sertad1 is required to initiate the apoptotic cell cycle pathway since its knockdown blocks subsequent pathway events. Finally, we find that Sertad1 expression is required for developmental neuronal death in the cerebral cortex. Sertad1 thus appears to be essential for neuron death in trophic support deprivation in vitro and in vivo and in models of DNA damage and Alzheimer's disease. It may therefore be a suitable target for therapeutic intervention.
Introduction
Neuronal loss by apoptosis is a physiological process during development (Oppenheim, 1991) and a pathological hallmark of many neurodegenerative disorders such as Alzheimer's disease (AD) and of additional insults to the nervous system such as DNA damage (Park et al., 1997a). There are striking and mutually informative similarities between the molecular mechanisms that govern neuron death under these various conditions (Greene et al., 2004, 2007). However, the molecular events, particularly those that initiate death of neurons during development and disease/injury, are incompletely understood.
One major focus regarding the mechanisms of developmental and disease-associated neuron death has been the aberrant activation of cell cycle-related proteins (Becker and Bonni, 2004; Greene et al., 2004, 2007; Herrup et al., 2004). Past studies have indicated a sequential and multistep pathway that is activated by various apoptotic insults including nerve growth factor (NGF) deprivation, DNA damage, and β-amyloid (Aβ) exposure and that is required for neuron death. The first described step is rapid activation of the G1/S kinase cyclin-dependent kinase 4 (Cdk4). This in turn hyperphosphorylates members of the Rb family, leading to dissociation of complexes comprised of Rb family members and E2F transcription factors. Ultimately, these events lead to induction of proapoptotic genes such as Bim and to activation of the core apoptotic machinery (Greene et al., 2007).
An important and currently unresolved issue about the apoptotic cell cycle pathway is how Cdk4 is activated in neurons by apoptotic stimuli. Understanding this will not only further elaborate how apoptotic stimuli lead to neuron death but may also identify additional molecular targets for therapeutic intervention. In addition, it must be recognized that the majority of evidence that causally links the steps in the apoptotic cell cycle pathway has been generated by in vitro studies. Thus, it remains important to demonstrate that the elements that make up this pathway are relevant in vivo.
The protein Sertad1, also known as p34(SEI-1) or Trip-Br1, has been implicated as a regulator of Cdk4 activity. Sertad1 was first identified as an antagonist of p16 INK4a that facilitates the formation and activation of cyclin D–Cdk4 complexes (Sugimoto et al., 1999). Additional studies revealed that it directly binds and activates Cdk4 in a concentration-dependent manner (Li et al., 2004). Functions in addition to regulation of Cdk4 have been described for Sertad1 including stimulation of the transcriptional activities of E2F1 (Hsu et al., 2001) and p53 (Watanabe-Fukunaga et al., 2005). Sertad1 was also additionally reported to exhibit antiapoptotic activity by stabilizing X-linked inhibitor of apoptosis protein (XIAP) in cancer cells (Hong et al., 2009).
In a screen for genes regulated in neurons after DNA damage, we identified Sertad1 transcripts as being robustly induced. Accordingly, we examined the potential role of Sertad1 in neuron death induced by apoptotic stimuli relevant both to normal development and to neurodegeneration. We find that Sertad1 is required for neuronal apoptosis both in vitro and in vivo. Furthermore, our findings indicate that Sertad1 is essential for initiating the Cdk4-dependent cascade of cell cycle events in neuronal cells that follow from trophic factor deprivation.
Materials and Methods
Materials.
Platinum TaqDNA polymerase, V5 antibody, green fluorescent protein (GFP) antibody, and Lipofectamine 2000 were from Invitrogen; anti-human NGF antiserum and anti-β-actin antibody were from Sigma-Aldrich; anti-Sertad1 antibody and Bim antibody were from Abcam; anti-ERK1, phospho-Rb, and C-Myb antibodies were from Santa Cruz Biotechnology; Zsgreen antibody was from Clontech; and the In Situ Cell Death Detection kit, TMR red was from Roche Applied Science. pSIREN vector was from BD Biosciences. Human recombinant NGF was a kind gift from Genentech. Camptothecin was obtained from Sigma-Aldrich. Bis-isopropylthiomethyl-K-252a (CEP11004) was obtained from Cephalon. E2F-1 and control short interfering RNAs (siRNAs) and E2F-1 antibody were purchased from Santa Cruz Biotechnology. p53 mice were genotyped according to published protocols (Aleyasin et al., 2004).
Cell culture.
PC12 cells were cultured and neuronally differentiated as previously described (Greene and Tischler, 1976). For NGF deprivation, after a week of NGF treatment, the cultures were washed with NGF-free medium twice, and anti-NGF antibody (1:100) was added. Control cells were washed with serum-free medium and maintained in RPMI 1640 medium supplied with NGF without serum. Neonatal rat superior cervical ganglion sympathetic (SCG) neurons were cultured as previously described (Park et al., 1998). HEK293 cells were cultured in DMEM with 10% fetal bovine serum. Embryonic rat and mouse cortical neurons were cultured as previously described (Park et al., 1998).
Microarray.
Total RNA was extracted from cortical neuron cultures using Trizol reagent according to the manufacturer's instructions (Invitrogen). RNA was sent to the Ottawa Genomics Innovation Centre Microarray Facility for processing and expression analysis using the Affymetrix Mouse 430 array (Affymetrix). Probe signals were scaled and normalized according to standard facility procedures.
Semiquantitative reverse transcriptase-PCR.
Total RNA was extracted using TriPure isolation reagent (Roche Applied Science). Fifty nanograms of total RNA were used for cDNA synthesis and gene amplification reactions using SuperScript One-Step RT-PCR kit (Invitrogen). cDNA synthesis was performed at 48°C for 45 min, followed by a 2 min initial denaturation step at 94°C. This was followed by 30 cycles (Sertad1) or 25 cycles (S12) at 94°C for 30 s, melting temperature (Tm) 60°C for 30 s, and 72°C for 1 min. Targeting primers were as follows: 5′-CGCAAGCGGGAGGAGGAGAC-3′ and 5′-AGGGGCTGGGGGCTGGATGG-3′ for Sertad1, 5′-GGAAGGCATAGCTGCTGG-3′ and 5′-CCTCGATGACATCCTTGG-3′ for S12. Transcript levels were normalized against S12 signals, and results were reported as times fold increase in reference to untreated control values. Data are presented as mean ± SEM of three independent experiments.
Reverse transcription-quantitative PCR.
Each sample of total RNA was isolated from cultured neurons by using TRI reagent (Molecular Research Center). cDNA was transcribed from total RNA with Superscript RT II (Invitrogen). The primers used for PCR amplification of rat Sertad1 were 5′-GCCTCCTGGAAGATCTCAGTC-3′ and 5′-CATTCTCAGGGACAGGTTTGA-3′. The primers for α-tubulin were 5′-ATGAGGCCATCTATGACATC-3′ and 5′-TCCACAAACTGGATGGTAC-3′. Equal amounts of cDNA template were used for each PCR analysis of Sertad1 or α-tubulin. Quantitative PCR was performed using a Cepheid SmartCycler following the manufacturer's specifications. α-Tubulin was used for Sertad1 transcript normalization. cDNA was added to a 25 μl volume reaction mix containing OmniMix HS master mix (Cepheid) and SYBR Green I (Invitrogen) together with appropriate primers at 0.2 μm each. Analyses of growth curves of real-time fluorescence and of melting curves were performed as described previously (Troy et al., 2000).
Western immunoblotting.
Neuronal PC12 cells were lysed and protein was analyzed by Western immunoblotting as described previously (Biswas and Greene, 2002). For mouse cortical neurons, Sertad1 was detected using a chicken IgY antibody against Sertad1 (1:1000; Genway). Goat anti-chicken HRP (1:3000) was used as secondary antibody.
Plasmids.
Rat Sertad1 was generated by reverse transcription (RT)-PCR of PC12 cDNA. The primers for the amplification were 5′-AGGATGCTGAGCAAAGGTCT-3′ and 5′-GCGCCCAGGTCCTGGTGGCC-3′. The PCR product was gel purified and cloned into pCDNA3.1 vector (Invitrogen), and then verified by sequencing. Sertad1 was also subcloned into pCMS-EGFP vector (Clontech) by using primers 5′-GATCTCGAGACCATGCTGAGCAAAGGTCTG-3′ and 5′-CTAGTCGACCTAGCGCCCAGGTCCTGGTGG-3′.
Preparation of short hairpin RNA.
Sertad1 short hairpin RNAs (shRNAs) were prepared in the pSIREN vector by using BD Knockout RNAi Systems according to the manufacturer's instructions (BD Biosciences) based on the following sequences: 5′-CCGTGGCTTCTAGCTCTCT-3′ (#2), 5′-GCTCCACCACAGCCTTCGG-3′ (#3), 5′-CCAGACCTCCGACACCTGG-3′ (#4), 5′-GATCTCAGTCATATTGAGG-3′ (#5). pSIREN-shRNA-RAND-Zsgreen was as described previously (Sproul et al., 2009). For in utero electroporation (see below), GFP constructs of Sertad1 shRNA and control shRNA were prepared by subcloning the shRNA expression cassette from pSIREN vector into pCMS-EGFP backbone sequence. The (CMV promoter)-MCS sequence in pCMS-EGFP was substituted with the (U6 promoter-shRNA) sequence from pSIREN-RetroQ-zsGreen by subcloning with BglII and EcoRI restriction enzymes. The control shRNA is an inactive mutant of the primary siRNA knockdown construct for GATA2: 5′-GCACCTGATGTCTTCTTCAACC-3′.
Transfections.
DNA was prepared with Plasmid Maxi kits (QIAGEN). Neuronal PC12 cells were cotransfected with 0.5 μg of plasmid pCDNA-V5, pCDNA-Sertad1-V5, pCMS-EGFP, pCMS-Sertad1-EGFP, pSIREN-shRNA-Sertad1-Zsgreen (#2, #3, #4, or #5), pSIREN-shRNA-Rand-Zsgreen, or pSIREN-shRNA-Luc-Zsgreen in 500 μl of serum-free medium per well in 24-well dishes using Lipofectamine 2000. Six hours later, medium with Lipofectamine 2000 was replaced with fresh complete medium. HEK293 cells were transfected as previously described (Xu et al., 2001). E2F-1 siRNA were transfected as previously described (Zhang et al., 2006).
Sertad1 shRNAs and viruses.
Mouse Sertad1-specific shRNA oligos (Applied Biosystems) were cloned into pSilencer3.0-H1 vector (Applied Biosystems). The shRNA fragments containing the H1 promoter were subcloned into the pAdTrack vector. shRNA adenoviruses and DN-c-Jun adenoviruses were constructed as previously described (He et al., 1998).
Preparation of amyloid.
Lyophilized, HPLC-purified Aβ1-42 was purchased from American Peptide, and dodecamer Aβ1-42 was prepared as described previously (Barghorn et al., 2005). Briefly, Aβ1-42 was reconstituted in 100% 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) to 1 mm, and HFIP was removed by evaporation in a Speed Vac, and then resuspended to 5 mm in anhydrous DMSO. This stock was then diluted with PBS to a final concentration of 400 μm. SDS was added to a final concentration of 0.2% and the resulting solution was incubated at 37°C for 18–24 h. The preparation was diluted again with PBS to a final concentration of 100 μm and incubated at 37°C for 18–24 h.
In utero electroporation.
Sprague Dawley rats (Charles River Laboratories) were housed, cared for, and electroporated under the guidelines established by Columbia University Medical Center Institutional Animal Care and Use Committee. Timed pregnant rats [embryonic day 16 (E16)] were anesthetized with ketamine/xylazine (100/10 mixture; 0.1 mg/g body weight, i.p.). The uterine horns were exposed, and the left lateral ventricles of embryos were injected with DNA constructs of a control shRNA or Sertad1 shRNA (1–5 μg/μl) and Fast Green (2 mg/ml; Sigma-Aldrich) by using pulled glass capillaries (Sutter Instrument). Electroporation was accomplished with a BTX electro square electroporator, model ECM830 (BTX). The head of each embryo was held between tweezer-type circular electrodes (Harvard Apparatus) across the uterus wall, and five electrical pulses (amplitude, 50 V; duration, 50 ms; intervals, 100 ms) were delivered. Brains from postnatal pups of 5 d of age were fixed in 4% paraformaldehyde (PFA) by cardiac perfusion.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay and immunohistochemistry.
Rat pups were anesthetized and perfused transcardially with 4% PFA in PBS, pH 7.4. After perfusion, brains were dissected out from the skull and postfixed overnight in fresh fixative. Then brains were washed with PBS, pH 7.4, and cryoprotected in 30% sucrose phosphate buffer. They were frozen with OCT and dry ice and sectioned (20 μm thick) in the coronal plane with a cryostat. To visualize nuclei with DNA cleavage, residues of fluorescein-labeled nucleotides were catalytically added to DNA fragments by terminal deoxynucleotidyl transferase (TdT). Briefly, sections were fixed in fresh 4% PFA/PBS at room temperature for 20 min, washed in PBS three times for 5 min, permeabilized with proteinase K for 5 min on ice, and incubated with nucleotide mix and TdT (In Situ Cell Death Detection kit, TMR red) at 37°C for 1 h. Apoptotic cells exhibit strong, nuclear red fluorescence. To visualize GFP-positive cells, sections were then immunostained with rabbit anti-GFP antibody (1:1000; Invitrogen) in 3% nonimmune goat serum overnight at 4°C, followed by secondary labeling with goat anti-rabbit antibody (1:4000; Alexa Fluor 488; Invitrogen) for 1 h.
Survival assays.
Neuronal PC12 cells, sympathetic neurons, or cortical neurons were transfected with either pCMS-Sertad1-EGFP, pCMS-EGFP, pSIREN-Sertad1-shRNA, pSIREN-Luc-shRNA, or a Random pSIREN-ZsGreen, and then 48 h later deprived of NGF (in case of neuronal PC12 cells and sympathetic neurons) or treated with 1.25 μm dodecamer Aβ (in case of cortical neurons). The numbers of surviving transfected (green) cells per well were assessed just after treatment and at 24 and 48 h after NGF deprivation or Aβ exposure as described previously (Biswas et al., 2007). Data represent means ± SEM of three experiments performed in triplicate.
Immunostaining.
Neuronal PC12 cells were transfected as described above with appropriate constructs of shRNA. Forty-eight hours later, cells were subjected to NGF withdrawal for 18 h and then immunostained as described by Angelastro et al. (2003). Briefly, PC12 cells were fixed with 4% paraformaldehyde for 10 min. After three washes with PBS, cells were blocked in 3% nonimmune goat serum for 2 h. The cultures were immunolabeled with rabbit anti-Bim (1:1000; Abcam) antibody, rabbit C-Myb antibody (1:500; Santa Cruz Biotechnology), or rabbit p-Rb antibody (1:100; Santa Cruz Biotechnology) in 3% nonimmune goat serum overnight at 4°C, followed by secondary labeling with goat anti-rabbit antibody (1:1000; Alexa Fluor 568; Invitrogen) for 1 h. For Sertad1 knockdown experiment, the cultures were immunostained with anti-Sertad1 antibody (1:200; Genway) followed by secondary labeling with goat anti-chicken antibody (1:500; Invitrogen) for 1 h.
Results
Sertad1 is rapidly induced in response to DNA damage and NGF deprivation
In an initial microarray screen for genes induced in cortical neurons by treatment with the DNA-damaging agent camptothecin (8 h), we identified Sertad1 as a candidate regulated gene. Induction was 46-fold over controls (Table 1). In the same screen, we detected increases in the Puma and Noxa genes as expected and previously reported (Aleyasin et al., 2004; Cregan et al., 2004). In contrast, β-tubulin did not change (Table 1). To verify the array data, we performed a semiquantitative reverse transcriptase-PCR assay. Consistent with the array data, the RT-PCR results demonstrated a robust (∼27-fold) increase of Sertad1 message as early as 2 h after camptothecin exposure. This increase persisted even at 8 h after camptothecin treatment (Fig. 1A). A similar magnitude of increase was obtained by quantitative PCR (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Next, we asked whether the DNA damage-induced elevation in Sertad1 message is associated with an increase in protein level. As revealed by Western blotting (Fig. 1B), Sertad1 protein expression was significantly induced by 6 h (∼3.2-fold) of camptothecin exposure and gradually increased until 18 h (∼7.2-fold). Under the conditions of our experiments, neuronal death first becomes apparent by 8–10 h of camptothecin exposure and 50% of neurons die within 16–20 h. Thus, Sertad1 induction is observed early and before overt signs of death.
Table 1.
Sertad1 upregulation in gene chip analysis
| Untreated | Camptothecina | Camptothecin/untreated signal ratio | p value | |
|---|---|---|---|---|
| Sertad1 | 135.8 | 6264.2 | 46.1 | 0.000167 |
| Noxa | 542.8 | 2141.6 | 3.95 | 0.000273 |
| Puma | 1124.2 | 3640.7 | 3.24 | 0.00013 |
| Tubb3b | 22,718.3 | 21,207.1 | 0.93 | NSc |
Microarray analysis of RNA extracted from cortical neurons after campothecin (10 m) treatment for 8 h or with vehicle (0.1% DMSO). Fold change represents the ratio of signal in campothecin-treated neurons relative to vehicle-treated neurons for each probe set.
aCortical neurons were treated with camptothecin for 8 h.
bTubb3 is β3-tubulin.
cNot significant.
Figure 1.

Sertad1 mRNA and protein levels are upregulated after DNA damage and NGF deprivation. A, B, Total mRNA or whole-cell lysates were extracted from cortical neurons treated with camptothecin (10 μm) for the indicated times and subjected to RT-PCR (A, top) or Western blot (B, top), respectively. Densitometry of all signals was performed using NIH ImageJ software and Sertad1 signals were normalized to S12 (A, bottom) or actin (B, bottom). Data are presented as fold increase relative to untreated control. Each bar represents the mean ± SEM from four independent experiments (one-way ANOVA/Tukey's least significant difference test; significance comparisons with untreated controls, *p < 0.05, **p < 0.01). C, D, Sertad1 mRNA levels are increased in response to NGF deprivation. Neuronal PC12 cells (C) and sympathetic neurons (D) were subjected to NGF deprivation for the indicated times, and total RNA was isolated, reverse transcribed, and amplified by PCR using specific primers for Sertad1 and α-tubulin. Data represent means ± SEM of three experiments (neuronal PC12 cells) or two experiments (SCG neurons), each performed with three replicate cultures. The asterisks denote statistically significant differences from 0 h control: *p < 0.05; **p < 0.001. E, NGF withdrawal elevates Sertad1 protein levels. Neuronal PC12 cells were subjected to NGF withdrawal for the indicated times, and cell proteins were subjected to Western immunoblotting using enhanced chemiluminescence for the detection of Sertad1 and ERK 1 (loading control). The right panel shows quantification of Sertad1 signals, normalized against ERK1 expression. Data represent means ± SEM of five experiments (except 6 h which is a single experiment). The asterisks denote statistically significant differences from 0 h control: *p < 0.05; **p < 0.001.
To identify upstream signals that mediate upregulation of Sertad1 mRNA in response to DNA damage, we tested three potential candidates: p53, E2F1, and c-Jun N-terminal kinases (JNKs). As shown in Figure 2A, germ line deficiency of p53 does not affect camptothecin-induced upregulation of Sertad1 message in cultured cortical neurons. In contrast, siRNA-mediated knockdown of E2F-1 blocks Sertad1 mRNA upregulation by ∼50% (Fig. 2B). Finally, neither JNK inhibitor CEP11004 nor dominant-negative c-Jun expression affect camptothecin-induced Sertad1 upregulation (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Together, these findings indicate that E2F1, but not p53 or the JNK pathway at least partially regulates Sertad mRNA expression after DNA damage.
Figure 2.

A, p53 deficiency does not inhibit camptothecin-induced upregulation of Sertad1. Cortical neurons from p53+/+ and p53−/− mice were treated with and without camptothecin for 8 h. Left, Sertad1 levels were then analyzed by RT-PCR. S12 was shown as a control for equal input. Right, Densitometric analysis of Sertad1 signals normalized against S12 signals. B, E2F-1 downregulation partially blocks camptothecin-induced upregulation of Sertad1. Cortical neurons were transfected with E2F-1 and control siRNA and were treated with and without camptothecin for 8 h. Left, Sertad1 levels were then analyzed by RT-PCR. S12 was shown as a control for equal input. Right, Densitometric analysis of Sertad1 signals normalized against S12 signals. Significance comparisons with untreated controls: *p < 0.05. Error bars indicate SEM. C, Western blot analyses showing downregulation of E2F-1 in cells treated with E2F-1 siRNA compared with control siRNA. Actin is provided as loading control.
Because neuronal death caused by DNA damage shares with death evoked by trophic factor deprivation a reliance on cell cycle proteins (Park et al., 1996, 1997a,b; Liu and Greene, 2001; Zhang et al., 2006; Gonzalez et al., 2008), we also investigated whether Sertad1 expression is regulated in response to NGF withdrawal. For this purpose, we used neuronally differentiated PC12 cells and primary cultures of SCG neurons. Both cell types undergo apoptosis (evident starting at ∼18 h) in response to NGF deprivation (Rukenstein et al., 1991; Xu et al., 2001). Similar to DNA damage, a time course performed by quantitative PCR revealed that Sertad1 mRNA levels were elevated in neuronal PC12 cells as early as one-half an hour after NGF deprivation and were consistently and significantly increased in SCG neurons after 2 h of such treatment (Fig. 1C,D). Sertad1 protein expression was also elevated by 2–4 h in response to NGF deprivation, and this change was similar in magnitude (by approximately twofold) to that of mRNA (Fig. 1E). Thus, both Sertad1 mRNA and protein expression are induced in response to DNA damage and trophic factor withdrawal, although this response is more modest in the case of NGF deprivation.
Sertad1 overexpression is not sufficient to trigger neuron death but enhances death in response to NGF deprivation
Next, we examined whether elevated expression of Sertad1 is sufficient in itself to trigger neuron death in the presence of trophic support. Expression of Sertad1 alone did not induce death of neuronal PC12 cells (Fig. 3A), cortical neurons (supplemental Fig. S3, available at www.jneurosci.org as supplemental material), or cerebellar granule neurons (data not shown). However, overexpression of Sertad1 significantly enhanced the level of neuronal cell death that occurs in response to NGF deprivation (Fig. 3B).
Figure 3.

Overexpression of Sertad1 is not sufficient to cause death but enhances death in response to NGF withdrawal. A, Neuronal PC12 cells were transfected with pCMS-EGFP or pCMS-Sertad1-EGFP. At 24 and 48 h after transfection, surviving transfected (green) cells were counted. Data represent mean of a representative experiment performed with three replicate cultures per point. Similar results were obtained in an independent experiment. B, Neuronal PC12 cells were transfected with pCMS-EGFP or pCMS-Sertad1-EGFP. After 48 h, they were maintained with or deprived of NGF and surviving transfected cells were counted at 24 and 48 h. Data represent mean ± SEM of a representative experiment performed with three replicate cultures per point. Similar results were obtained in an independent experiment. The asterisks denote statistically significant differences from control (GFP) in absence of NGF: *p < 0.05; **p < 0.01.
Sertad1 is required for neuron death induced by NGF deprivation and DNA damage
Because of the observed upregulation of Sertad1 in multiple neuronal death paradigms and the sensitization to death with Sertad1 overexpression, we next examined whether neuron death in our systems requires Sertad1. We first studied the role of Sertad1 in NGF deprivation. To achieve this, we prepared several shRNAs specifically targeted to rat Sertad1 and identified three that substantially reduced expression of the overexpressed protein (Fig. 4A). Each of the effective shRNAs significantly protected neuronal PC12 cells from death induced by NGF withdrawal (Fig. 4B,C). Moreover, most of the Sertad1 shRNA-expressing cells preserved overall neuron morphology even after 2 d of NGF deprivation (Fig. 4B). This is reminiscent of the preservation of neuronal morphology after NGF deprivation that is achieved with small molecule Cdk inhibitors (Park et al., 1996, 1997b) or Cdk4 shRNAs (Biswas et al., 2005). Similar experiments with SCG neurons also showed that downregulation of Sertad1 by two independent shRNAs significantly blocks death and preserves overall neuron morphology after NGF deprivation (Fig. 4D) (data not shown).
Figure 4.

shRNAs targeted to Sertad1 specifically block gene expression and neuron death in response to NGF deprivation. A, Sertad1 shRNAs suppress Sertad1 expression. HEK293 cells were cotransfected with pCDNA-Sertad1-V5 along with pSIREN-Luc (Luciferase)-Zsgreen shRNA or pSIREN-Sertad1-Zsgreen shRNAs. After 48 h, cells were lysed and subjected to Western immunoblotting using enhanced chemiluminescence for the detection of V5 and Zsgreen. B, Sertad1 knockdown prevents neuronal degeneration and death after NGF deprivation. Neuronal PC12 cells were transfected with pSIREN-Random-Zsgreen shRNA (control shRNA) or pSIREN-Sertad1#5-ZsGreen shRNA (Sertad1 shRNA), maintained for 48 h, and then washed twice with RMPI 1640 medium and maintained with or without NGF. Representative images of transfected cells were taken in each case before and after 24 and 48 h of treatment. C, D, Sertad1 shRNAs provide protection against death evoked by NGF deprivation of neuronal PC12 cells or SCG neurons. Neuronal cells were transfected with pSIREN-Luc-Zsgreen shRNA (shLuc), pSIREN-Sertad1-ZsGreen shRNA (shRnad), or pSIREN-Sertad1-ZsGreen shRNAs (shSertad1#2, shSertad1#4, and shSertad1#5), maintained for 48 h, and then deprived of NGF as indicated. Numbers of surviving transfected (green) cells were counted just before NGF deprivation and after 24 and 48 h of NGF deprivation. Data are from one of three independent experiments, each with comparable results and are shown as means ± SEM, performed in triplicates. The asterisks denote statistically significant differences from control (shLuc or shRand): *p < 0.05; **p < 0.001.
Similarly, we used a shRNA knockdown strategy to assess whether Sertad1 also plays a required role in neuronal death after DNA damage. In this case, we used adenoviruses to deliver shRNAs to cultured mouse cortical neurons. We screened two potential shRNA sequences directed against mouse Sertad1 to knockdown endogenous Sertad1. As shown in Figure 5, A and B, infection with both Sertad1 shRNA viruses, but not a control virus, significantly reduced the Sertad1 immunostaining signal (54 and 33%, respectively) after camptothecin treatment. Survival assays in response to camptothecin treatment were performed using these two Sertad1 shRNAs along with a control shRNA virus. As shown in Figure 5C, expression of the two different Sertad1 shRNAs resulted in significant protection from camptothecin at times up to 48 h. This finding is consistent with our observations in the NGF deprivation model and indicates that Sertad1 plays a required role in the neuronal death process.
Figure 5.
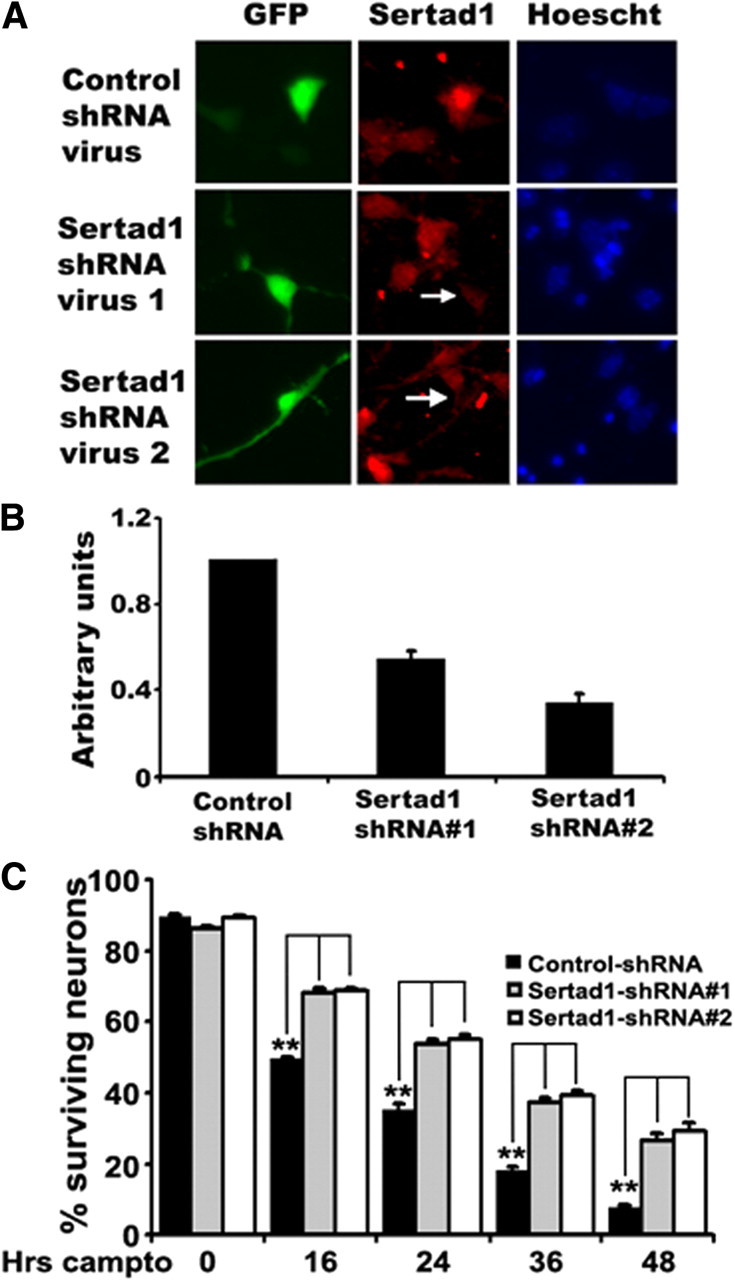
Sertad1 downregulation by adenoviral shRNAs protects cortical neurons from DNA damage-evoked cell death. A, B, shRNA-mediated knockdown of Sertad1 in cortical neurons. Cortical neurons were infected with two adenoviral shRNAs targeting mouse Sertad1 gene or a control virus at the time of plating, and 24 h after infection cells were treated with camptothecin for 16 h to induce Sertad1 expression. Cells were fixed, and immunostaining was performed using Sertad1 chicken IgY antibody. A, Both Sertad1 shRNA viruses repress the upregulation of Sertad1 in cortical neurons treated with camptothecin (arrows indicate Sertad1 shRNA-expressing cells). B, Quantification of fluorescence intensity of Sertad1-immunostained neurons infected with control and Sertad1 shRNA viruses. Values are mean ± SEM (n = 30–35). C, Expression of two Sertad1 shRNAs diminishes death of primary cultured cortical neurons treated with camptothecin. Cortical neurons were infected with control and Sertad1 shRNA viruses (multiplicity of infection, 10) at the time of plating. Two days after plating, cells were treated with 10 μm camptothecin for 16, 24, 36, and 48 h, fixed, and stained with Hoechst 33258 (0.25 μg/ml). GFP-positive cells were counted based on nuclear integrity. Significance comparisons with camptothecin-treated controls: **p < 0.01. Data represent the mean ± SEM from three independent experiments.
Sertad1 is induced and required for neuron death in a model of Alzheimer's disease
We additionally examined the importance of Sertad1 in a culture model directly relevant to neurodegeneration. A substantial body of evidence has implicated cell cycle molecules in neuron death associated with Alzheimer's disease (Herrup et al., 2004; Greene et al., 2007; Zhu et al., 2007; Copani et al., 2008). Moreover, there are striking similarities between the molecular mechanisms of neuron death induced by NGF deprivation, DNA damage, and Aβ exposure (Greene et al., 2007), and recent findings support a mechanism in which neuron death triggered by NGF deprivation is mediated by elevated production and release of Aβ (Matrone et al., 2008a,b). We therefore examined whether Sertad1 is induced and is necessary for death of cultured cortical neurons induced by exposure to aggregated Aβ. Sertad1 transcripts were increased by nearly fourfold within 3 h of Aβ treatment (Fig. 6A). Downregulation of Sertad1 by shRNA significantly protected the cortical neurons from Aβ induced death (Fig. 6B). Moreover, Sertad1 shRNA-expressing cells showed preservation of overall neuron morphology in presence of aggregated Aβ (Fig. 6C). Together, these findings indicate that Sertad1 is induced and plays a required role in in vitro paradigms of both development and pathological neuron death.
Figure 6.
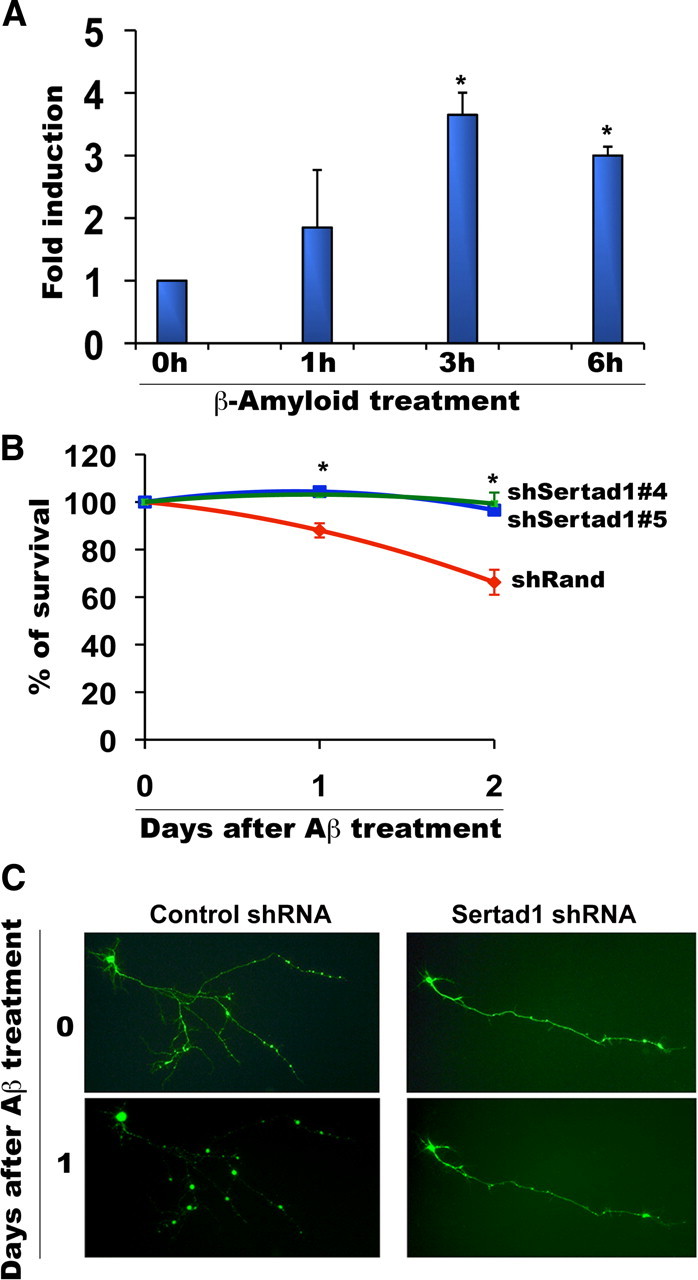
Sertad1 is induced and is required for degeneration of cultured cortical neurons in response to Aβ treatment. A, Sertad1 mRNA levels are increased in response to Aβ. Cortical neurons were treated with 1.25 μm dodecamer Aβ for the indicated times and total RNA was isolated, reverse transcribed, and amplified by PCR using Sertad1- and α-tubulin-specific primers. Data represent means ± SEM of three independent experiments. The asterisks denote statistically significant differences from control (0 h): *p < 0.01. B, C, Sertad1 knockdown prevents neuronal degeneration and death after Aβ treatment. Cortical neurons were transfected with pSIREN-Random-shRNA-Zsgreen (shRand) or pSIREN-Sertad1-shRNA-ZsGreen (shSertad1-#4, #5), maintained for 48 h and then treated with 1.25 μm dodecamer Aβ. Numbers of surviving transfected (green) cells were counted just before treatment and after 24 and 48 h with or without treatment. Data are from one of three independent experiments, each with comparable results, and are shown as means ± SEM, performed in triplicates. The asterisks denote statistically significant differences from control (shRand): *p < 0.01. Representative images of transfected neurons for pSIREN-Random-Zsgreen shRNA (control ShRNA) and pSIREN-Sertad1#5-ZsGreen shRNA (Sertad1 shRNA) were taken before and after 24 h of treatment.
Sertad1 is required for developmental death of cortical neurons in vivo
The above evidence indicates the importance of Sertad1 in in vitro models of neuronal death. However, its involvement in vivo is unknown. Critically, this uncertainty extends generally to whether the cell cycle pathways may be of importance in developmental death in vivo. Because limiting levels of target-derived trophic support appear to regulate neuron death in vivo, our in vitro data led us to test whether Sertad1 is essential for developmental neuron death in the early postnatal cerebral cortex. We used in utero electroporation to deliver DNA encoding Sertad1 shRNA and GFP or a control DNA expressing random shRNA and GFP into the left lateral ventricles of E16 rat embryonic brains. DNA delivered in this manner is taken up by ventricular zone progenitor cells that subsequently differentiate and migrate toward the pial surface. Because maximum cortical neuron death occurs during the first week of development (Spreafico et al., 1995), we killed the DNA-electroporated rat pups on postnatal day 5 (P5) and then analyzed their fixed cortices by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assays coupled with immunohistochemistry for GFP to assess the proportions of transfected neurons undergoing death. Past studies have established that TUNEL staining faithfully reports apoptotic neurons in developing brain as judged by nuclear morphology and caspase 3 activation (Sophou et al., 2006). Transfected, morphologically identified neurons were mostly present in cortical layers II, III, and IV of the electroporated side of the brain (Fig. 7A; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Counts of TUNEL-positive cells indicated that ∼14 per 1000 transfected neurons (1.4 ± 0.13%; N = 3) were apoptotic in these layers of the cerebral cortex for those animals electroporated with DNA expressing control shRNA (Fig. 7A,B). This is consistent with a past study that reported ∼15 apoptotic (TUNEL-positive) nuclei per 1000 neurons in the same cortical layers of normal P5 brains (Spreafico et al., 1995). In contrast, we found an average of only 6 apoptotic nuclei per 1000 Sertad1 shRNA-expressing neurons (0.6 ± 0.03%; N = 3) in the same three layers of the cerebral cortex (Fig. 7B,C). There were no evident effects of the Sertad1 shRNA on the migration or morphology of the transfected neurons. Together, our findings indicate that Sertad1 plays an essential role in neuron death during cortical development in vivo.
Figure 7.

shRNAs targeted to Sertad1 block cortical neuron death in vivo during development. A, Distribution of transfected neurons in cortex of the electroporated brain. GFP-expressing DNA constructs of control shRNA or Sertad1 shRNA #4 were electroporated into the left lateral ventricles of E16 rat embryos, which were subsequently harvested at P5. The brains were fixed and subjected to staining for eGFP expression. Morphologically identified eGFP-expressing neurons are mostly present in cortical layers II, III, and IV of the electroporated side of the brain in each case. B, Location of TUNEL-positive eGFP-expressing neurons in cortices of brains electroporated with control and Sertad1 shRNA. Note that TUNEL-positive eGFP-expressing neurons are encircled in each case. C, Sertad1 shRNA inhibits developmental cell death of cortical neurons. The cortices of animals electroporated with control and Sertad1 shRNAs were evaluated for proportions of transfected cells (eGFP positive) that show TUNEL staining. Data represent means (SEM of 3 animals, each from an independent electroporation). Approximately 2000–3000 cells were evaluated per animal. The asterisks denote statistically significant differences between the two mean values: *p < 0.01.
Sertad1 is required for Rb phosphorylation and consequent myb and Bim induction in response to NGF deprivation
Our past studies (Park et al., 1997b; Liu and Greene, 2001; Liu et al., 2004, 2005; Biswas et al., 2005, 2007; Greene et al., 2007) have established a sequential pathway for neuron death caused by NGF deprivation and Aβ exposure in which (1) activated Cdk4 phosphorylates members of Rb family of transcription-regulating proteins; (2) such phosphorylation leads to dissociation of repressor complexes containing Rb family members, E2F transcription factor proteins, and chromatin modifiers; (3) there is consequent de-repression and elevated expression of E2F-responsive genes including the transcription factors B- and C-Myb; and (4) the induced mybs activate transcription of the gene encoding the proapoptotic BH3-only protein Bim. If Sertad1 acts as anticipated at the proximal end of this pathway by binding and activating Cdk4, we would predict that the downstream responses such as Rb phosphorylation, Myb induction, and Bim induction should be blocked by Sertad1 downregulation. We first assessed the effects of shRNA-mediated Sertad1 knockdown on Rb phosphorylation after NGF deprivation. As shown in Figure 8A and consistent with our past findings, NGF deprivation (18 h) raised the proportion of cells with a high level of phospho-Rb immunostaining from ∼8 to 65%. Transfection with Sertad1 shRNA reduced this to an average of 12.5%. Similar findings were achieved for expression of C-Myb and its downstream target Bim (Fig. 8B,C). In each case, shRNAs targeted to Sertad1 strongly suppressed the large increases in the proportions of transfected cells that showed high staining for C-Myb and Bim after NGF deprivation. Together, these results support the conclusion that Sertad1 is required for the activation of a neuronal apoptotic pathway in response to NGF deprivation that depends on Cdk-mediated Rb family phosphorylation and consequent induction of the E2F-responsive gene Myb and its proapoptotic target Bim.
Figure 8.
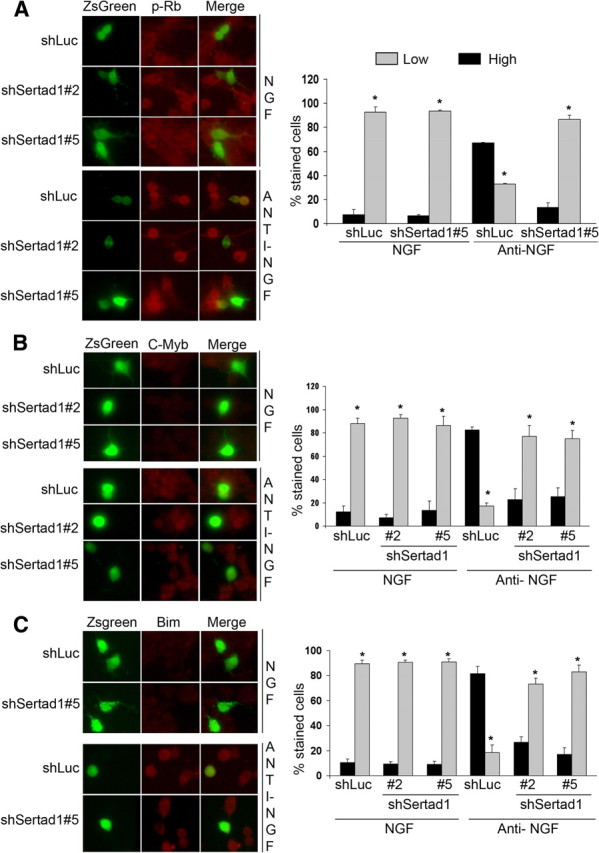
shRNAs targeted to Sertad1 block Rb phosphorylation and repress upregulation of endogenous Myb and Bim in PC12 cells subjected to NGF deprivation. A–C, Left panels show neuronal PC12 cells that were transfected with the indicated constructs, maintained for 48 h, and then deprived of NGF for 18 h after which they were immunostained with antibodies against phospho-Rb (p-Rb), C-Myb, or Bim. The percentage of stained cells shown in the right panels pertain to the proportions of transfected cells (as indicated by Zs-Green expression) that show p-Rb, C-Myb, or Bim staining at either high (more than that of vast majority of NGF-maintained cells) or low (equal or less than that of vast majority of NGF-maintained cells) levels. Data represent means (SEM of 3 independent experiments, each performed in triplicate). Approximately 50 cells were evaluated per culture. The asterisks denote statistically significant differences between low-staining cells and high-staining cells: *p < 0.001.
Discussion
Cell cycle proteins have been implicated as required elements in the mechanisms of postmitotic neuron death, both during normal development and in response to injury, stroke, and a range of neurodegenerative diseases (Smith et al., 2004; Neve and McPhie, 2006; Greene et al., 2007; Nunomura et al., 2007; Rashidian et al., 2007). However, the proximal events that initiate the involvement of cell cycle proteins, particularly the activation of Cdk4, have been unclear. Here, we performed experiments that now implicate the cell cycle regulatory protein Sertad1 in three different paradigms of neuron death: NGF deprivation, DNA damage, and Aβ exposure. We find that Sertad1 expression is rapidly elevated in cultured neurons in all three apoptotic paradigms and that downregulation of Sertad1 by shRNA is protective in each case. We further show that downregulation of Sertad1 inhibits the increase in Rb phosphorylation and consequent induction of Myb and Bim that mediate neuron death caused by NGF deprivation. These findings thus place Sertad1 proximal to the other defined events in the Cdk4-dependent apoptotic cell cycle pathway that is triggered by loss of trophic support.
The steps that comprise the apoptotic cell cycle pathway described here have been determined mostly by in vitro experimentation. We used in vivo electroporation to extend our studies to the developing cortex and found that, in this instance also, Sertad1 plays an essential role in normally occurring neuronal death. Such findings thus implicate not only Sertad1 but also events downstream of it in developmental neuron death.
Sertad1 has been found to directly bind and activate Cdk4 as well as to render active Cdk4–cyclinD1 complexes resistant to inhibition by p16(INK4a) (Sugimoto et al., 1999). Such actions are entirely consistent with our findings that knockdown of Sertad1 protects neurons in our three death paradigms, each of which requires Cdk4 activation (Greene et al., 2004, 2007). Our studies of the effects of Sertad1 knockdown on cell cycle events triggered by NGF deprivation also support the idea that the capacity of Sertad1 to activate Cdk4 is crucial to its death-promoting actions in neurons. For example, this mechanism would explain why Sertad1 knockdown blocks hyperphosphorylation of Rb, a major cellular target of Cdk4 as well as its inhibition of the subsequent induction of Myb and of Bim. In contrast, other known functions of Sertad1 seem less likely to account for its role in neuron death caused by NGF withdrawal. For instance, Sertad1 binds the E2F1 partner DP-1, thereby enhancing the transactivation of DP-1/E2F1 complexes (Hsu et al., 2001). However, our past findings showed that gene de-repression rather than transactivation is required for neuron death triggered by NGF deprivation (Liu and Greene, 2001). Such a mechanism would also not account for our observations that Sertad1 knockdown blocks Rb hyperphosphorylation and induction of Myb (which is repressed by E2F complexes) and Bim. Another potentially relevant action of Sertad1 is its capacity to stimulate p53 transcriptional activity (Watanabe-Fukunaga et al., 2005). However, the major proapoptotic targets of p53, Noxa and Puma, do not appear to be either induced or required for death in response to NGF deprivation (S. C. Biswas and L. A. Greene, unpublished observations), and it is unclear that p53 plays a major role in death associated with this paradigm (Sadoul et al., 1996). In addition to activating Cdk4, Sertad1 was recently reported to stabilize XIAP and thus disinhibit caspases in cancer cells (Hong et al., 2009). Such an action of Sertad1, if occurred in neurons, would have an antiapoptotic effect and does not seem to be consistent with the role found here of being proapoptotic in response to NGF deprivation, DNA damage, and Aβ. However, in our death paradigms, either camptothecin or Aβ evokes a series of death pathways that are both caspase-dependent and caspase-independent (Stefanis et al., 1999; Selznick et al., 2000; Cregan et al., 2002). Even if Sertad1 confers antiapoptotic properties, delayed neuronal death still occurs in a caspase-independent manner in these paradigms.
Although Cdk4 activity is essential for neuron death induced by DNA damage, this paradigm of death differs from that caused by trophic factor withdrawal in that it requires both E2F1 and p53 as well as induction of Puma (Park et al., 2000; Wyttenbach and Tolkovsky, 2006). This raises the possibility that, in addition to its role in activating Cdk4, the capacities of Sertad to activate E2F1 and p53 may be relevant to neuronal death triggered by DNA damage. Additionally, we found that E2F1 at least partially mediates death induced by camptothecin.
Although Sertad1 overexpression enhanced neuronal death caused by NGF deprivation, it was not sufficient to promote neuron death when overexpressed in healthy neurons. This observation indicates that Sertad1 is necessary, but not sufficient for neuron death and that additional events are required to initiate the death pathway. This could include, for instance, modification of Sertad1 triggered by apoptotic stimuli, induction of additional proteins essential for Cdk4 activation, or promotion of Sertad1-independent events such as phosphorylation/dephosphorylation of Cdk4 that are needed for its full activation. A past study has shown that activation of Cdc25A is required for Cdk4 activation and neuron death in response to DNA damage (Zhang et al., 2006), raising the possibility for its involvement at least in the case of this death paradigm.
Past studies have described strong parallels between the mechanisms by which neurons die in response to NGF deprivation and Aβ treatment (Greene et al., 2007). This also appears to be the case for the vulnerable populations of neurons in AD that coexpress elevated levels of Cdk4, hyperphosphorylated Rb, and Bim protein (Biswas et al., 2007). A unifying explanation has recently been provided by the observations that neurotrophic factor deprivation causes the enhanced formation and release of Aβ, which then interacts with cells to trigger an apoptotic pathway (Matrone et al., 2008a,b). In this light, it is significant that Sertad1, as in the case of trophic deprivation, is also required for neuron death triggered by exposure to aggregated Aβ. Such findings further support a common pathway for the two apoptotic stimuli and raise the possibility that Sertad1 may be a therapeutic target for amelioration of neuron death and degeneration in AD. In addition to AD, activation of cell cycle proteins has also been implicated in a number of neurodegenerative diseases and nervous system insults associated with neuron death (Becker and Bonni, 2004; Greene et al., 2004, 2007; Herrup et al., 2004). For example, Sertad1 was among those genes that were induced in a cellular model of Parkinson's disease (Ryu et al., 2005), raising the possibility of its involvement at least in this disorder and perhaps in others.
Footnotes
This work was supported in part by grants from the National Institute of Neurological Disorders and Stroke (L.A.G.), Alzheimer's Disease Research Center/Taub pilot grant (Columbia University; S.C.B.), Indian Institute of Chemical Biology (S.C.B.), Canadian Institute of Health Research (R.S.S., D.S.P.), Heart and Stroke Foundation Ontario (R.S.S., D.S.P.), and World Class University Program through National Research Foundation of Korea Grant R31-2008-000-20004-0 (D.S.P.). We thank Janet Peterson for technical assistance. We also thank Dr. Julio Pozueta for providing us dodecamer β-amyloid.
References
- Aleyasin H, Cregan SP, Iyirhiaro G, O'Hare MJ, Callaghan SM, Slack RS, Park DS. Nuclear factor-κB modulates the p53 response in neurons exposed to DNA damage. J Neurosci. 2004;24:2963–2973. doi: 10.1523/JNEUROSCI.0155-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelastro JM, Ignatova TN, Kukekov VG, Steindler DA, Stengren GB, Mendelsohn C, Green LA. Regulated expression of ATF5 is required for the progression of neural progenitor cells to neurons. J Neurosci. 2003;23:4590–4600. doi: 10.1523/JNEUROSCI.23-11-04590.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner RS, Harlan J, Barlow E, Ebert U, Hillen H. Globular amyloid beta-peptide oligomer—a homogenous and stable neuropathological protein in Alzheimer's disease. J Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- Becker EB, Bonni A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog Neurobiol. 2004;72:1–25. doi: 10.1016/j.pneurobio.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Biswas SC, Greene LA. Nerve growth factor (NGF) down-regulates the Bcl-2 homology 3 (BH3) domain-only protein Bim and suppresses its proapoptotic activity by phosphorylation. J Biol Chem. 2002;277:49511–49516. doi: 10.1074/jbc.M208086200. [DOI] [PubMed] [Google Scholar]
- Biswas SC, Liu DX, Greene LA. Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J Neurosci. 2005;25:8349–8358. doi: 10.1523/JNEUROSCI.1570-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SC, Shi Y, Vonsattel JP, Leung CL, Troy CM, Greene LA. Bim is elevated in Alzheimer's disease neurons and is required for β-amyloid-induced neuronal apoptosis. J Neurosci. 2007;27:893–900. doi: 10.1523/JNEUROSCI.3524-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copani A, Guccione S, Giurato L, Caraci F, Calafiore M, Sortino MA, Nicoletti F. The cell cycle molecules behind neurodegeneration in Alzheimer's disease: perspectives for drug development. Curr Med Chem. 2008;15:2420–2432. doi: 10.2174/092986708785909030. [DOI] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Arbour NA, Maclaurin JG, Callaghan SM, Fortin A, Cheung EC, Guberman DS, Park DS, Slack RS. p53 activation domain 1 is essential for PUMA upregulation and p53-mediated neuronal cell death. J Neurosci. 2004;24:10003–10012. doi: 10.1523/JNEUROSCI.2114-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez YR, Zhang Y, Behzadpoor D, Cregan S, Bamforth S, Slack RS, Park DS. CITED2 signals through peroxisome proliferator-activated receptor-gamma to regulate death of cortical neurons after DNA damage. J Neurosci. 2008;28:5559–5569. doi: 10.1523/JNEUROSCI.1014-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Biswas SC, Liu DX. Cell cycle molecules and vertebrate neuron death: E2F at the hub. Cell Death Differ. 2004;11:49–60. doi: 10.1038/sj.cdd.4401341. [DOI] [PubMed] [Google Scholar]
- Greene LA, Liu DX, Troy CM, Biswas SC. Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim Biophys Acta. 2007;1772:392–401. doi: 10.1016/j.bbadis.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24:9232–9239. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SW, Kim CJ, Park WS, Shin JS, Lee SD, Ko SG, Jung SI, Park IC, An SK, Lee WK, Lee WJ, Jin DH, Lee MS. p34SEI-1 inhibits apoptosis through the stabilization of the X-linked inhibitor of apoptosis protein: p34SEI-1 as a novel target for anti-breast cancer strategies. Cancer Res. 2009;69:741–746. doi: 10.1158/0008-5472.CAN-08-1189. [DOI] [PubMed] [Google Scholar]
- Hsu SI, Yang CM, Sim KG, Hentschel DM, O'Leary E, Bonventre JV. TRIP-Br: a novel family of PHD zinc finger- and bromodomain-interacting proteins that regulate the transcriptional activity of E2F-1/DP-1. EMBO J. 2001;20:2273–2285. doi: 10.1093/emboj/20.9.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Melvin WS, Tsai MD, Muscarella P. The nuclear protein p34SEI-1 regulates the kinase activity of cyclin-dependent kinase 4 in a concentration-dependent manner. Biochemistry. 2004;43:4394–4399. doi: 10.1021/bi035601s. [DOI] [PubMed] [Google Scholar]
- Liu DX, Greene LA. Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron. 2001;32:425–438. doi: 10.1016/s0896-6273(01)00495-0. [DOI] [PubMed] [Google Scholar]
- Liu DX, Biswas SC, Greene LA. B-myb and C-myb play required roles in neuronal apoptosis evoked by nerve growth factor deprivation and DNA damage. J Neurosci. 2004;24:8720–8725. doi: 10.1523/JNEUROSCI.1821-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DX, Nath N, Chellappan SP, Greene LA. Regulation of neuron survival and death by p130 and associated chromatin modifiers. Genes Dev. 2005;19:719–732. doi: 10.1101/gad.1296405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrone C, Di Luzio A, Meli G, D'Aguanno S, Severini C, Ciotti MT, Cattaneo A, Calissano P. Activation of the amyloidogenic route by NGF deprivation induces apoptotic death in PC12 cells. J Alzheimers Dis. 2008a;13:81–96. doi: 10.3233/jad-2008-13109. [DOI] [PubMed] [Google Scholar]
- Matrone C, Ciotti MT, Mercanti D, Marolda R, Calissano P. NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc Natl Acad Sci U S A. 2008b;105:13139–13144. doi: 10.1073/pnas.0806133105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RL, McPhie DL. The cell cycle as a therapeutic target for Alzheimer's disease. Pharmacol Ther. 2006;111:99–113. doi: 10.1016/j.pharmthera.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Moreira PI, Lee HG, Zhu X, Castellani RJ, Smith MA, Perry G. Neuronal death and survival under oxidative stress in Alzheimer and Parkinson diseases. CNS Neurol Disord Drug Targets. 2007;6:411–423. doi: 10.2174/187152707783399201. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Park DS, Farinelli SE, Greene LA. Inhibitors of cyclin-dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem. 1996;271:8161–8169. doi: 10.1074/jbc.271.14.8161. [DOI] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Greene LA, Geller HM. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci. 1997a;17:1256–1270. doi: 10.1523/JNEUROSCI.17-04-01256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Levine B, Ferrari G, Greene LA. Cyclin-dependent kinase inhibitors and dominant-negative cyclin-dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997b;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 1998;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Morris EJ, Bremner R, Keramaris E, Padmanabhan J, Rosenbaum M, Shelanski ML, Geller HM, Greene LA. Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J Neurosci. 2000;20:3104–3114. doi: 10.1523/JNEUROSCI.20-09-03104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidian J, Iyirhiaro GO, Park DS. Cell cycle machinery and stroke. Biochim Biophys Acta. 2007;1772:484–493. doi: 10.1016/j.bbadis.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Rukenstein A, Rydel RE, Greene LA. Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci. 1991;11:2552–2563. doi: 10.1523/JNEUROSCI.11-08-02552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu EJ, Angelastro JM, Greene LA. Analysis of gene expression changes in a cellular model of Parkinson disease. Neurobiol Dis. 2005;18:54–74. doi: 10.1016/j.nbd.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Sadoul R, Quiquerez AL, Martinou I, Fernandez PA, Martinou JC. p53 protein in sympathetic neurons: cytoplasmic localization and no apparent function in apoptosis. J Neurosci Res. 1996;43:594–601. doi: 10.1002/(SICI)1097-4547(19960301)43:5<594::AID-JNR9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Selznick LA, Zheng TS, Flavell RA, Rakic P, Roth KA. Amyloid beta-induced neuronal death is bax-dependent but caspase-independent. J Neuropathol Exp Neurol. 2000;59:271–279. doi: 10.1093/jnen/59.4.271. [DOI] [PubMed] [Google Scholar]
- Smith PD, O'Hare MJ, Park DS. Emerging pathogenic role for cyclin dependent kinases in neurodegeneration. Cell Cycle. 2004;3:289–291. [PubMed] [Google Scholar]
- Sophou S, Dori I, Antonopoulos J, Parnavelas JG, Dinopoulos A. Apoptosis in the rat basal forebrain during development and following lesions of connections. Eur J Neurosci. 2006;24:573–585. doi: 10.1111/j.1460-9568.2006.04929.x. [DOI] [PubMed] [Google Scholar]
- Spreafico R, Frassoni C, Arcelli P, Selvaggio M, De Biasi S. In situ labeling of apoptotic cell death in the cerebral cortex and thalamus of rats during development. J Comp Neurol. 1995;363:281–295. doi: 10.1002/cne.903630209. [DOI] [PubMed] [Google Scholar]
- Sproul AA, Xu Z, Wilhelm M, Gire S, Greene LA. Cbl negatively regulates JNK activation and cell death. Cell Res. 2009;19:950–961. doi: 10.1038/cr.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L, Park DS, Friedman WJ, Greene LA. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M, Nakamura T, Ohtani N, Hampson L, Hampson IN, Shimamoto A, Furuichi Y, Okumura K, Niwa S, Taya Y, Hara E. Regulation of CDK4 activity by a novel CDK4-binding protein, p34(SEI-1) Genes Dev. 1999;13:3027–3033. doi: 10.1101/gad.13.22.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. Caspase-2 mediates neuronal cell death induced by β-amyloid. J Neurosci. 2000;20:1386–1392. doi: 10.1523/JNEUROSCI.20-04-01386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe-Fukunaga R, Iida S, Shimizu Y, Nagata S, Fukunaga R. SEI family of nuclear factors regulates p53-dependent transcriptional activation. Genes Cells. 2005;10:851–860. doi: 10.1111/j.1365-2443.2005.00881.x. [DOI] [PubMed] [Google Scholar]
- Wyttenbach A, Tolkovsky AM. The BH3-only protein Puma is both necessary and sufficient for neuronal apoptosis induced by DNA damage in sympathetic neurons. J Neurochem. 2006;96:1213–1226. doi: 10.1111/j.1471-4159.2005.03676.x. [DOI] [PubMed] [Google Scholar]
- Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol. 2001;21:4713–4724. doi: 10.1128/MCB.21.14.4713-4724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Qu D, Morris EJ, O'Hare MJ, Callaghan SM, Slack RS, Geller HM, Park DS. The Chk1/Cdc25A pathway as activators of the cell cycle in neuronal death induced by camptothecin. J Neurosci. 2006;26:8819–8828. doi: 10.1523/JNEUROSCI.2593-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Lee HG, Perry G, Smith MA. Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta. 2007;1772:494–502. doi: 10.1016/j.bbadis.2006.10.014. [DOI] [PubMed] [Google Scholar]