

Abstract
The straightforward interpretation of solution state residual dipolar couplings (RDCs) in terms of internuclear vector orientations generally requires prior knowledge of the alignment tensor, which in turn is normally estimated using a structural model. We have developed a protocol which allows the requirement for prior structural knowledge to be dispensed with as long as RDC measurements can be made in three independent alignment media. This approach, called Rigid Structure from Dipolar Couplings (RSDC), allows vector orientations and alignment tensors to be determined de novo from just three independent sets of RDCs. It is shown that complications arising from the existence of multiple solutions can be overcome by careful consideration of alignment tensor magnitudes in addition to the agreement between measured and calculated RDCs. Extensive simulations as well applications to the proteins ubiquitin and Staphylococcal protein GB1 demonstrate that this method can provide robust determinations of alignment tensors and amide N-H bond orientations often with better than 10° accuracy, even in the presence of modest levels of internal dynamics.
Keywords: residual dipolar couplings, alignment tensor, protein structure determination, protein GB1, ubiquitin
Introduction
Residual dipolar couplings (RDCs) measured under weakly aligning conditions are sensitive probes of internuclear vector orientation relative to a common molecule fixed frame. However, the relationship between experimental measurements and internuclear vector orientation is not immutable, but rather is governed by the specific details of molecular alignment, described by five parameters which make up the alignment tensor. It is very straightforward to estimate the alignment tensor from the experimental RDCs if a structural model is available, and this route provides a powerful means for purpose of validation or subsequent refinement of a structural model. In the absence of prior structural information, the situation becomes substantially more complicated. In addition to the ambiguity resulting from the cone-like continuum of possible internuclear vector orientations which correspond to a single measured RDC, the problem is compounded by an inability to even establish the correct cone of orientations due to lack of knowledge of the alignment tensor. As such, the development of methods to circumvent these difficulties have been the focus of numerous investigations (Bax 2003; Griesinger et al. 2004; Prestegard et al. 2004; Blackledge 2005; Tolman and Ruan 2006; Bouvignies et al. 2007).
One of the earliest suggestions for overcoming the RDC underdetermination problem was to utilize a second, different alignment medium (Ramirez and Bax 1998). Although this approach still required prior structural information for estimation of alignment tensors, it was shown that possible internuclear vector orientations corresponding to a single measured RDC could be restricted to a discrete number of possibilities corresponding to the intersection of the two cones describing the orientational solutions in each of the two alignment media. Alternatively, sets of RDCs measured for distinct sub-fragments of known structure could be used to estimate the alignment tensor for that specific fragment and then orient the different fragments relative to one another (Weaver and Prestegard 1998; Al-Hashimi et al. 2000; Fowler et al. 2000; Hus et al. 2000; Skrynnikov et al. 2000; Hus et al. 2001; Tolman et al. 2001; Giesen et al. 2003; Skrynnikov 2004). More recently, several approaches have been proposed in which the structure of individual helices or beta-strands are parameterized and then fit to the experimental RDCs along with alignment parameters (Mesleh et al. 2003; Mesleh and Opella 2003; Wang and Donald 2004; Chen and Tjandra 2007; Wang et al. 2007). In principle, solutions for individual elements of secondary structure can then be built up into a complete model. In practice, the success of these approaches depends strongly on one or several factors such as the ability to measure RDCs corresponding to many different dipolar interactions with a high level of completeness and the accuracy of structural fragments employed in the analysis.
An alternative approach is to make RDC measurements utilizing a large number of different alignment media. Instead of measuring RDCs for many different dipolar interactions and then using either idealized or real structural models to allow a coupled interpretation of these data, the multi-alignment approach seeks to overcome the fundamental ambiguity inherent in RDC analysis by exploiting the complementary information which results when the alignment tensor changes. It has been demonstrated that if RDCs can be measured in five different alignment media, one can dispense with the need for prior structural information entirely as well as characterize motions of the internuclear vector (Meiler et al. 2001; Hus and Bruschweiler 2002; Peti et al. 2002; Tolman 2002; Briggman and Tolman 2003; Lakomek et al. 2006). However, the applicability of these approaches remains limited due to the experimental difficulties associated with acquisition of five RDC datasets of sufficient independence. This has led to the development of hybrid approaches, in which one takes advantage of the additional information content of several independent RDC datasets, but renders the problem more tractable by utilizing structural and dynamic modeling. For example, Clore and Schweiters (Clore and Schwieters 2004) have introduced an ensemble simulated annealing approach which can allow refinement of a small number of conformers in order to account for dynamic averaging of RDCs. Blackledge and coworkers have introduced a method which utilizes 2 or 3 independent sets of RDCs and a set of structural coordinates in order to characterize Gaussian Axial Fluctuations (GAF) motions of individual peptide plane moieties along the backbone (Bernado and Blackledge 2004; Bouvignies et al. 2005; Bouvignies et al. 2008).
The acquisition of five independent alignments remains experimentally challenging due to the lack of experimental control over alignment (Ulmer et al. 2003; Ruan and Tolman 2005). In many cases it may be much easier to acquire RDC data in just three independent media, for example by choosing media which are neutrally, positively and negatively-charged, respectively. As such, the question which arises is whether in this case the RDC data may be interpreted if prior structural information is not available. It is demonstrated herein that the measurement of RDCs utilizing three independent alignment media allows for the de novo determination of internuclear vector orientations. The method proceeds by least squares optimization of both internuclear vector orientations and alignment tensors starting from an appropriately chosen initial ‘guess’ and is referred to as RSDC (Rigid Structure from Dipolar Couplings). While in principle the approach appears very straightforward, in practice there are a couple of pitfalls which must be avoided. Key to the robustness of the approach are the method for arriving at the initial guesses for the alignment tensors and the likelihood filtering of best fit alignment tensors based on the average magnitude. Although RSDC explicitly assumes that dynamics can be neglected, simulations indicate that the modest presence of dynamic averaging can be tolerated, although with correspondingly reduced precision of determination of vector orientations and alignment tensors. The methods are illustrated with applications to human ubiquitin and the first IgG-binding domain of Streptococcal protein G.
Theoretical background
Under the assumption that molecular structure and dynamics are invariant to changes in alignment medium and assuming that motions and alignment are uncorrelated, the multi-alignment RDC problem can be concisely expressed as a matrix equation (Tolman 2002),
| (1) |
The matrix D is formed directly from the RDC measurements with dimensions N x M, where N is the number of measured RDCs, and M is the number of distinct datasets. As such, the element Dij denotes the RDC of the ith internuclear vector measured in the jth alignment media. The matrix à = κA contains the alignment tensors scaled by the interaction constant κ such that elements of the alignment tensors will take on values in Hz which are directly comparable to the measured RDCs. Each of the M columns of the matrix A contains the irreducible tensorial description of each respective alignment tensor, expressed as follows in terms of elements of the Saupe order tensor (Tolman 2002),
| (2) |
The matrix B in Eq. 1 contains the motionally averaged irreducible tensorial descriptions corresponding to each of the internuclear vectors. The ith row of B, B⃗i, can be written as,
| (3) |
where the spherical angles θi and φi describe the orientation of the ith internuclear vector relative to an arbitrary molecule fixed reference frame.
Construction of Orthogonal Linear Combination (OLC)-RDC datasets
The success of any multi-alignment RDC study will depend strongly on the ability to measure the required independent datasets. This objective is complicated by the fact that one has minimal control over alignment and thus a group of experimental RDC datasets usually have a substantial degree of linear dependence. As such, it is desirable to be able to quantify the extent of linear independence of the data and then work with a group of RDC datasets which exhibit perfect linear dependence. Assessment of linear independence is accomplished by means of a singular value decomposition (SVD) of the RDC data, according to (Press 1992; Tolman 2002; Tolman and Ruan 2006),
| (4) |
The diagonal matrix W, containing the singular values of the data matrix D, reports on the relative weights of different orthogonal combinations within the data as a whole. RDC datasets which exhibit perfect linear independence can be constructed according to (Ruan and Tolman 2005; Gebel et al. 2006),
| (5) |
Note that the above equation differs slightly from previous formulations by a constant scaling factor. We refer to these independent RDC datasets as orthogonal linear combination (OLC)-RDCs.
Each individual OLC-RDC dataset will have a very different magnitude according to its representation among the directly recorded RDC datasets. As a consequence, the decision as to how many independent RDC datasets are present within the data requires consideration of the signal to noise ratio for the weaker OLC-RDC datasets. To aid in this assessment, we define a Qnoise parameter representing the contribution from random errors and thus a lower bound for the Q value if the RDCs in question were to be fit to a set of structural coordinates. For any individual RDC dataset, Qnoise is defined as,
| (6) |
in which N is the number of internuclear vectors included in the analysis, σD is the experimental error and the element di refers to the measured RDC for the ith internuclear vector. The derivation of the Qnoise parameter and its relationship to the commonly employed Q value is described in the Appendix.
Estimation of alignment tensor magnitude
A recurrent problem that arises in the analysis of RDCs is the determination of the absolute magnitude of the alignment tensor. This problem arises due to the presence of dynamics, the limited and often non-uniform distribution of internuclear vector orientations, and experimental errors in the RDC measurements themselves. With the exception of experimental errors, these effects invariably lead to underestimation of the actual magnitude of alignment. This is because there is a certain minimum magnitude of alignment necessary in order to produce the observed RDCs. On the other hand, it is quite possible to invoke a degree of alignment which is much larger than reality and still account, however erroneously, for observation. Our purpose here is to establish, under the simplifying conditions of no dynamics and a uniform distribution of vectors, an upper and lower bound for the magnitude of the alignment tensor based on the observed extrema for the residual dipolar couplings, with the larger magnitude coupling defined as dmax and the other as dmin. Note that dmax and dmin will normally have opposite sign. An abbreviated description is included below with the full derivation included in the appendix.
An absolute magnitude of alignment can be specified in terms of the generalized degree of order (GDO), ϕ, as follows (Tolman et al. 2001),
| (7) |
in which the elements Aii correspond to the three eigenvalues of the 3×3 Saupe matrix describing alignment. Neglecting random errors, the observed values for dmin and dmax underestimate the true magnitude of Ayy and Azz.
| (8) |
Nevertheless, an estimate of ϕ can be obtained from dmin and dmax as follows,
| (9) |
We consider the simplified case in which dynamics are negligible and the distribution of internuclear vectors is uniform. Under these conditions upper and lower bounds can be established on permissible values for the magnitude of alignment (ϕ). Recalling the expression for ϕest in Eq. 9, the lower bound is given by,
| (10) |
and the upper bound is given by,
| (11) |
where N is the total number of internuclear vectors, σϕ is the propagated uncertainty in ϕ (defined in the appendix Eq. A9), and δϕmax is the maximum difference between the estimated and true values of GDO, as derived in the appendix Eqs. A14 to A16.
Materials and Methods
Preparation of [15N]-labeled streptococcal Protein G B1 Domain
The host strain Escherichia coli (BL21), harboring the plasmid construct (gB1) under control of the T7 promoter, was used for overexpression of the B1 domain of protein G and was generously supplied by Prof. Blake Hill. The initial culture growth was performed at 37°C, until an optical density of 0.7–0.8 (600 nm) was reached (generally 3–5 hrs). The growth was centrifuged at 6,000g at 4°C for 20 minutes, and the pellets resuspended in M9 minimal medium containing glucose and 15NH4Cl. The expression of protein G was induced with 0.5 mM IPTG at 37°C and reinduced four hours later with 0.25 mM IPTG. Cells were harvested after 8 hours by centrifugation at 6000g at 4°C for 20 minutes. The cell pellets were resuspended in 20 mM Tris buffer (pH=8.0) in the ratio of 1.0 g of cell paste/5 mL of buffer, and lysed using a French press. The protein of interest was isolated on a FPLC system using a QFF anion-exchange column, and further purified using a 3,000 MWCO filter.
Acquisition of RDC data
The amide 15N-1H RDC datasets for ubiquitin are taken from the literature (Ottiger and Bax 1998; Briggman and Tolman 2003). Protein GB1 samples (1mM) were prepared to contain 10mM phosphate (pH 6.6 except note specifically), 0.05% NaN3 and 5% D2O. Following acquisition of isotropic reference data, samples were prepared using the following alignment media: 35 mg/ml bacteriophage Pf1 (Hansen et al. 2000) with 50mM NaCl, 5% w/v bicelles (Tjandra and Bax 1997), 5.7% bicelles doped with 0.2% CTAB (Losonczi and Prestegard 1998), 5% bicelles doped with Eu3+ (Prosser et al. 1996), 1.8% CPBr with 90mM NaBr (Barrientos et al. 2000), 5% PEG (Ruckert and Otting 2000), 3.75% PEG with 0.86% CPBr and 78mM NaBr, and ether bicelles (DIODPC:DIOHPC) doped with 1:20 (molar ratio) SDS at pH 3.3 (Ottiger and Bax 1999). The isotropic and charged bicelle data were acquired in Varian Inova 600MHz and 500MHz spectrometers, respectively. All other NMR experiments were carried out on a Bruker Avance spectrometer operating at a 1H resonance frequency of 600MHz and equipped with a triple resonance probe. All experiments were carried out at 35°C, with amide 15N-1H RDCs obtained by difference between one-bond 15N-1H couplings measured under isotropic and aligned conditions. All 1JNH coupling measurements were performed using the IPAP-HSQC (Ottiger et al. 1998) experiment. Total experimental acquisition times ranged between 12 and 17hrs. Data processing was carried out using NMRPipe software and PIPP (Delaglio et al. 1995; Garrett et al. 1995).
Generation of synthetic data
Synthetic RDC data was generated using Eq. 1 based on a set of provided alignment tensors A and a matrix B describing a set of internuclear vectors averaged according to the specified level of internal dynamics. The alignment tensors were generated randomly with the magnitude restricted such that the maximum magnitude of the RDCs produced was 15 Hz. The vector orientations comprising the matrix B were randomly generated with a variable total number of internuclear vectors. From a set of four randomly generated RDC datasets, three synthetic OLC-RDC datasets were extracted after an SVD analysis. Synthetic OLC-RDC data were not employed unless all three OLC-RDCs had a Qnoise < 0.4. This corresponds to a relative error of measurement which is greater than 20% and thus it represents a liberal lower bound for quality of data. Random errors drawn from a Gaussian distribution with specified standard deviation were subsequently added to the calculated RDCs. The effect of dynamic averaging was simulated by direct modification of the eigenvalues of the specific residual dipolar tensor according to the desired generalized order parameter with a randomly generated motional asymmetry parameters η (Tolman 2002). The diagonal residual dipolar tensor was then rotated back into the proper frame with the Wigner γ angle randomly generated and α and β angles taken from the spherical angles describing the orientation of the specific internuclear vector.
Results and Discussion
It is well established that when two independent RDC datasets are available, the possible orientations for individual internuclear vectors are restricted to the intersection between two cones representing the continuous range of possible vector orientations relative to the principal axes of alignment. Implicit in this picture, however, is that the orientation of the PASs of alignment (and thus of the cones) is known a priori. In the event that details of the alignment are not available, then the intersection between the two cones becomes entirely dependent on the choice of the respective alignment PASs, and the problem remains underdetermined. By extension it seems plausible that utilization of a third independent RDC dataset would allow internuclear vector orientations and alignment tensors to become overdetermined under the assumption that effects due to dynamics are negligible. As illustrated in Figure 1, such a determination might be carried out in practice by requiring any feasible set of internuclear vectors and alignment tensors be internally consistent such that the corresponding 3 cones calculated for each internuclear vector share a common intersection with allowance made for experimental precision of measurement. This intuitive picture forms the basis for the design of the Rigid Structure from Dipolar Couplings (RSDC) protocol described below.
Figure 1.
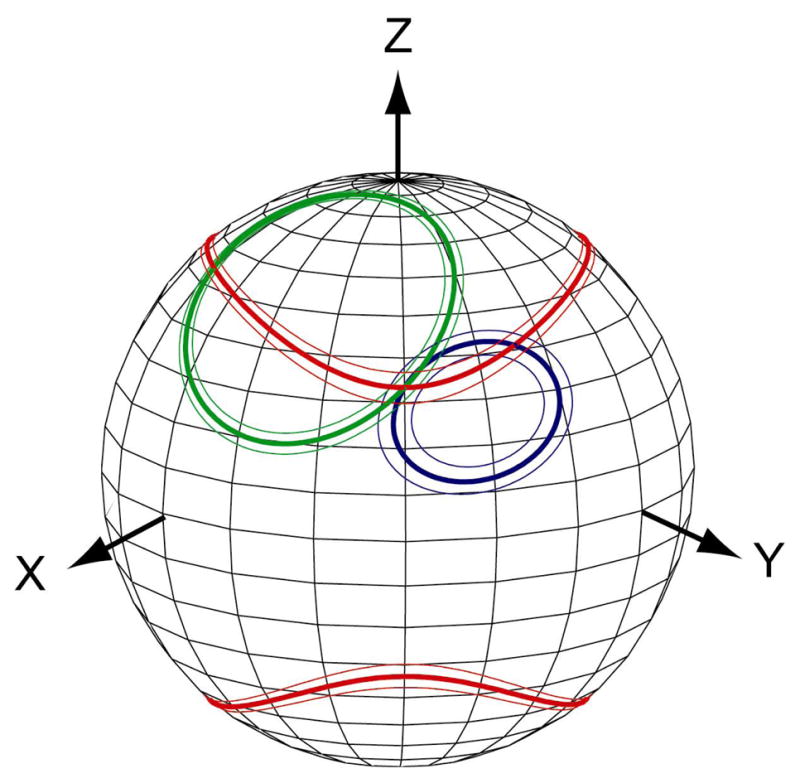
Measurement of residual dipolar couplings in one or two alignment media places restraints on internuclear vector orientations only to the extent to which the corresponding alignment tensors are known. Addition of a third independent set of RDC data places restrictions on possible alignment tensors because in the absence of dynamics the three cones must share a common intersection relative to a molecule fixed frame.
The RSDC protocol
The overall scheme of the RSDC protocol is summarized in Figure 2. The RSDC protocol is composed of three distinct stages: 1) Generation of initial guesses for the alignment tensors, 2) minimization of vector orientations and alignment tensors to convergence, and 3) Choice of the ‘best’ overall solution according to defined selection criteria.
Figure 2.
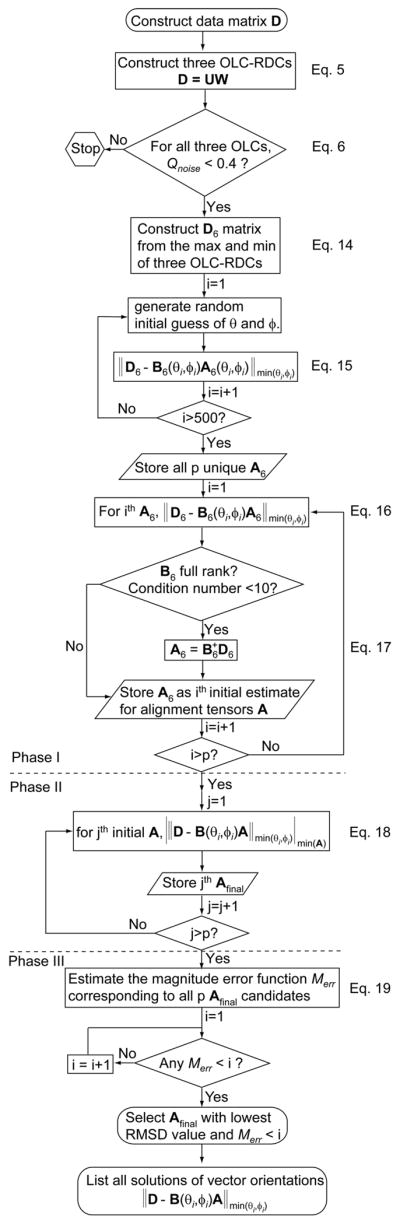
Flow chart describing the Rigid Structure from Dipolar Couplings (RSDC) protocol.
Phase I: Generation of initial guesses for the alignment tensors
For each set of measured RDCs, one can always identify two measurements which correspond to the most positive and most negative observed couplings. The coupling of largest absolute magnitude can be used to estimate the principal magnitude of alignment Azz while the other coupling provides an estimate for Ayy (Clore et al. 1998). An estimate for the asymmetry parameter η can be obtained from these two values according to,
| (12) |
In the idealized case, these two couplings will also correspond to internuclear vectors which lie precisely along the y and z principal axes of alignment. Under this simplifying assumption, and utilizing the irreducible forms of A and B expressed in Eqs. 2 and 3, one arrives at the following expression for the jth alignment tensor written in its PAS,
| (13) |
in which the row vectors B⃗Z and B⃗Y correspond to vectors lying along the z and y principal axes of alignment. Although the above equation was derived in the PAS of alignment for simplicity, note that it remains valid in an arbitrary coordinate frame. Given the above results, if consideration is restricted to the six internuclear vectors which correspond to maximum and minimum observations in all three media, then one can arrive at the following matrix equation,
| (14) |
Note that the minimum and maximum observed dipolar couplings are indicated in bold and the corresponding internuclear vectors and alignment tensors are written in terms of spherical coordinates according to Eqs. 2 and 3. As the above formulation depends on 9 unknown angles and 18 measured RDCs, the nine angles can be determined by non-linear least squares minimization (the Levenberg-Marquardt algorithm is utilized for all minimizations) given initial guesses for the nine angles, according to,
| (15) |
where the subscript 6 indicates the use of matrices of reduced dimensionality according to Eq. 14. In practice, more than one set of best fit values for the nine angles will be obtained depending on the initial guesses. This arises because the angles appear solely in terms of their cos and sin functions with consequent loss of information concerning phase. On the other hand, the number of distinct possibilities remains limited due to the relative simplicity of the associated trigonometric functions. For example, noting that B1Z and B1Y can always be placed unambiguously simply by choice of reference frame, any third vector can be placed with fourfold ambiguity. This would suggest that there could exist as many as 44 = 256 different combinations of the nine angles which would minimize to different solutions. However, this neglects the additional restraints contained within D6 which do not involve the vectors B1Z or B1Y. Indeed, B3Z will be uniquely determined given specific vectors B1Z and B2Z. Taking this into account, and maintaining the ambiguity in B2Y and B3Y due to uncertainties in η, the maximum uncertainty can be reduced to 64 distinct cases with the expectation that here will still be substantially fewer in reality. Rather then attempt to analytically derive all possible cases, 500 random initial guesses are generated for the nine angles (θi, φi) and all unique solutions stored. As anticipated, experience has consistently shown that not more than a few dozen unique solutions result. The number of unique solutions obtained is denoted by p in Figure 2. Given the resultant p estimates for the alignment tensors A, a correction for the non-perfect collinearity of the 6 internuclear vectors with the corresponding principal axes of alignment is achieved by an additional minimization step with the alignment tensors determined from Eq. 15 held fixed,
| (16) |
The final matrix A, to be used as one of the p initial guesses, is then obtained by an unrestrained best fit of the six internuclear vectors to the corresponding RDCs according to,
| (17) |
provided that the condition number for the matrix B6, defined as the ratio of its largest to smallest singular values, is less than 10. The condition number is checked in order to ensure that A is not estimated from a near-singular matrix B6, in which case the original matrix A6 determined in Eq. 15 remains a better estimate for A.
Phase II: Determination of the best-fit internuclear vector orientations and alignment tensors
As a result of the above described procedure, one typically generates up to as many as 40 different initial guesses for the alignment tensors specified in the form of the matrix A. In the second step of the RSDC protocol, an iterative minimization procedure is carried out utilizing all of the RDC data in order to find the best-fit solution for the alignment tensors and vector orientations corresponding to each individual initial guess for A. This procedure is carried out with iterative application of the following nested minimization,
| (18) |
in which minimization of the matrix B is performed row by row using the parameterization in terms of θi and φi according to Eq. 3. The minimization of the matrix A is carried out in the PAS of the first alignment tensor but otherwise unrestrained over the remaining 12 free parameters. Note that the inner minimization amounts to a rigid reorientation of individual vector orientations to best fit the RDCs, while the outer minimization is identical to the best fit determination of alignment tensors based on a set of structural coordinates. The degree to which a set of vector orientations can be found which agree with the RDC measurement is reflected in RMSD between measured and best fit couplings.
Phase III: Choice of solution
In general, distinct local minima will be encountered after minimization according to Eq. 18 depending on the initial guess for the alignment tensors A. Each of these distinct local minima will correspond to a unique set of alignment tensors A and internuclear vectors B. That multiple local minima are encountered is not surprising as the minimization is over a total of 2N + 12 degrees of freedom, where N is the number of internuclear vectors. One possible approach for dealing with this ambiguity would be to simply select the solution which exhibits the lowest final RMSD between calculated and measured couplings after minimization. This approach is quite logical given that the RMSD reports directly on how well a joint set of internuclear vectors and alignment tensors can replicate the measured RDCs. However, while it is clear that a sufficiently good initial guess for A will lead to a good solution for B and hence a low final RMSD, a question which arises is whether a bad guess for A can combine with a bad set of vector orientations to produce a comparably low final RMSD. As shown later, this scenario is indeed possible and occurs with non-negligible frequency. To avoid this situation we propose that the best fit solution be chosen by means of a joint consideration of the final RMSD and a function of the average generalized magnitude of the final best fit alignment tensors. This function, Merr, is defined in terms of the generalized degree of order (ϕi) for final computed alignment tensors, as follows,
| (19) |
where the values for ϕest and ϕupper are defined in Eqs. 9 and 11, respectively. When the average magnitude of the best fit alignment tensors does not exceed the estimated upper bound, Merr will assume a value less than 1. Such a situation is one in which the final matrix A is in complete conformity with estimates derived from the observed RDCs. In such circumstances, the set of minimized internuclear vectors which exhibit the lowest RMSD will be the set chosen to be the best fit solution. However, it may be that none of the solutions have an associated Merr ≤ 1. This may be due to dynamics or an unusually anisotropic distribution of internuclear vectors, as discussed in the subsequent section. In this case, the solution with the lowest RMSD and an Merr < 2 would be selected. In the event that there are still no solutions, then the threshold for Merr is incremented in steps of 1 until a suitable solution is found according to the scheme outlined in Figure 2.
Performance of the RSDC protocol using synthetic RDC data
The performance of the RSDC protocol was subjected to fourteen distinct test cases using synthetic data. For each case, a random distribution of vector orientations was drawn and RDCs calculated either assuming rigidity or with some level of dynamic averaging included. Four alignment tensors were randomly generated with the magnitude of each fixed such that the maximum observed RDC cannot exceed 15 Hz. An SVD analysis was then performed on the four synthetic datasets and the three strongest OLC-RDCs were then submitted to the RSDC protocol provided that Qnoise < 0.4 for all three OLC-RDC datasets. Fifty separate test runs were carried out and various statistics computed for each case with a specific number of vectors (N) and added random errors σD. These results are summarized in Table 1. Considering first the mean angular deviation (θ̄), it is clear that RSDC can robustly determine vector orientations to a very good precision (< 10° on average for nearly all cases considered) depending strongly on the level of experimental error. The final agreement between the calculated and measured RDCs is consistently better than experimental precision, which is expected given that three data points are being used to estimate two parameters. Agreement is also very good for the final alignment tensors, although not as good as for individual vectors because they are much more strongly overdetermined. Note that variation in the number of vectors, N, has a surprisingly small effect on the average performance, with an exception being for smaller distributions such as the N = 50 cases. Given that the vector orientations and alignment tensors are being determined simultaneously, it is expected that there will be a minimum threshold for N in order for RSDC to produce acceptable results. Our experience indicates that this threshold is ca. 25–30 vectors. Increases in N above 50 produce only modest improvements, primarily in the quality of best fit alignment tensors. The quality of initial calculated RDCs and alignment tensors, which result from the initial guess phase of RSDC, are actually remarkably good. The ability to produce good initial guesses is an important feature underlying the robustness of RSDC.
Table 1.
RSDC test results for synthetic data
| Na | σD (Hz)b | Dynamicc | RMSD(Dinit)d | RMSD(Dfinal)e | PM>1(%)f | RMSD(Ãinit)g | RMSD(Ãfinal)h | Θ̄(°)i | Multiple local minima (%)j |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| localk | 1σ | 2σ | 3σ | |||||||||
| 50 | 0.5 | N/A | 0.67 ± 0.17 | 0.24 ± 0.06 | 2 | 1.3 ± 0.3 | 0.56 ± 0.55 | 3.8 ± 1.8 | 13 ± 10 | 15 ± 9 | 30 ± 14 | 38 ± 16 |
| 50 | 1.5 | N/A | 0.99 ± 0.15 | 0.56 ± 0.07 | 24 | 1.9± 0.5 | 2.6± 1.1 | 11.6 ± 3.2 | 31 ± 12 | 33 ± 14 | 63 ± 18 | 76 ± 17 |
| 100 | 0.2 | N/A | 0.54 ± 0.20 | 0.14 ± 0.07 | 0 | 0.91 ± 0.41 | 0.12 ± 0.13 | 1.2 ± 0.5 | 4 ± 6 | 6 ± 5 | 13 ± 8 | 18 ± 10 |
| 100 | 0.5 | N/A | 0.62 ± 0.16 | 0.26 ± 0.05 | 2 | 1.1 ± 0.4 | 0.29 ± 0.24 | 3.0 ± 0.8 | 10 ± 8 | 13 ± 8 | 26 ± 14 | 33 ± 16 |
| 100 | 1.0 | N/A | 0.82 ± 0.14 | 0.49 ± 0.06 | 6 | 1.4 ± 0.5 | 0.82 ± 0.57 | 6.2 ± 1.9 | 16 ± 11 | 21 ± 11 | 42 ± 20 | 52 ± 22 |
| 100 | 1.5 | N/A | 1.0 ± 0.1 | 0.64 ± 0.08 | 24 | 1.7 ± 0.4 | 1.6 ± 0.7 | 9.1 ± 1.8 | 24 ± 10 | 33 ± 15 | 61 ± 19 | 72 ± 18 |
| 150 | 0.5 | N/A | 0.58 ± 0.14 | 0.27 ± 0.03 | 2 | 0.95 ± 0.30 | 0.24 ± 0.20 | 2.9 ± 0.8 | 10 ± 6 | 15 ± 9 | 29 ± 14 | 37 ± 17 |
| 200 | 0.5 | N/A | 0.57 ± 0.12 | 0.28 ± 0.03 | 0 | 0.91 ± 0.28 | 0.23 ± 0.18 | 2.9 ± 0.6 | 10 ± 6 | 15 ± 8 | 29 ± 14 | 37 ± 16 |
| 200 | 1.5 | N/A | 1.1 ± 0.2 | 0.72 ± 0.06 | 16 | 1.9 ± 0.4 | 1.2 ± 0.6 | 8.2 ± 1.5 | 20 ± 12 | 28 ± 12 | 52 ± 18 | 62 ± 18 |
| 100 | 0.5 | 0 | 0.65 ± 0.14 | 0.37 ± 0.06 | 0 | 1.1 ± 0.3 | 0.59 ± 0.41 | 4.8 ± 1.4 | 14 ± 7 | 13 ± 6 | 31 ± 11 | 43 ± 15 |
| 100 | 0.5 | 5% | 0.72 ± 0.13 | 0.42 ± 0.06 | 4 | 1.3 ± 0.4 | 0.68 ± 0.33 | 5.1 ± 1.2 | 15 ± 9 | 11 ± 7 | 26 ± 12 | 38 ± 15 |
| 100 | 0.5 | 10% | 0.75 ± 0.17 | 0.45 ± 0.08 | 6 | 1.3 ± 0.3 | 0.87 ± 0.48 | 6.1 ± 1.8 | 16 ± 9 | 11 ± 6 | 28 ± 12 | 39 ± 16 |
| 100 | 0.5 | 15% | 0.79 ± 0.14 | 0.49 ± 0.10 | 12 | 1.4 ± 0.4 | 0.97 ± 0.51 | 6.6 ± 1.7 | 17 ± 7 | 10 ± 6 | 27 ± 13 | 39 ± 14 |
| 100 | 0.5 | 20% | 0.79 ± 0.15 | 0.50 ± 0.10 | 12 | 1.4 ± 0.3 | 1.3 ± 0.6 | 7.7 ± 2.4 | 22 ± 12 | 10 ± 6 | 27 ± 12 | 40 ± 15 |
Number of internuclear vectors.
Random errors added to the synthetic RDCs.
Percentage of vectors synthesized with S2 values ranging from 0.16 to 0.64 and with motional asymmetry parameters η between 0 to 1.0. The remaining vectors were synthesized with S2 from 0.64 to 1.0 and η from 0 to 0.3.
RMSD between the true synthetic RDCs and those back-calculated using the initial guess for the alignment tensors.
RMSD between the true synthetic RDCs and those back-calculated using the final best-fit alignment tensors.
Percentage of cases in which the lowest RMSD (Dfinal) had an Merr >1.
RMSD between the true synthetic alignment tensors and the initial estimates for the tensors.
RMSD between the true synthetic alignment tensors and the final best-fit alignment tensors.
The mean angular deviation between the true synthetic vector orientations and the final best-fit vector orientations.
The percentage of cases for which vector orientations are in agreement within the specified multiple of σD.
Percentage of cases where a local minimum (not the global one) corresponds to a solution closest to the true vector orientation.
The need for two separate criteria for evaluating solutions
In the course of development of the RSDC protocol, it was discovered that the RMSD (or Q value), is not a sufficient metric for evaluating the quality of a specific solution. In other words, cases arise in which the global minimum obtained when comparing experimental versus calculated RDCs actually corresponds to a solution which is strongly inferior to other solutions which exhibit a higher RMSD between experimental and calculated RDCs. The percentage of cases for which this situation occurred during the simulations is reported in Table 1 under the PM>1(%) column. Two such cases are illustrated in Figure 3. One case is drawn from the simulations without dynamics, and the second is a synthetic case based on the X-ray coordinates of calmodulin (CaM; PDB 1CLL) (Chattopadhyaya et al. 1992) with dynamics added. Plotted are the final RMSDs between calculated and measured RDCs versus the average angular deviation of the final vector orientations from the true orientation for all unique solutions obtained from RSDC. In both cases, the global minimum in terms of the RMSD between measured and calculated couplings exhibits deviations from the actual vector orientations of nearly 20° as opposed to the best solutions which are in the vicinity of 10°. In the synthetic CaM case, there are actually six solutions which exhibit a better RMSD than the ‘good’ solution. This situation arises because the RMSD (or Q value) does not provide any direct restraint on the alignment tensors themselves. As such, under certain circumstances the final best fit alignment tensors can assume magnitudes which are strongly unrealistic. In response to this problem we have defined in Eq. 19 a parameter, Merr, which quantifies the extent to which the average magnitude of the final alignment tensors conform with a derived upper bound for the magnitude of alignment. Merr will assume values between 0 and 1 when the average magnitude of alignment is within expectation, and will increase linearly with increasing deviations in alignment magnitude from expectations. In referring back to Figure 3, note that in both cases the ‘good’ solution exhibits an Merr less than 1, while the spurious solutions exhibit Merr values which are greater than 1 and in most cases greater than 3. For all cases encountered in the simulations, consideration of Merr allowed the correct solution to be successfully selected even when it was not the global best fit to the experimental couplings.
Figure 3.

Plot of the mean angular deviation of final vector orientations from actual orientations versus the corresponding RMSD between the synthetic and the calculated RDCs. Results shown are for test cases using synthetic RDCs derived either from a set of randomly generated vector orientations (N = 100, σD = 1.0 Hz) (A) or from X-ray structural coordinates of Calmodulin (Chattopadhyaya et al. 1992) (PDB 1CLL) (B) For the calmodulin case, the effects of dynamic averaging was included as follows: 18% of vectors were assigned generalized order parameters S between 0.4 and 0.8 while the other 82% had S values lying between 0.8 and 1.0.
What is the origin of these spurious minima? A closer analysis reveals that these spurious minima are due to distortions of the distribution of internuclear vectors towards greater anisotropy, which is then compensated for by increases in alignment magnitudes. This can be seen in Figure 4A, which show further details for the synthetic CaM case illustrated in Figure 3B. Plotted with filled circles is the correlation between the computed values of Merr and the condition number obtained for the matrix B. The condition number is computed as the ratio of largest to smallest singular values of the matrix B, and thus it is a measure of deviation of vector orientations from isotropy. The correlation is quite strong. In addition, the corresponding angular deviation from the true solution is denoted by attached open circles. What is striking is that all solutions except one show poor agreement with the true vector orientations, which may be expected given that there are many ways to distort the distribution, but only one correct distribution. It is important to note that solutions with high Merr values and yet very low RMSDs still technically remain viable solutions. However, this appears to be exceedingly unlikely given that estimates of alignment magnitudes from the RDC data will nearly always underestimate the true magnitude, and thus the minimization is strongly biased towards finding solutions of higher rather than lower anisotropy for the vector distribution. Shown in Figure 4B are condition numbers calculated for a number of different proteins with structures deposited in the PDB. As is evident, most proteins do not exhibit strong anisotropies in their NH vector distributions, with condition numbers around 2. Even for the deliberately chosen difficult case of a four helix bundle, the condition number is only 4.
Figure 4.

(A) Correlation plot of Merr versus the condition number of the matrix B (●) and the mean angular deviation of vector orientations from their true values (○) for each of the ten unique solutions obtained in the RSDC test case using synthetic calmodulin data with dynamics added (see text). The dotted lines denote that two different correlations are being displayed for each RSDC solution. (B) Calculated condition numbers for the B matrix constructed using all amide N-H bonds taken from the indicated PDB entry. The condition number is the ratio of the largest to smallest singular values resulting from a singular value decomposition (SVD) of the matrix B formed as described in the text.
Solutions for vector orientations are multi-valued
Although the RSDC protocol arrives at an unambiguous solution for the alignment tensors with an associated set of best-fit internuclear vector orientations, not all of the final internuclear vector orientations are uniquely determined within experimental uncertainty. This is not particularly surprising given that individual vector orientations are being determined from only three data points with their own associated experimental errors. The two most typical outcomes for individual vectors are illustrated in Figure 5 and statistics summarizing the prevalence of multiple minima during the simulations are compiled in the last four columns of Table 1. The column labeled local in Table 1 refers to the percentage of cases in which the vector orientation which produces the best fit to the data in terms of RMSD does not correspond to the minimum which lies closest to the true orientation. The remaining three columns list the percentage of residues exhibiting more than one minimum which agree with the RDC data within the specified multiple of σD. Clearly a substantial fraction of vectors have more than one viable solution with the actual percentage strongly dependent on the level of experimental error.
Figure 5.

The RSDC protocol produces a single solution for the majority of cases (A), however, a sizable fraction of internuclear vectors have two viable solutions (B). Plotted is the RMSD between experimental and couplings calculated using the best fit alignment tensors, for two illustrative cases taken from the simulations.
Robustness to dynamics
Given that an underlying assumption for RSDC is that dynamic averaging effects are negligible, a set of simulations was carried out to probe the performance of RSDC for a dynamic protein. To mimic the effect of dynamics, a modest level of motion was randomly assigned to all residues (S2 ranging between 0.64 and 1.0), except for a minority percentage of residues which were assigned much greater amplitudes of motion (S2 between 0.16 and 0.64). The intent was to simulate the presence of some highly mobile loop regions. The results are summarized in the last five rows of Table 1. Clearly the presence of dynamics leads to a general degradation in the performance of RSDC, but what is striking is that RSDC remains robust in the presence of dynamics, with the cost being a reduction in precision of the determined vector orientations and alignment tensors.
Application to ubiquitin and the B1 domain of protein G
An experimental test of the RSDC protocol was carried out using existing RDC data for the protein ubiquitin and new RDC data acquired for the B1 domain of protein G (GB1). RDC data for the two proteins consisted of 11 datasets for ubiquitin and 8 for protein GB1. After SVD analysis of the RDC data, the three OLC-RDC datasets of largest magnitude were selected and provided as input to the RSDC protocol. Summarized in Table 2 are the magnitudes of each of the OLC-RDC datasets and the associated Qnoise and Q values relative to the X-ray coordinates (1UBQ and 1PGB) (Vijaykumar et al. 1987; Gallagher et al. 1994). Plots of solutions resulting from all unique initial guesses for the alignment tensors are shown in Figure 6 for both ubiquitin and GB1. Note that in both cases, the global best fit corresponds to alignment tensors which lie within prediction (Merr < 1). Upon comparison with the X-ray structures, the average angular deviation of the RSDC vector orientations from the X-ray orientations is 6.5° and 8.9° for ubiquitin and GB1 respectively. In Figure 7, residue specific results are depicted for all solutions which agree with experimental data within 3σD (0.6 and 2.1 Hz for ubiquitin and GB1, respectively). For ubiquitin, residues 8 and 12 have best fit solutions which lie outside of the 3σD range. This can be explained by the fact that those two residues are adjacent to a flexible loop and are thus subject to substantial dynamic averaging. Notably the RSDC results for ubiquitin are better than obtained for GB1. This is due to smaller experimental errors in the case of Ub, and to the fact that only 39 vectors are available for GB1 compared to 53 for ubiquitin. For both the Ub and GB1 applications, the final best fit alignment tensors are in excellent agreement with alignment tensors calculated from X-ray or NMR coordinates (Figure 8).
Table 2.
SVD analysis of the experimental RDC datasets of ubiquitin and protein GB1 measured in multiple aligning conditionsa
| 1 | 2 | 3 | 4 | 5 | 6 | ||
|---|---|---|---|---|---|---|---|
| ubiquitin | Singular values | 125.5 | 69.44 | 22.08 | 20.79 | 9.129 | 1.633 |
| Qnoise | 0.012 | 0.021 | 0.066 | 0.070 | 0.159 | 0.892 | |
| Q valuesb | 0.171 | 0.170 | 0.295 | 0.207 | 0.311 | 0.982 | |
| GB1 | Singular values | 278.5 | 68.49 | 36.91 | 10.11 | 8.984 | 5.030 |
| Qnoise | 0.016 | 0.064 | 0.118 | 0.432 | 0.487 | 0.869 | |
| Q valuesc | 0.204 | 0.287 | 0.363 | 0.904 | 0.634 | 0.864 | |
A total of 11 and 8 RDC datasets were acquired for ubiquitin and GB1, respectively. Only the first six of the singular values and the corresponding Qnoise and Q values are listed for comparison.
Computed using the X-ray coordinates (PDB: 1UBQ).
Computed using the X-ray coordinates (PDB: 1PGB).
Figure 6.

Plot of all unique solutions resulting from application of the RSDC protocol to the first 3 OLC-RDCs of ubiquitin (A) and GB1 (B). The RMSD between the experimental RDCs and those determined by RSDC is utilized to select the best solution under the restraint that optimal tensor magnitude be maintained (0 ≤ Merr ≤ 1). The structural accuracy is reported as the mean angular deviation for each RSDC solution from the relevant X-ray structures for ubiquitin (1UBQ) (Vijaykumar et al. 1987) and protein GB1 (1PGB) (Gallagher et al. 1994).
Figure 7.

Residue by residue comparison of the final RSDC vector orientations with the ubiquitin (A) and protein GB1 (B) X-ray structures (1UBQ and 1PGB, respectively). Shown are both the best fit orientations (●) in best agreement with the X-ray structures as well as all other orientations (○) which agree with the experimental RDC data within 3σD.
Figure 8.

Comparison of the best-fit alignment tensors determined from RSDC and those derived from a best fit to the X-ray coordinates (○)(Vijaykumar et al. 1987; Gallagher et al. 1994)or NMR coordinates (x) (Cornilescu et al. 1998; Kuszewski et al. 1999) in the case of ubiquitin (A) and GB1 (B). For the case of ubiquitin, the triangles indicate agreement with the mean amide N-H bond orientations determined using the DIDC procedure (Briggman and Tolman 2003), which enables simultaneous determination of bond orientations and dynamics.
Conclusions
Our results indicate that given three independent RDC datasets of sufficient quality, the RSDC protocol proposed herein can robustly determine both alignment tensors and internuclear vector orientations de novo. In most cases vector orientations are determined with better than 10° accuracy. Furthermore, the method is robust to the presence of modest levels of dynamics, although the precision of determination of vector orientations is concomitantly decreased. Depending on the level of experimental errors, a sizable minority (and rarely a majority) of internuclear vectors will exhibit more than one orientational solution which is within experimental error. The results of our simulations indicate that in certain circumstances solutions may be obtained which agree well with the RDC data yet exhibit magnitudes of alignment well outside of expectation and with correspondingly poorer agreement with the actual vector orientations. Although this phenomenon was not observed in either the ubiquitin or GB1 applications, it appears that these cases arise due to a complex interplay between the orientational distribution of vectors and alignment tensors. These problems can be avoided by filtering solutions based on the conformity of associated alignment tensor magnitudes with expectations based on the observed RDC data.
The most significant implication of the current work is the ability to cleanly separate contributions to measured RDCs arising due to overall molecular alignment from those relating to vector orientations in the absence of prior knowledge or assumptions about structure. Typically, RDCs can only be employed fruitfully given a preliminary structural model, which in turn will depend heavily on NOE data. Notwithstanding the expected contributions of RSDC towards the development of robust RDC-dominated methods for structure determination, its greatest impact will likely be for systems in which traditional NOE-based methods begin to fail due to an insufficient density of restraints. In these cases, the ability to specify the alignment tensors in advance could allow the RDC data to be deployed during the critical early stages of structure determination when the global fold is not yet defined. Alternatively, the internuclear vector solutions could be recast into dihedral restraints (Wang and Donald 2004) or fit to fragment of peptide backbone in a fashion similar to that employed by the molecular fragment replacement approach (Delaglio et al. 2000).
Supplementary Material
Acknowledgments
The authors would like to acknowledge support from the NIH (GM075310) and NSF (MCB-0615786).
Appendix
Separation of the Q value into components arising from structural quality and noise
The Q value is used to assess the level of agreement between a structural model and a single RDC dataset. It can be written as follows:
| (A1) |
where || || denotes the norm and d⃗ is a column vector consisting of the RDC measurements. The matrix B is of dimension N × 5 where N is the number of dipolar interactions for which RDC measurements have been made and the matrix B+ is its Moore-Penrose pseudoinverse. Each row of the matrix B contains the irreducible tensorial description of the specific dipolar interaction tensor. Contributions to a computed Q value can arise from errors in the measured RDCs themselves or structural and dynamic deviations from the coordinates embodied in B. To distinguish, we write the set of measured couplings d⃗ = d⃗′ + ε, in which d⃗′ is the set of true couplings and ε is a vector containing the experimental errors. Substitution into Eq. A1 leads to,
| (A2) |
in which I is the identity matrix. Note that if there are no experimental errors, ε = 0, then the Q value depends only on the first term in the numerator and is solely an assessment of structural quality. On the other hand if the structural model B is perfect then only the second term will be non-zero and it will be solely related to the magnitude of experimental errors. From Eq. A2, one can arrive at the following relationship under the assumption that experimental errors are uncorrelated with the structural model B,
| (A3) |
with
| (A4) |
It is the value of Qstruct that is normally desired and thus it would be useful if Qnoise could be estimated. We start by writing the error vector ε in terms of a normalized vector ε0 and the estimated random error specified by σD. Given a normalized N-dimensional vector, its elements form a distribution with σ = 1/sqrt(N). This leads to the following expression for ε.
| (A5) |
Considering that B is rank 5 and that BB+ represents an orthogonal projector (Albert 1972) which projects an N dimensional vector onto a 5 dimensional subspace, the following relationships can be derived,
| (A6) |
given that ε0′ and ε0″ are both normalized N-dimensional vectors. This leads to the desired expression for Qnoise.
| (A7) |
Errors in estimation of alignment tensor magnitudes based on observed dmin and dmax
Recalling the expression for the estimated generalized degree of order (GDO) from the observed values of dmin and dmax,
| (A8) |
we note that in the absence of experimental errors, ϕest represents an absolute lower bound for the actual value of ϕ. In the presence of experimental errors, the lower bound, ϕlower, will be reduced below that of ϕest according to the propagated uncertainty in ϕ from the measurements dmin and dmax. The expression for σϕ is obtained by evaluation of,
| (A9) |
under the assumption of axial symmetry (η = 0), which produces the maximum propagation of error into ϕ Finally, one obtains the desired expression for σϕ,
| (A10) |
Recalling the expression for ϕest in Eq. A8, this allows a lower bound for ϕ to be established as follows,
| (A11) |
Establishing an upper bound requires an additional piece of information. Namely, the upper limit on the extent to which ϕest underestimates the actual value of ϕ due to noncoincidence of internuclear vectors with the Z and Y principal axes of alignment corresponding to Azz and Ayy. To do this a uniform distribution of internuclear vector orientations will be assumed. Under this assumption, the extent of solid angle on the unit sphere occupied by one of a set of N internuclear vectors is equal to 4π/N and the semiangle for a cone spanning that solid angle can be described by the angle λ, which satisfies the following equation,
| (A12) |
This leads to the following result for λ,
| (A13) |
Thus one can say that each internuclear vector inhabits its own cone on the surface of the unit sphere with a semi-angle given by λ. While it is not geometrically possible to cut a sphere up into perfect cones, the deviation from this simplified picture is expected to be very small. For a uniform distribution of vectors, each vector can thus be considered to lie at the center of its respective cone and choice of a random vector on the sphere cannot deviate from one of the preexisting N vectors by more than the angle λ. Within this framework, the maximum possible underestimation of Azz and Ayy occurs for vectors which have spherical coordinates (λ, 90) and (90-λ, 90), respectively, relative to the true principal axes,
| (A14) |
From the above expressions, it is apparent that the largest possible underestimation occurs for cases of highest asymmetry (η = 1). As estimation of the asymmetry is subject to greater uncertainty than for Azz, we derive an expression for the maximum possible underestimation in the GDO for the case of η = 1,
| (A15) |
This leads to the following expression for the maximum difference between the estimated and true values of the GDO assuming a uniform distribution of internuclear vectors and the absence of dynamic averaging,
| (A6) |
An estimate for the upper bound in the magnitude of alignment can then be obtained after some algebraic simplification utilizing results shown in Eqs. A10, A15, and A16,
| (A17) |
Footnotes
Supporting information available: Included in the supporting information is more detailed output resulting from application of RSDC to protein GB1 and ubiquitin. The RSDC program is available from the authors upon request.
References
- Al-Hashimi HM, Valafar H, Terrell M, Zartler ER, Eidsness MK, Prestegard JH. Variation of molecular alignment as a means of resolving orientational ambiguities in protein structures from dipolar couplings. J Magn Reson. 2000;143(2):402–406. doi: 10.1006/jmre.2000.2049. [DOI] [PubMed] [Google Scholar]
- Albert A. Regression and the Moore-Penrose Pseudoinverse. Academic Press; New York: 1972. [Google Scholar]
- Barrientos LG, Dolan C, Gronenborn AM. Characterization of surfactant liquid crystal phases suitable for molecular alignment and measurement of dipolar couplings. J Biomol NMR. 2000;16(4):329–337. doi: 10.1023/a:1008356618658. [DOI] [PubMed] [Google Scholar]
- Bax A. Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci. 2003;12(1):1–16. doi: 10.1110/ps.0233303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernado P, Blackledge M. Anisotropic Small Amplitude Peptide Plane Dynamics in Proteins from Residual Dipolar Couplings. J Am Chem Soc. 2004;126(15):4907–4920. doi: 10.1021/ja036977w. [DOI] [PubMed] [Google Scholar]
- Blackledge M. Recent progress in the study of biomolecular structure and dynamics in solution from residual dipolar couplings. Prog Nucl Magn Reson Spectrosc. 2005;46(1):23–61. [Google Scholar]
- Bouvignies G, Bernado P, Blackledge M. Protein backbone dynamics from N-HN dipolar couplings in partially aligned systems: a comparison of motional models in the presence of structural noise. Journal of magnetic resonance (San Diego, Calif: 1997) 2005;173(2):328–38. doi: 10.1016/j.jmr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Bouvignies G, Markwick Phineus RL, Blackledge M. Characterization of protein dynamics from residual dipolar couplings using the three dimensional Gaussian axial fluctuation model. restaurants Proteins. 2008;71(1):353–63. doi: 10.1002/prot.21703. [DOI] [PubMed] [Google Scholar]
- Bouvignies G, Markwick PRL, Blackledge M. Simultaneous definition of high resolution protein structure and backbone conformational dynamics using NMR residual dipolar couplings. Chem Phys Chem. 2007;8(13):1901–1909. doi: 10.1002/cphc.200700353. [DOI] [PubMed] [Google Scholar]
- Briggman KB, Tolman JR. De novo determination of bond orientations and order parameters from residual dipolar couplings with high accuracy. J Am Chem Soc. 2003;125:10164–10165. doi: 10.1021/ja035904+. [DOI] [PubMed] [Google Scholar]
- Chattopadhyaya R, Meador WE, Means AR, Quiocho FA. Calmodulin structure refined at 1.7. ANG resolution. J Mol Biol. 1992;228(4):1177–92. doi: 10.1016/0022-2836(92)90324-d. [DOI] [PubMed] [Google Scholar]
- Chen K, Tjandra N. Top-down approach in protein RDC data analysis: de novo estimation of the alignment tensor. J Biomol NMR. 2007;38(4):303–313. doi: 10.1007/s10858-007-9168-4. [DOI] [PubMed] [Google Scholar]
- Clore GM, Gronenborn AM, Bar A. A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information. J Magn Reson. 1998;133(1):216–221. doi: 10.1006/jmre.1998.1419. [DOI] [PubMed] [Google Scholar]
- Clore GM, Schwieters CD. How Much Backbone Motion in Ubiquitin Is Required To Account for Dipolar Coupling Data Measured in Multiple Alignment Media as Assessed by Independent Cross-Validation? J Am Chem Soc. 2004;126(9):2923–2938. doi: 10.1021/ja0386804. [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquide crystalline phase. J Am Chem Soc. 1998;120(27):6836–6837. [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe - a Multidimensional Spectral Processing System Based On Unix Pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Kontaxis G, Bax A. Protein structure determination using molecular fragment replacement and NMR dipolar couplings. J Am Chem Soc. 2000;122(9):2142–2143. [Google Scholar]
- Fowler CA, Tian F, Al-Hashimi HM, Prestegard JH. Rapid determination of protein folds using residual dipolar couplings. J Mol Biol. 2000;304:447–460. doi: 10.1006/jmbi.2000.4199. [DOI] [PubMed] [Google Scholar]
- Gallagher T, Alexander P, Bryan P, Gilliland GL. Two crystal structures of the B1 immumoglobulin-binding domain of streptococcal protein G and comparison with NMR. Biochem. 1994;33:4721–4729. [PubMed] [Google Scholar]
- Garrett DS, Gronenborn AM, Clore GM. Automated and Interactive Tools For Assigning 3d and 4d Nmr- Spectra of Proteins - Capp, Stapp and Pipp. J Cell Biochem. 1995:71–71. [Google Scholar]
- Gebel EB, Ruan K, Tolman JR, Shortle D. Multiple alignment tensors from a denatured protein. J Am Chem Soc. 2006;128:9310–9311. doi: 10.1021/ja0627693. [DOI] [PubMed] [Google Scholar]
- Giesen AW, Homans SW, Brown JM. Determination of protein global folds using backbone residual dipolar coupling and long-range NOE restraints. J Biomol NMR. 2003;25(1):63–71. doi: 10.1023/a:1021954812977. [DOI] [PubMed] [Google Scholar]
- Griesinger C, Peti W, Meiler J, Bruschweiler R. Projection angle restraints for studying structure and dynamics of biomolecules. Methods in molecular biology (Clifton, NJ) 2004;278:107–21. doi: 10.1385/1-59259-809-9:107. [DOI] [PubMed] [Google Scholar]
- Hansen MR, Hanson P, Pardi A. Filamentous bacteriophage for aligning RNA, DNA, and proteins for measurement of nuclear magnetic resonance dipolar coupling interactions. Method Enzymol. 2000;317:220–240. doi: 10.1016/s0076-6879(00)17017-x. [DOI] [PubMed] [Google Scholar]
- Hus JC, Bruschweiler R. Reconstruction of interatomic vectors by principle component analysis of nuclear magnetic resonance data in multiple alignments. J Chem Phys. 2002;117(3):1166–1172. [Google Scholar]
- Hus JC, Marion D, Blackledge M. De novo determination of protein structure by NMR using orientational and long-range order restraints. J Mol Biol. 2000;298(5):927–936. doi: 10.1006/jmbi.2000.3714. [DOI] [PubMed] [Google Scholar]
- Hus JC, Marion D, Blackledge M. Determination of protein backbone structure using only residual dipolar couplings. J Am Chem Soc. 2001;123(7):1541–1542. doi: 10.1021/ja005590f. [DOI] [PubMed] [Google Scholar]
- Kuszewski J, Gronenborn AM, Clore GM. Improving the Packing and Accuracy of NMR Structures with a Pseudopotential for the Radius of Gyration. J Am Chem Soc. 1999;121(10):2337–2338. [Google Scholar]
- Lakomek NA, Carlomagno T, Becker S, Griesinger C, Meiler J. A Thorough Dynamic Interpretation of Residual Dipolar Couplings in Ubiquitin. J Biomol NMR. 2006;34(2):101–115. doi: 10.1007/s10858-005-5686-0. [DOI] [PubMed] [Google Scholar]
- Losonczi JA, Prestegard JH. Improved dilute bicelle solutions for high-resolution NMR of biological macromolecules. J Biomol NMR. 1998;12(3):447–451. doi: 10.1023/a:1008302110884. [DOI] [PubMed] [Google Scholar]
- Meiler J, Prompers JJ, Peti W, Griesinger C, Bruschweiler R. Model-free approach to the dynamic interpretation of residual dipolar couplings in globular proteins. J Am Chem Soc. 2001;123(25):6098–6107. doi: 10.1021/ja010002z. [DOI] [PubMed] [Google Scholar]
- Mesleh MF, Lee S, Veglia G, Thiriot DS, Marassi FM, Opella SJ. Dipolar Waves Map the Structure and Topology of Helices in Membrane Proteins. J Am Chem Soc. 2003;125(29):8928–8935. doi: 10.1021/ja034211q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesleh MF, Opella SJ. Dipolar Waves as NMR maps of helices in proteins. J Magn Reson. 2003;163(2):288–299. doi: 10.1016/s1090-7807(03)00119-8. [DOI] [PubMed] [Google Scholar]
- Ottiger M, Bax A. Determination of relative N-H-N N-C′, C-alpha-C′, and C(alpha)-H-alpha effective bond lengths in a protein by NMR in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120(47):12334–12341. [Google Scholar]
- Ottiger M, Bax A. Bicelle-based liquid crystals for NMR measurement of dipolar couplings at acidic and basic pH values. J Biomol NMR. 1999;13(2):187–191. doi: 10.1023/a:1008395916985. [DOI] [PubMed] [Google Scholar]
- Ottiger M, Delaglio F, Bax A. Measurement of J and dipolar couplings from simplified two- dimensional NMR spectra. J Magn Reson. 1998;131(2):373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- Peti W, Meiler J, Bruschweiler R, Griesinger C. Model-free analysis of protein backbone motion from residual dipolar couplings. J Am Chem Soc. 2002;124:5822–5833. doi: 10.1021/ja011883c. [DOI] [PubMed] [Google Scholar]
- Press WHT, SA, Vetterling WT, Flannery BP. Numerical recipies in C. Cambridge University Press; Cambridge, UK: 1992. [Google Scholar]
- Prestegard JH, Bougault CM, Kishore AI. Residual Dipolar Couplings in Structure Determination of Biomolecules. Chem Rev. 2004;104(8):3519–3540. doi: 10.1021/cr030419i. [DOI] [PubMed] [Google Scholar]
- Prosser RS, Hunt SA, DiNatale JA, Vold RR. Magnetically aligned membrane model sysytems with positive order parameter: switching the sign of Szz with parmagnetic ions. J Am Chem Soc. 1996;118:269–270. [Google Scholar]
- Ramirez BE, Bax A. Modulation of the alignment tensor of macromolecules dissolved in a dilute liquid crystalline medium. J Am Chem Soc. 1998;120(35):9106–9107. [Google Scholar]
- Ruan K, Tolman JR. Composite alignment media for the measurement of independent sets of NMR residual dipolar couplings. J Am Chem Soc. 2005;127:15032–15033. doi: 10.1021/ja055520e. [DOI] [PubMed] [Google Scholar]
- Ruckert M, Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J Am Chem Soc. 2000;122(32):7793–7797. [Google Scholar]
- Skrynnikov NR. Orienting molecular fragments and molecules with residual dipolar couplings. Comptes Rendus Physique. 2004;5(3):359–375. [Google Scholar]
- Skrynnikov NR, Goto NK, Yang DW, Choy WY, Tolman JR, Mueller GA, Kay LE. Orienting domains in proteins using dipolar couplings measured by liquid-state NMR: Differences in solution and crystal forms of maltodextrin binding protein loaded with beta-cyclodextrin. J Mol Biol. 2000;295(5):1265–1273. doi: 10.1006/jmbi.1999.3430. [DOI] [PubMed] [Google Scholar]
- Tjandra N, Bax A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278(5340):1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- Tolman JR. A novel approach to the retrieval of structural and dynamic information from residual dipolar couplings using several oriented media in biomolecular NMR spectroscopy. J Am Chem Soc. 2002;124:12020–12030. doi: 10.1021/ja0261123. [DOI] [PubMed] [Google Scholar]
- Tolman JR, Al-Hashimi HM, Kay LE, Prestegard JH. Structural and dynamic analysis of residual dipolar coupling data for proteins. J Am Chem Soc. 2001;123(7):1416–1424. doi: 10.1021/ja002500y. [DOI] [PubMed] [Google Scholar]
- Tolman JR, Ruan K. NMR residual dipolar couplings as probes of biomolecular dynamics. Chem Rev. 2006;106:1720–1736. doi: 10.1021/cr040429z. [DOI] [PubMed] [Google Scholar]
- Ulmer TS, Ramirez BE, Delaglio F, Bax A. Evaluation of Backbone Proton Positions and Dynamics in a Small Protein by Liquid Crystal NMR Spectroscopy. J Am Chem Soc. 2003;125(30):9179–9191. doi: 10.1021/ja0350684. [DOI] [PubMed] [Google Scholar]
- Vijaykumar S, Bugg CE, Cook WJ. Structure of Ubiquitin Refined At 1.8 a Resolution. J Mol Biol. 1987;194(3):531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- Wang J, Walsh JD, Kuszewski J, Wang Y-X. Periodicity, planarity, and pixel (3P): A program using the intrinsic residual dipolar coupling periodicity-to-peptide plane correlation and phi/psi angles to derive protein backbone structures. J Magn Reson. 2007;189(1):90–103. doi: 10.1016/j.jmr.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Wang L, Donald BR. Exact Solutions for Internuclear Vectors and Backbone Dihedral Angles from NH Residual Dipolar Couplings in Two Media, and their Application in a Systematic Search Algorithm for Determining Protein Backbone Structure. J Biomol NMR. 2004;29(3):223–242. doi: 10.1023/B:JNMR.0000032552.69386.ea. [DOI] [PubMed] [Google Scholar]
- Weaver JL, Prestegard JH. Nuclear magnetic resonance structural and ligand binding studies of BLBC, a two-domain fragment of barley lectin. Biochemistry. 1998;37(1):116–128. doi: 10.1021/bi971619p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.