

Abstract
Disease progression modelling can provide information about the time course and outcome of pharmacological intervention on the disease. The basic PK/PD principles of proliferative and circular systems within the context of modelling disease progression and the effect of treatment thereupon are illustrated with the goal to better understand/predict eventual clinical outcome. Circular/proliferative systems can be very complex. To facilitate the understanding of how a dosing regimen can be defined in such systems we have shown the derivation of a system parameter named the Reproduction Minimum Inhibitory Concentration (RMIC) which represents the critical concentration at which the system switches from growth to extinction. The RMIC depends on two parameters (RMIC = (R 0 − 1) × IC50): the basic reproductive ratio (R 0) a fundamental parameter of the circular/proliferative system that represents the number of offspring produced by one replicating species during its lifespan, and the IC50, the potency of the drug to inhibit the proliferation of the system. The RMIC is constant for a given system and a given drug and represents the lowest concentration that needs to be achieved for eradication of the system. When exposure is higher than the RMIC, success can be expected in the long term. Time varying inhibition of replicating species proliferation is a natural consequence of the time varying inhibitor drug concentrations and when combined with the dynamics of the circular/proliferative system makes it difficult to predict the eventual outcome. Time varying inhibition of proliferative/circular systems can be handled by calculating the equivalent effective constant concentration (ECC), the constant plasma concentration that would give rise to the average inhibition at steady state. When ECC is higher than the RMIC, eradication of the system can be expected. In addition, it is shown that scenarios that have the same steady state ECC whatever the dose, dosage schedule or PK parameters have also the same average R 0 in the presence of the inhibitor (i.e. R 0-INH) and therefore lead to the same outcome. This allows predicting equivalent active doses and dosing schedules in circular and proliferative systems when the IC50 and pharmacokinetic characteristics of the drugs are known. The results from the simulations performed demonstrate that, for a given system (defined by its RMIC), treatment success depends mainly on the pharmacokinetic characteristics of the drug and the dosing schedule.
Keywords: PK/PD, Disease modelling, Circular systems, Proliferative systems, Basic reproductive ratio (R0), Reproduction Minimum Inhibitory Concentration (RMIC)
Introduction
The role of modelling in the science of clinical pharmacology is undisputed [1, 2]. In parallel with the increasingly widespread application of the art of modelling, the models themselves have become more complex; physiologically based pharmacokinetic (PBPK) models in pharmacokinetics (PK) [3] and more mechanistic pharmacokinetic/pharmacodynamic (PK/PD) models [4–8] are increasingly common. In some sense there is the impression that mechanism based PK/PD and disease modelling is rapidly moving from an empirical approach [9] towards the bottom-up approach usually associated with systems biology [10].
Disease progression modelling in combination with modelling the influence of drug action on the parameters of the disease progression model can provide information about the time course and outcome of pharmacological intervention on the disease. Disease progression modelling has been reviewed by Chan and Holford [11] and Danhof et al. [8]. The incorporation of circular/proliferative systems within the field of disease modelling offers further potential to advance the mechanistic development and application of disease models. One of the most advanced circular/proliferative system is the predator/prey example, independently introduced by Lotka in 1925 [12] and Volterra in 1926 [13] and adapted for viral dynamics by Nowak and Bonhoeffer [14–17]. In the predator/prey system, the predators multiply when there are many prey, but, the predators eventually outgrow their food supply and consequently decline in number. As the predator population declines, the prey population increases again. These dynamic interactions continue in cycles of growth and decline. Intuitively, it can be seen that proliferative systems such as infectious diseases (viral, bacterial and fungal) and cancer share at least the same circular aspect in which the organisms of each generation are produced from identical organisms of the previous generation. Another therapeutic area where models for circular/proliferative systems could be applied is immunology, specifically to describe allergy and inflammation.
A fundamental parameter of the circular/proliferative system is the basic reproductive ratio (R 0). This parameter is a system specific parameter and is independent of the drug being used to treat the illness. Simply put it is the number of offspring produced by one replicating species during its lifespan. The R 0 is a concept that has been used within the epidemiological field for some time and has been reviewed by Heffernan et al. [18].
Combination of circular and proliferative systems with PK/PD principles leads to specific system behaviour that can have therapeutic implications. This article focuses on introducing the basic PK/PD (concentration/effect) principles of proliferative and circular systems within the context of modelling disease progression and the effect of treatment thereupon. The importance of the ability to define the R 0 for the system will be illustrated, with respect to identifying if treatment intervention can be successful or not. Further, which pharmacokinetic parameter is most appropriate to use when comparing different doses and dosage schedules within circular/proliferative systems will be examined. Furthermore, how short term observations (studies) can be used to predict long term clinical outcome will be demonstrated. In the spirit of generality a simple circular/proliferative system will be used to illustrate the principles presented. A more complex viral infection model will also be used to highlight the same principles.
The ultimate aim of this body of work is to show generally how the circular/proliferative models can be linked to both pharmacodynamic disease models and the pharmacokinetics of the drug under study, to better understand/predict dosing regimens and eventual clinical outcome.
Theory
In this section two circular/proliferative systems will be presented in order to introduce the R 0 concept. The first circular/proliferative system is a simple one and is described to present the theoretical concepts. The second circular/proliferative system is more complex but has greater practical relevance.
A simple circular/proliferative system model
Consider the simple circular/proliferative system model illustrated in Fig. 1. The system is simple in that there is no interaction of B with potential partners in the system (e.g. bacteria can grow by themselves, they do not need host cells to reproduce). In Fig. 1, km is the production rate of new B and kd is the death rate of B. If km > kd then the system will grow. If km < kd, the system will go to extinction. If km equals kd then the system will just survive.
Fig. 1.

A simple circular/proliferative system model with inhibitor present that causes inhibition (INH)
The R 0 is defined as the ratio of km to kd and is the number of new Bs produced by one B during its lifespan. By simple extension, if R 0 is greater than 1, the system will grow. If R 0 is less than 1, the system will go to extinction. If R 0 equals 1 then the system will just survive.
Consider now the introduction of an inhibitor of km into the system, also illustrated in Fig. 1. The degree of inhibition caused by the inhibitor (INH) can be expressed as shown in Eq. 1 [19].
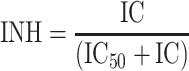 |
1 |
where IC is the plasma concentration of the inhibitor and IC50 is the plasma concentration of inhibitor that results in 50% inhibition. The rate of change of B with respect to time t, in the presence of inhibitor, is illustrated in Eq. 2.
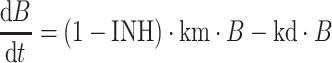 |
2 |
The basic reproductive ratio in the presence of the inhibitor (R 0-INH) can be obtained from
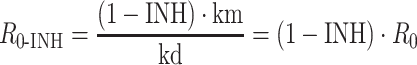 |
3 |
Again, if R 0-INH is greater than 1, the system will continue to grow; if R 0-INH is less than 1, the system will die and if R 0-INH equals 1, then the system will just survive.
Proliferative systems in the presence of an inhibitor are characterized by the Reproduction Minimum Inhibitory Concentration (RMIC). When inhibition is such that the system just survives, then
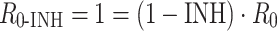 |
4 |
Therefore,
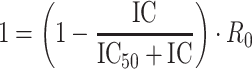 |
5 |
which rearranges to
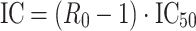 |
6 |
Since Eq. 6 was derived under circumstances where the system just survives, then, in this case, IC is equivalent to RMIC. Therefore
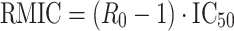 |
7 |
In Eq. 7 R 0 is a system specific parameter and IC50 is a drug (inhibitor) specific parameter.
A viral dynamic circular/proliferative system model
The simple system defined above can now be extended to the more complex viral dynamic model illustrated in Fig. 2 and used in HIV [17]. The system is more complex since the virus (B in the simple model) now needs to interact with target cells in order to reproduce. The model originally included five different types of cells (Fig. 2): target CD4 cells (T), actively infected cells (A), latently infected cells (L) which eventually can be reactivated to actively infected cells, persistently infected cells (P) with a very long half-life (≥1,000 days), defectively infected cells (D), as well as the virus particles (V). However, in the present treatise the persistently and defectively infected cells have been removed from the model because they do not significantly contribute to the viral load at equilibrium or at initiation of antiviral treatment.
Fig. 2.

The viral dynamic model based on the predator–prey system described by Lotka–Volterra. Pathways in grey are not included in the present analyses. INH indicates the site of drug action. Parameter definitions can be found in the text
The interaction between the target cells and the virus is described by differential equations in this mathematical model:
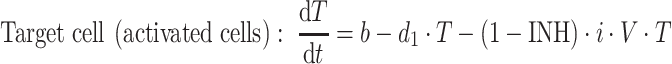 |
8 |
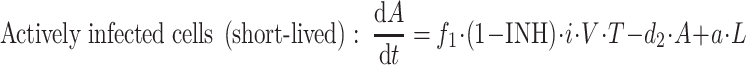 |
9 |
 |
10 |
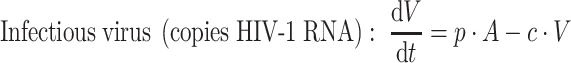 |
11 |
where b is the activation rate constant of healthy target cells (T); d 1 the death rate constant of T cells; i the infection rate constant of T cells; V the number of virus particles; f 1 the fraction of healthy T cells which become short-lived infected T cells (A); d 2 the death rate constant of short-lived infected T cells; f 2 (=1 − f 1) the fraction of healthy T cells becoming long-lived infected T cells (L); d 3 the death rate constant of latently infected resting cells; a the reactivation rate constant of latently infected resting cells; p the viral production rate constant of short-lived infected T cells; c the death rate constant of virus; and INH is the inhibition of the infection rate by the drug.
This more complex system is also characterized by a R 0. Here the R 0 is defined as the average number of secondary viruses generated by viruses introduced into an uninfected environment.
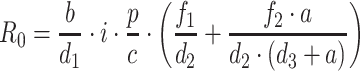 |
12 |
where 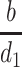 is the number of activated Target cells in the absence of virus, i is the infection rate,
is the number of activated Target cells in the absence of virus, i is the infection rate,  is the amount of circulating virus per infected cell (at steady state), and
is the amount of circulating virus per infected cell (at steady state), and 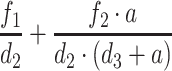 is the factor for living, actively infected cells.
is the factor for living, actively infected cells.
An inhibitory E max model decreasing the infection rate is usually used to describe the effect of antiretroviral compounds acting before DNA replication, leading to the same forms for the R 0-INH and RMIC formulae as described above for the simple model;
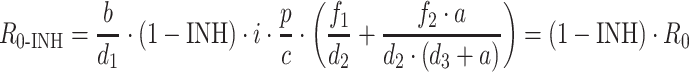 |
13 |
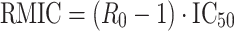 |
7 |
Attributes of circular/proliferative systems and pharmacodynamic disease models
The concepts presented above give rise to the following attributes of circular/proliferative system models with respect to R 0 and RMIC, and for binary outcomes analysis. The various attributes presented below will be illustrated later by means of simulation.
R0 and RMIC
When the inhibitory concentration equals the RMIC the system is in dynamic equilibrium; the replicating species neither increase nor decrease in number.
- Depending on R 0, system survival (i.e. R 0-INH = 1) can occur at different levels of inhibition:
- For R 0 = 2, RMIC = IC50
- For R 0 = 10, RMIC = 9 * IC50 = IC90.
If in vitro and in vivo R 0 are different, in vitro and in vivo RMIC will also be different.
From Eq. 7 it can be seen that the RMIC is a joint distribution of R 0 and IC50 in the population.
Binary outcomes analysis
The treatment is regarded as a success if the system is eradicated and a failure if the system persists. Consequently,
- Inhibition of circular/proliferative systems naturally leads to binary outcomes and logistic analysis so an:
- IC greater than RMIC leads to success (the system is eradicated).
- IC less than RMIC leads to failure (the system continues to grow/survive).
The time of failure is a function of the IC/RMIC ratio; for failure (when the ratio is below 1), the smaller the ratio, the sooner the failure will occur.
Mechanistically, logistic regression of binary outcomes such as failure/success rates as a function of inhibitor exposure (IC) is an expression of the RMIC distribution across the population.
The link between pharmacokinetics and the circular/proliferative and pharmacodynamic disease models
After drug administration, plasma concentration varies as a function of time and pharmacokinetic models are commonly used to describe the time varying profiles. Time varying inhibition of the proliferation of replicating species is a natural consequence of the time varying drug concentrations. The time varying aspect of the inhibition combined with the dynamics of the circular/proliferative system makes it difficult to predict the eventual outcome without the help of PK–PD-Disease simulation models. However, time varying inhibition of proliferative systems can be handled by calculating the equivalent effective constant concentration (ECC) [20]. ECCss is the constant plasma concentration that would give rise to the same average inhibition of proliferation at steady state as the time varying concentration. The ECCss is obtained by first calculating the time varying viral inhibition profile;
 |
14 |
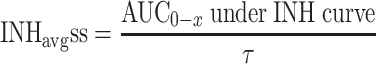 |
15 |
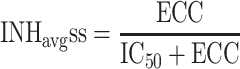 |
16 |
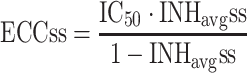 |
17 |
where C ss(TAD) is the steady state concentration at a given time after dose (TAD). The ECCss is the calculated concentration that gives rise to INHavgss across the dosing interval.
Attributes of the link between pharmacokinetics and circular/proliferative and pharmacodynamic disease models
To be equally efficacious at steady state (i.e. same proliferation rate), two treatments (e.g. QD vs. BID) should give rise to the same average R 0 in the presence of the inhibitor, at steady state (i.e. R 0-INHss) and therefore should have the same ECC.
At R 0-INHss equal to 1, ECC is equal to RMIC.
Methods
Simulations
Parameter values of the simple circular/proliferative dynamic model used in the simulations are presented in Table 1.
Table 1.
Parameter values used for the simulations with the simple circular/proliferative system model
| Parameter | Parameter description | Value | CVa (%) |
|---|---|---|---|
| km | Production rate (h−1) | 0.5 | |
| kd | Death rate (h−1) | 0.1 | |
| R 0 | Basic reproductive ratio | 5 | 30 |
| IC50 | Plasma concentration of inhibitor giving 50% inhibition (ng/ml) | 10 | 30 |
| RMIC | Reproduction minimum inhibitory concentration (ng/ml) | 40 |
aCoefficient of variation
Parameters of the more complex viral dynamic model used in the simulations were obtained from the literature [17, 20] and slightly adapted for didactic purposes (e.g. R 0 and IC50). The values of the relevant model parameters are given in Table 2.
Table 2.
Parameter values used for the simulations with the viral dynamic model
| Parameter | Parameter description | Mean |
|---|---|---|
| R 0 | Reproductive ratio before virus infection and treatment | 5.0 |
| b | Uninfected cell activation rate (day−1) | 1.0 |
| d 2 | Actively infected cell death rate (day−1) | 0.693 |
| IC50 | IC50 after in vitro to in vivo scale factor (ng/ml) | 10.0 |
| d 1 | Uninfected cell death rate (day−1) | 0.0060 |
| f 1 | Fraction of infected cells actively versus latently infected | 0.96 |
| d 3 | Latently infected cell death rate (day−1) | 0.0132 |
| a | Rate of conversion from latently to actively infected (day−1) | 0.0370 |
| p/c | Ratio of birth to death rate of virus | 35.4 |
Various effect (e.g. viral load) time profiles, under different conditions, were simulated by implementing the simple and viral dynamic models with the parameter estimates given in Tables 1 and 2 in Trial Simulator TS2 version 2.1.2. (Pharsight, Mountain View, CA, USA). The results of the simulations were analysed in S-PLUS 6.1 (Insightful Corporation, Seattle, WA, USA).
Viral load-time profiles in a typical subject (i.e. using model mean parameter values) were also simulated for various constant levels of inhibition (INHavg of 0, 0.1, 0.3, 0.5, 0.6, 0.7, 0.75, 0.8, 0.85 and 0.9) starting at day 0 and lasting for a period of 10,000 days.
Simulations were also performed to evaluate the interaction between dosing regime, i.e. QD or BID dosing, and elimination rate on viral eradication (causing R
0-INH to be below 1). Simulations were performed for the steady state condition. Plasma concentrations were simulated for a one compartment model [21] where bioavailability was complete (F = 1), Ka = 1 h−1, V = 70 l, and where Ke = 0.05, 0.1, 0.15 and 0.2 h−1, giving rise to terminal half lives of 13.9, 6.93, 4.62 and 3.47 h, respectively (Table 3). Data were simulated after a range of total daily doses (1–40 mg), given once or twice daily. In conjunction with the pharmacokinetic parameter values, parameter values of the simple circular/proliferative model (Table 1) and the viral kinetic model (Table 2) were used when deriving the inhibition (INH) and R
0-INH. In these simulations, several metrics were calculated at steady state (ss) for a dosing interval: C
maxss, C
minss, area under the concentration–time curve (AUCss), area under the inhibition-time curve (AUCINHss), area under the R
0–time curve 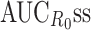 ), average concentration (Css_avg = AUCss/τ), average inhibition (INHss_avg = AUCINHss/τ), average R
0-INH (
), average concentration (Css_avg = AUCss/τ), average inhibition (INHss_avg = AUCINHss/τ), average R
0-INH (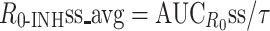 ) and ECCss. These metrics were then compared with the observed viral outcome.
) and ECCss. These metrics were then compared with the observed viral outcome.
Table 3.
PK/PD parameters used in the simulations shown in Fig. 6 together with the corresponding total daily dose and PK metrics that lead to the same viral load outcome (middle graphs) for both QD and BID regimes
| Schedule | Parameter description | QD | BID |
|---|---|---|---|
| Daily dose (mg) | Daily dose | 19.6 | 11.5 |
| Ka (h−1) | First order rate constant for absorption | 1 | 1 |
| Ke (h−1) | First order rate constant for elimination | 0.15 | 0.15 |
| V (l) | Volume of distribution | 70 | 70 |
| IC50 (ng/ml) | Concentration that results in 50% inhibition | 10 | 10 |
| R 0 | Basic reproductive ratio | 5 | 5 |
| R 0-INHss_avg | R 0 in the presence of the inhibitor, at steady state | 1.00 | 1.00 |
| C minss (ng/ml) | Minimum concentration at steady state | 9.3 | 19.2 |
| C maxss (ng/ml) | Maximum concentration at steady state | 207 | 72.8 |
| AUC24hss (ng h/ml) | Area under the curve, 0 to 24 h, at steady state | 1,866 | 1,096 |
| C avgss (ng/ml) | Average concentration at steady state | 77.8 | 45.7 |
| ECCss (ng/ml) | Effective constant concentration at steady state | 40.0 | 40.0 |
The dosing regimes were chosen to give an R 0-INHss_avg of 1. It can then be seen that all other parameters are different except ECCss (and which now equals RMIC)
Lastly, simulations were performed using parameters of Table 2 where the probability of virological success (eradication of virus) was simulated as a function of drug concentration (ECCss). R 0 and IC50 were sampled for 1,000 individuals assuming an inter-subject variability of 30% CV in the population (Table 1). Corresponding individual RMIC were calculated using Eq. 7. Success probability was estimated for a given ECC by calculating the cumulative density of subjects with RMIC below the ECC divided by 1,000. To simulate the trial outcome, 1,000 ECC values between 1 and 1,000 ng/ml were sampled (with replacement) and assigned to the 1,000 subjects. When the ECC was higher than the RMIC, the patient was considered to have a treatment success (and assigned a value of 1). When ECC was lower the patients was considered to have a treatment failure (and assigned a value of 0). Simulated success as a function of the ECC was analyzed by logistic regression.
Results
Circular/proliferative viral dynamic model
Viral load-time profiles in a typical subject (i.e. using model mean parameter values) simulated for various constant levels of inhibition starting at day 0 and lasting for a period of 10,000 days, are presented in Fig. 3. The results show that at inhibitor concentrations higher than the RMIC (e.g. IC >40 ng/ml) the viral load decreases rapidly and does not rebound, whereas at lower levels (e.g. IC <40 ng/ml) the viral load decreases rapidly initially, but then returns to a new steady state level close to the baseline value. It can also be seen that viral load rebounds faster at lower inhibitor concentration levels (e.g. IC = 15 vs. IC = 35 ng/ml).
Fig. 3.

Simulated viral load–time profiles in a typical subject exposed to various constant long-term inhibitor concentrations (IC) corresponding to constant inhibition (INH) fractions (R 0 = 5, IC50 = 10 ng/ml, RMIC = 40 ng/ml, ‘breakout’ inhibition = 0.8)
The simulations presented in Fig. 3 have been summarized in Fig. 4. Here the viral load-time profiles are now depicted for the first 10 days of treatment and at equilibrium (e.g. day 10,000) for the different INHavg values indicated. These results highlight that the initial drop of the viral load (e.g. at day 10) is not particularly indicative/predictive of viral load at equilibrium. It is only when the inhibition reaches a critical high INHavg value (break point) that the system goes to extinction. At INHavg values (slightly) lower than the break point, the virus (and the system) adjusts to the constant inhibition and, after an initial drop, viral load rebounds to equilibrate at a new level towards the baseline before treatment. Therefore it is almost impossible to identify the INHavg break point value using viral load information at day 10 only.
Fig. 4.

Simulated viral load–time profiles in a typical subject, up to day 10 and at equilibrium, given various constant long-term inhibition concentrations (R 0 = 5, IC50 = 10 ng/ml, RMIC = 40 ng/ml, ‘breakout’ inhibition = 0.8)
In Fig. 5 the predicted viral loads at equilibrium shown in Fig. 4 have been plotted against the ECC for the various regimens that give rise to the INHavg values used in Fig. 4. The resulting relationship is linear. Extrapolation of the relationship shown in Fig. 5 shows that, for this hypothetical example, if the ECC is maintained above 40 ng/ml then viral eradication should ensue in the long term.
Fig. 5.

Viral load at equilibrium (as a fraction of baseline) versus ECC
Influence of dosing regime/elimination half life
Whatever the dose, dosage schedule or PK parameters, scenarios that have the same average R 0-INHss_avg (or same ECCss) in the presence of inhibitor lead to the same outcome. This is illustrated in Fig. 6 which shows that when the R 0-INH_avg is just higher than 1 (i.e. ECC < RMIC) in both QD and BID dosage regimes the viral load rebounds after an initial drop whereas when the R 0-INH_avg is just lower than 1 (i.e. ECC > RMIC) the viral load continuously decreases. Interestingly, in the example presented in Fig. 6 the daily doses that were necessary to reach the same average R 0 (e.g. average R 0-INHss_avg = 1) were significantly different between the QD (e.g. 19.6 mg) and BID (e.g. 2 × 5.75 = 11.50 mg) dosage regimes (Table 3). In addition, as illustrated in Table 3, the same viral load outcome in both QD and BID treatments was not associated with the same Cmaxss, Cminss, AUC24hss or Cavgss. However, as is expected from the theory, the same ECCss was observed and corresponds to the calculated RMIC. Identical observations were made with the simple circular/proliferative model (not shown).
Fig. 6.

Simulated viral load–time profiles during treatment with a drug administered QD (top graphs) and BID (bottom graphs) at doses that lead to average R 0 just above 1 (left hand side graphs), equal to 1 (middle column graphs) or just below 1 (right hand side graphs)
The influence of dosing regime and half life is depicted further in Fig. 7. The solid lines indicate the R 0-INHss at the total daily dose, given QD. The dashed lines, in the same colour for a given elimination rate, represent the R 0-INHss when the total daily dose was given as a BID regime. The solid horizontal black line at an R 0-INHss of 1 is the cut off point below which a particular dosing regime/elimination rate would be predicted to result in viral eradication. Consideration of the solid and dashed blue lines shows that a total single daily dose of just under 20 mg is equivalent to a total daily dose of around 12 mg when given as 6 mg BID, in terms of when R 0-INH equals 1. Figure 7 also indicates that to have the same viral load effect (same average R 0-INHss), compounds with short half-lives require higher daily doses when given QD than when given BID, whereas compounds with long half-lives would require the same total daily dose. Figure 7 also confirms that whatever the dose, dosage schedule or PK parameters, scenarios that have the same ECC have the same R 0-INHss and therefore are expected to lead to the same outcome.
Fig. 7.

Simulated average R 0-INH at steady state versus daily dose (top graph) and ECC (bottom graph) for QD and BID dosing and for different elimination half lives. The horizontal black lines indicate an average R 0-INH of 1. The vertical line indicates the RMIC
Insights into long term exposure response
As has already been stated, the RMIC is a joint distribution of R 0 and IC50 in the population. Knowing the distribution of the RMIC in the population and the individual subject’s inhibitor concentration, then the chance/probability of success can be estimated for each individual. These results are presented in Fig. 8. Building on this further, the probability curve of success as a function of the inhibitor concentration (ECC) (Fig. 9, bottom left graph) can be constructed if we know the RMIC distribution in the population (Fig. 9, top graph) or the RMIC distribution in the population can be derived if we know the success rate as a function of the concentration (Fig. 9 bottom right hand graph). This type of approach was applied to maraviroc, an anti-HIV drug of the entry inhibitor class, and the outcome is presented in Fig. 10. Inspection of Fig. 10 shows that the probability of virological failure based on the RMIC distribution estimated in the Phase IIa studies (i.e. short term monotherapy) corresponded well with the probability of failure (defined as a viral load higher than 50 copies/ml) as a function of the ECC estimated in Phase IIb/III studies in treatment experienced HIV positive patients receiving maraviroc on top of an optimized background therapy.
Fig. 8.

Histograms (blue bars) of the simulated distribution of RMIC in a population with different individual IC values superimposed (vertical red lines). The particular individual IC values (25, 50, 75 and 100 ng/ml) have been used to calculate the associated probability of success of viral suppression (area on the left hand side of the red bar divided by the total area)
Fig. 9.

Simulated distribution of RMIC in the population (top graph) and derived predicted probability of success in function of inhibitor concentration (bottom left hand side graph). Simulated observed success rate in function of inhibitor concentration in a study (bottom right hand side graph) and derived RMIC distribution in the population (top graph)
Fig. 10.

The relationship between maraviroc equivalent constant concentration (ECC) and the estimated probability of virological failure. The dotted line shows the probability based on the RMIC distribution estimated from Phase IIa data utilising a circulatory viral kinetic model. The blue area is the 95% confidence interval of the probability of virological failure in the Phase IIb/III studies estimated by generalized additive logistic modelling (24 week data)
Discussion
Disease progression modelling in combination with modelling the influence of drug action on the parameters of the disease progression model can provide useful information about the time course and outcome of pharmacological intervention on the disease. The obvious ultimate goal is to best use new/existing pharmacological agents for the benefit of the patient. The basic principles of circular/proliferative models for both a simple example where the organism does not interact with the host at a cellular level, and, a more complex viral example where the organism needs to interact intimately with the host cell to reproduce, have been presented. The circular/proliferative viral dynamic model is based on the prey and predator principle introduced by Lotka/Volterra [12, 13] almost one hundred years ago and adapted for viral dynamics by Nowak and Bonhoeffer much more recently [14–17]. This viral dynamic model describes the interaction between the virus and the target cells by means of differential equations and has been used to predict individual viral load-time profiles after short as well as long term treatment [22].
The R 0 concept that is central to the circular/proliferative systems described is not new. Indeed, it has long a familiar concept in the epidemiological world where it is used to gauge the risk of spread of infection with a pathogen and compare that to other more well known pathogens [18]. The combination of R 0 with PK/PD models is rather new and has been discussed by several authors [23–26]. The combination leads to principles specific to circular/proliferative systems such as the somewhat unexpected linear concentration/effect relationship (on a linear axis) presented in Fig. 5.
In the presence of a drug that inhibits the growth of the system the R 0 in combination with RMIC, gives an indication of how much drug will be needed to cause the system to be eradicated in the long term. In case of inhibition that can be described by an inhibitory Emax model acting on one of the circular aspects of the system, the relationship between R 0 and RMIC (Eq. 7) is simple and is useful at a high level in understanding what concentration/dose of drug is needed in order to eradicate the system. This relationship shows that the critical concentration that switches the system from growth to extinction not only depends on the potency of the drug (i.e. IC50), but also on the capacity of the system to grow (i.e. R 0); the higher the capacity for growth, the higher the critical concentration needed to eradicate the system. This is of particular importance for in vitro in vivo extrapolation of efficacious concentrations. If in vitro and in vivo systems have different R 0, then the RMIC will also be different in vitro and in vivo. Equation 7 can be used to correct for this difference. In terms of clinical study design short term studies are (reasonably) required early on in the development of new compounds; the efficacy and safety information thus obtained is used to support/design the performance of more long term studies. There are many examples in the literature for viral infections in short term studies where outcome, in terms of viral load drop, is said to predict long term clinical outcome [27–31]. The present work demonstrates that the fall in viral load from short term studies alone cannot reliably predict long term clinical outcome. What can be gained from such studies is an estimate of IC50 for the drug under evaluation. Also if the study design is extended so that a follow up period after treatment cessation has been included and viral load re-growth is monitored, then applying models such as those described herein can provide information about R 0 [22] for the patient population under study. Under such conditions, with quantitative information about both the IC50 of the compound in question and the R 0 for the particular virus in the population, the RMIC can be calculated (Eq. 7) which gives an indication about the target exposure that needs to be reached to have a given chance/probability of success in the long term.
Figure 3 shows that the time of failure of an inhibitory drug treatment is a function of the [inhibitory drug]/RMIC ratio; for failure (when the ratio is below 1), the smaller the ratio, the sooner the failure will occur. Therefore the time of failure (in the absence of resistance) also provides a quantitative indication of how far the current exposure is from the critical RMIC and could be the basis for a therapeutic decision to either increase the dose of the inhibitor or switch to another inhibitor.
The incorporation of the pharmacokinetics into the combination of circular/proliferative models with disease models allows an understanding to be developed around what the outcome might be if dosing regimes are changed, and what measure of exposure best drives the circular/proliferative models and disease model system. It is frequently stated that efficacy is probably more related to AUC and side effects are probably driven by C max. In the therapeutic area of bacteriology this is taken further and effect is usually attributed to either AUC/MIC (concentration dependent) or the length of time that the concentration is above the MIC (time dependent or ‘concentration independent’), for different classes of drug and pathogens [32]. Karlsson et al. [33] developed a general time dissociated model for paclitaxel myelosuppression and showed that the commonly used terms of concentration dependent or concentration independent drug action are both specific cases of a more general model. In the present treatise different pharmacokinetic parameters have been explored to see which best relate to eventual outcome. Of these, R 0 (averaged over the dosing interval, at steady state, in the presence of the inhibitor) or ECC (at steady state in our simulations) in combination with RMIC were the measures that best predicted outcome for two different dosing regimes (Table 3). In the simulated examples, an inhibitory Emax model acting on one of the circular aspects of the systems has been used. It can be shown that, whatever the inhibitory function (e.g. linear, log-linear, exponential, sigmoid Emax, operational antagonism) used, if two treatments have the same average R 0 in the presence of the inhibitor, at steady state (i.e. R 0-INHss), then they will be equally efficacious, thus treatment success depends essentially on the pharmacokinetic characteristics of the drug and the dosing schedule. This approach allows calculation of equivalent effective doses for different compounds with different pharmacokinetics (e.g. volume or/and clearance) or/and pharmacodynamic (e.g. IC50) potencies or for different dosage schedules (e.g. QD vs. BID). However, with respect to the latter aspect, it must be kept in mind that the transient variations of the system during a dose interval (i.e. maximum and minimum effects) should not induce unacceptable, non-linear or irreversible outcomes.
Inspection of Fig. 9 shows that if the distribution of the RMIC in the population is known then the relationship between the probability of treatment success as a function of inhibitory drug exposure (using ECC) can be obtained. On the other hand, if the distribution of the RMIC in the population is not known (study design thus far has not been sufficient to allow characterisation of the RMIC distribution) then logistic regression analysis of a study where a binary outcome has been measured (clinical success/failure), plotted as a function of the inhibitory drug concentration (ECC) will allow the derivation of the RMIC distribution in the population. In this way logistic regression analysis which is usually viewed as a purely descriptive analysis can be used to gain mechanistic information about the biological system. Data presented in Fig. 10 show that both approaches led to the same information with regard to the probability of failure (or success) as a function of the inhibitory drug exposure and were confirmatory of each other.
Within the field of HIV treatment, modelling of disease progression and the effects of therapy has been relatively limited thus far. Some examples are Huang et al. [24] and Wu et al. [26] who both used a model similar to the complex viral model described herein and linked it to pharmacokinetic parameters (such as Cmax, AUC and Ctrough) and adherence in order to predict virological response. Labbé and Verotta [34] also used a system of differential equations to describe the dynamics of viral load together with adherence and took the approach one step further by demonstrating that increased adherence was associated with a lower R 0. However, they did not complete the circle by using the model to define if a treatment would be successful or not, in the long term (other authors have looked at adherence in the long term [23, 35]). A thorough review of the disease modelling work done thus far in the HIV field is not a goal of this work; nor is it even required from the perspective of illustrating the basic PK/PD principles of circular/proliferative systems. The reader is referred elsewhere for such a summary [36].
The circular/proliferative systems approach that has been presented here only presents the theoretical aspects of a dose–concentration–effect relationship in the absence of emerging resistance due to mutation and has also ignored the complications of using combination therapy and the role of the immune response. The reality is more complex and most of what has been theoretically described could be hidden in practice by resistance phenomena. However, the probability of the emergence of resistant mutations is a function of the average R 0-INH and could be incorporated into the model as well as extending the model to take account of multiple treatment agents on R 0-INH. The subject of resistance has been addressed by other authors [23, 37, 38] and is not covered in the present paper.
Other assumptions that have been made include that the HIV model presented has been simplified from the full model which included both defectively and persistently infected cells. The latter are the reason why viral eradication cannot be obtained despite a sustained R 0 lower than 1 in the presence of inhibitor. This is in contrast to what is observed for bacterial or HCV infections where, at a certain time in treatment, the numbers of bacteria or virus and infected cells can be brought below the critical value required to re-generate an infection and so the infection can be eradicated.
In conclusion, the basic PK/PD principles of circular/proliferative systems have been presented by means of a simple model and a more complex viral dynamic model. A fundamental parameter of the circular/proliferative system is the R 0. The combination of R 0 with PK/PD leads onto the model parameter RMIC which is the Reproduction Minimum Inhibitory Concentration. The importance of defining the R 0 and the RMIC for the particular infected system and inhibitor under consideration has been illustrated, with respect to identifying the dose and dosing schedule that can be successful or not in the long term.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Grasela TH, Dement CW, Kolterman OG, Fineman MS, Grasela DM, Honig P, Antal EJ, Bjornsson TD, Loh E. Pharmacometrics and the transition to model-based development. Clin Pharmacol Ther. 2007;82:137–142. doi: 10.1038/sj.clpt.6100270. [DOI] [PubMed] [Google Scholar]
- 2.Lalonde RL, Kowalski KG, Hutmacher MM, Ewy W, Nichols DJ, Milligan PA, Corrigan BW, Lockwood PA, Marshall SA, Benincosa LJ, Tensfeldt TG, Parivar K, Amantea M, Glue P, Koide H, Miller R. Model-based drug development. Clin Pharmacol Ther. 2007;82:21–32. doi: 10.1038/sj.clpt.6100235. [DOI] [PubMed] [Google Scholar]
- 3.Nestorov I. Whole body pharmacokinetic models. Clin Pharmacokinet. 2003;42(10):883–908. doi: 10.2165/00003088-200342100-00002. [DOI] [PubMed] [Google Scholar]
- 4.de Winter W, DeJongh J, Post T, Ploeger B, Urquhart R, Moules I, Eckland D, Danhof M. A mechanism based disease progression model for comparison of long term effects of pioglitazone, metformin and glicazide on disease processes underlying type 2 diabetes mellitus. J Pharmacokinet Pharmacodyn. 2006;33:313–343. doi: 10.1007/s10928-006-9008-2. [DOI] [PubMed] [Google Scholar]
- 5.Ribbing J (2007) Covariate model building in non-linear mixed effects models. Acta Universitatis Upsaliensis. Digital comprehensive summaries of Uppsala dissertations from the Faculty of Pharmacy 59, Uppsala, 77 pp. ISBN 978-91-554-6916-3
- 6.Perelson AS. Modelling viral and immune system dynamics. Nat Rev Immunol. 2002;2:28–36. doi: 10.1038/nri700. [DOI] [PubMed] [Google Scholar]
- 7.Jonsson EN, Macintyre F, James I, Krams M, Marshall S. Bridging the pharmacokinetics and pharmacodynamics of UK-279,276 across healthy volunteers and stroke patients using a mechanistically based model for target-mediated disposition. Pharm Res. 2005;22(8):1236–1246. doi: 10.1007/s11095-005-5264-x. [DOI] [PubMed] [Google Scholar]
- 8.Danhof M, de Jongh J, De Lange ECM, Della Pasqua O, Ploeger BA, Voskuyl RA. Mechanism-based pharmacokinetic/pharmacodynamic modeling: biophase distribution, receptor theory, and dynamical systems analysis. Annu Rev Pharmacol Toxicol. 2007;47:357–400. doi: 10.1146/annurev.pharmtox.47.120505.105154. [DOI] [PubMed] [Google Scholar]
- 9.Holford NHG, Chan PLS, Nutt JG, Kieburtz K, Shoulson I, Parkinson Study Group Disease progression and pharmacodynamics in Parkinson’s disease—evidence for functional protection with levodopa and other treatments. J Pharmacokinet Pharmacodyn. 2006;33:281–312. doi: 10.1007/s10928-006-9012-6. [DOI] [PubMed] [Google Scholar]
- 10.Wilson I (2007) Top-down versus bottom-up—rediscovering physiology via systems biology? Mol Syst Biol 3: Article number 113. doi:10.1038/msb4100154 [DOI] [PMC free article] [PubMed]
- 11.Chan PL, Holford NH. Drug treatment effects on disease progression. Annu Rev Pharmacol Toxicol. 2001;41:625–659. doi: 10.1146/annurev.pharmtox.41.1.625. [DOI] [PubMed] [Google Scholar]
- 12.Lotka AJ. Elements of physical biology. Baltimore: Williams & Wilkins Co; 1925. [Google Scholar]
- 13.Volterra V (1926) Variazioni e fluttuazioni del numero d’individui in specie animali conviventi. Mem R Accad Naz dei Lincei Ser VI, vol 2
- 14.Nowak MA, Bonhoeffer S, Shaw GM, May RM. Anti-viral drug treatment: dynamics of resistance in free virus and infected cell population. J Theor Biol. 1997;184:203–217. doi: 10.1006/jtbi.1996.0307. [DOI] [PubMed] [Google Scholar]
- 15.Bonhoeffer S, May RM, Shaw GM, Nowak MA. Virus dynamics and drug therapy. Proc Natl Acad Sci USA. 1997;94:6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonhoeffer S. Models of viral kinetics and drug resistance in HIV-1 infection. AIDS Patient Care STDS. 1998;12:769–774. doi: 10.1089/apc.1998.12.769. [DOI] [PubMed] [Google Scholar]
- 17.Funk GA, Fischer M, Joos B, Opravil M, Günthard H, Ledergerber B, Bonhoeffer S. Quantification of in vivo replicative capacity of HIV-1 in different compartments of infected cells. J Acquir Immune Defic Syndr. 2001;26(5):397–404. doi: 10.1097/00126334-200104150-00001. [DOI] [PubMed] [Google Scholar]
- 18.Heffernan JM, Smith RJ, Wahl LM. Perspectives on the basic reproductive ratio. J R Soc Interface. 2005;2(4):281–293. doi: 10.1098/rsif.2005.0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hill AV. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J Physiol. 1910;40:iv–vii. [Google Scholar]
- 20.Rosario MC, Poland B, Sullivan J, Westby M, van der Ryst E. A pharmacokinetic-pharmacodynamic model to optimize the phase IIa development program of maraviroc. J Acquir Immune Defic Syndr. 2006;42(2):183–191. doi: 10.1097/01.qai.0000220021.64115.37. [DOI] [PubMed] [Google Scholar]
- 21.Gibaldi M, Perrier D (1982) Chapter 1. In: Pharmacokinetics (2nd edn). Marcel Dekker Inc., New York
- 22.Jacqmin P, McFadyen L, Wade JR. A receptor theory-based semimechanistic PD model for the CCR5 noncompetitive antagonist maraviroc. Br J Clin Pharmacol. 2008;65(Suppl 1):95–106. doi: 10.1111/j.1365-2125.2008.03141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wahl LM, Nowak MA. Adherence and drug resistance: predictions for therapy outcome. Proc Biol Sci. 2000;267(1445):835–843. doi: 10.1098/rspb.2000.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Liu D, Wu H. Hierarchical bayesian methods for estimation of parameters in a longitudinal HIV dynamic system. Biometrics. 2006;62:413–423. doi: 10.1111/j.1541-0420.2005.00447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferguson NM, Donnelly CA, Hooper J, Ghani AC, Fraser C, Bartley LM, Rode RA, Vernazza P, Lapins D, Mayer SL. Adherence to antiretroviral therapy and its impact on clinical outcome in HIV-infected patients. J R Soc Interface. 2005;2:349–363. doi: 10.1098/rsif.2005.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu H, Huang Y, Acosta EP, Park JG, Yu S, Rosenkranz SL, Kuritzkes DR, Eron JJ, Perelson AS, Gerber JG. Pharmacodynamics of antiretroviral agents in HIV-1 infected patients: using viral dynamic models that incorporate drug susceptibility and adherence. J Pharmacokinet Pharmacodyn. 2006;33(4):399–419. doi: 10.1007/s10928-006-9006-4. [DOI] [PubMed] [Google Scholar]
- 27.Lepri AC, Miller V, Phillips AN, Rabenau H, Sabin CA, Taszewski S. The virological response to highly active antiretroviral therapy over the first 24 weeks of therapy according to the pre-therapy viral load and the weeks 4 ± 8 viral load. AIDS. 2001;15:47–54. doi: 10.1097/00002030-200101050-00008. [DOI] [PubMed] [Google Scholar]
- 28.Knolle PA, Kremp S, Höhler T, Krummenauer F, Schirmacher P, Gerken G. Viral and host factors in the prediction of response to interferon-alpha therapy in chronic hepatitis C after long-term follow-up. J Viral Hepat. 1998;5:399–406. doi: 10.1046/j.1365-2893.1998.00127.x. [DOI] [PubMed] [Google Scholar]
- 29.Kessler HH, Pierer K, Santner BI, Vellimedu SK, Stelzl E, Marth E, Fickert P, Stauber RE. Evaluation of molecular parameters for routine assessment of viremia in patients with chronic hepatitis C who are undergoing antiviral therapy. J Hum Virol. 1998;1:314–319. [PubMed] [Google Scholar]
- 30.Ampurdanés S, Olmedo E, Maluenda MD, Forns X, López-Labrador FX, Costa J, Sánchez-Tapias JM, de Anta MT, Rodés J. Permanent response to alpha-interferon therapy in chronic hepatitis C is preceded by rapid clearance of HCV-RNA from serum. J Hepatol. 1996;25:827–832. doi: 10.1016/S0168-8278(96)80285-6. [DOI] [PubMed] [Google Scholar]
- 31.Kitchen CM, Kitchen SG, Dubin JA, Gottlieb MS. Initial virological and immunologic response to highly active antiretroviral therapy predicts long-term clinical outcome. Clin Infect Dis. 2001;33:466–472. doi: 10.1086/321900. [DOI] [PubMed] [Google Scholar]
- 32.Gunderson BW, Ross GH, Ibrahim KH, Rotschafer JC. What do we really know about antibiotic pharmacodynamics? Pharmacotherapy. 2001;21:302S–318S. doi: 10.1592/phco.21.18.302S.33905. [DOI] [PubMed] [Google Scholar]
- 33.Karlsson MO, Molnar V, Bergh J, Freijs A, Larsson R. A general model for time-dissociated pharmacokinetic-pharmacodynamic relationships exemplified by paclitaxel myelosuppression. Clin Pharmacol Ther. 1998;63:11–25. doi: 10.1016/S0009-9236(98)90117-5. [DOI] [PubMed] [Google Scholar]
- 34.Labbé L, Verotta D. A non-linear mixed effect dynamic model incorporating prior exposure and adherence to treatment to describe long-term therapy outcome in HIV-patients. J Pharmacokinet Pharmacodyn. 2006;33(4):519–542. doi: 10.1007/s10928-006-9022-4. [DOI] [PubMed] [Google Scholar]
- 35.Huang Y, Rosenkranz SL, Wu H. Modeling HIV dynamics and antiviral responses with consideration of time-varying drug exposures, sensitivities and adherence. Math Biosci. 2003;184:165–186. doi: 10.1016/S0025-5564(03)00058-0. [DOI] [PubMed] [Google Scholar]
- 36.Csajka C, Verotta D. Pharmacokinetic-pharmacodynamic modelling: history and perspectives. J Pharmacokinet Pharmacodyn. 2006;33(3):227–279. doi: 10.1007/s10928-005-9002-0. [DOI] [PubMed] [Google Scholar]
- 37.Phillips AN, Youle M, Johnson M, Loveday C. Use of a stochastic model to develop understanding of the impact of different patterns of antiretroviral drug use on resistance development. AIDS. 2001;15(17):2211–2220. doi: 10.1097/00002030-200111230-00001. [DOI] [PubMed] [Google Scholar]
- 38.Rong L, Feng Z, Perelson AS. Emergence of HIV-1 drug resistance during antiretroviral treatment. Bull Math Biol. 2007;69(6):2027–2060. doi: 10.1007/s11538-007-9203-3. [DOI] [PubMed] [Google Scholar]