

Abstract
Cyclooxygenase-2 (COX-2) plays a significant role in tumor development and progression. Nonsteroidal anti-inflammatory drugs (NSAIDs) exhibit potent anticancer effects in vitro and in vivo by COX-2 dependent and independent mechanisms. In this study, we used microarray analysis to identify the change of expression profile regulated by a COX-2 specific NSAID NS-398 (0.01 and 0.1mM), a non-specific NSAID ibuprofen (0.1 and 1.5mM) and RNA interference-mediated COX-2 inhibition (COX-2 RNAi) in PC3 prostate cancer cells. A total of 3,362 differentially expressed genes with 2 fold change, and p<0.05 were identified. Low concentrations of NSAIDs and COX-2 RNAi altered very few genes (1-3%) compared to the higher concentration of NS-398 (17%) and ibuprofen (80%). Ingenuity Pathway Analysis (IPA) was used for distributing the differentially expressed genes into biological networks and for evaluation of functional significance. The top 3 networks for the both NSAIDs included functional categories DNA replication, recombination and repair, and gastrointestinal disease. Immune response function was specific to NS-398, and cell cycle, cellular movement were among the top functions for ibuprofen. IPA also identified renal and urological disease as a function specific for ibuprofen. This comprehensive study identified several COX-2 independent targets of NSAIDs which may help explain the antitumor and radiosensitizing effects of NSAIDs. However, none of these categories were reflected in the identified networks in PC3 cells treated with clinically relevant low concentrations of NS-398 and ibuprofen or with COX-2 RNAi suggesting the benefit to fingerprinting pre-clinical drug concentrations to improve their relevance to the clinical setting.
Keywords: microarray, NSAIDs, COX-2, NS-398, ibuprofen, COX-2 RNAi
Introduction
A number of preclinical and clinical studies have demonstrated that nonsteroidal anti-inflammatory drugs (NSAIDs) are effective chemopreventive and antitumor agents either alone or in combination with standard cancer therapies including radiotherapy (1-5). The best characterized targets of NSAIDs are cyclooxygenase enzymes COX-1 and COX-2 (6). COX-1 is constitutively expressed in most normal tissues and is responsible for the normal tissue homeostasis, while COX-2 is a stress response gene induced by inflammatory cytokines, oncogenes and growth factors (5, 6). COX-2 is selectively overexpressed at the site of inflammation and also in a variety of human tumors. Overexpression of COX-2 in human tumors is associated with poor prognosis and COX-2 is considered as one of the crucial targets for cancer therapy (2, 5, 7).
Nonspecific NSAIDs such as ibuprofen or indomethacin inhibit both COX-1 and COX-2. Although they are effective chemopreventive and antitumor agents, long-term use of the nonspecific NSAIDs can result in renal and gastric toxicity, attributed to the inhibition of prostaglandins (PG) derived from COX-1 (8, 9). COX-2 specific drugs were developed based on the hypothesis that selective COX-2 inhibition at the site of inflammation would lead to reduction in pain and inflammation sparing the normal tissue toxicity, and were considered as safer alternatives to the traditional NSAIDs to use in the clinic (10). However, recent studies revealed that the long-term use of COX-2 inhibitors led to increased risk of cardiovascular toxicity, a risk that will likely be of greater importance for long term use as chemoprevention agent than for short-term use as treatment modifiers (8, 11).
We have a long-standing interest in the use of NSAIDs to enhance the efficiency of radiation (1, 12, 13). Our novel approach of using NSAIDs to enhance the effects of radiation therapy was based on a clinical observation that ibuprofen ameliorated acute radiation-induced urinary symptoms in patients with prostate cancer (14). Although this observation was not confirmed in a randomized trial, prior to undertaking the clinical trial, we studied the efficacy of ibuprofen in vitro and in vivo to confirm that there was no tumor protection. Indeed, we found radiation sensitization of prostate carcinoma cells by ibuprofen (1, 12) and we have since pursued the potential mechanisms of action of NSAIDs.
Although NSAIDs inhibit prostaglandin synthesis at ≤ micromolar concentrations, the antitumor and radiosensitizing effects of NSAIDs are generally seen at higher concentrations. At higher concentrations NSAIDs inhibit a variety of cellular processes including signal transduction, transcription, and DNA repair; alter cell cycle distribution and inhibit cyclins; modulate Bcl-2 family proteins and induce apoptosis (5, 13, 15-19). NSAIDs also inhibit angiogenesis, an important factor necessary for tumor growth and survival (20). Microarray studies have demonstrated alterations in genes regulating metastasis (21), apoptosis (22, 23), cell cycle (24, 25), programmed cell death, cell proliferation and cell-cell communication (26) following treatment with nonspecific and/or COX-2 specific NSAIDs. Thus NSAIDs affect multiple cellular targets in COX-2-dependent and -independent manner (16, 19, 27).
We hypothesized that the gene expression profiles would differ for treatment with COX-2 specific and non-specific NSAIDs as they would for different concentrations of NSAIDs. In the present study we analyzed the global gene expression profile in PC3 human prostate carcinoma cells treated with NS-398, a COX-2-specific NSAID (0.01 and 0.1mM), and ibuprofen (0.1 and 1.5mM), a nonspecific NSAID that inhibits both COX-1 and COX-2. Pharmacokinetic studies indicate that oral administration of ibuprofen (400mg-3200mg per day) results in peak plasma concentrations in the range of 0.2 to 1.0mM (18, 28). While NS-398 is not in clinical use the COX-2 specific inhibitor, celocoxib, (400-800 mg) produces peak plasma concentration in the range of 3-8μmol/L (29). Thus, our low concentrations are close to clinically relevant molar concentrations. Although effective in inhibiting prostaglandin synthesis, the low concentrations in general, are less cytotoxic. The high concentrations chosen for this study are typically used in preclinical studies to demonstrate antitumor effects of NSAIDs. While pharmacological intervention by NSAIDs can result in alterations of COX-2 dependent and independent target genes, inhibition of COX-2 by RNAi is expected to reveal changes in COX-2 specific target genes. We knocked down the COX-2 gene by RNAi in order to evaluate the gene expression changes specific to COX-2 inhibition. In addition, we studied the effect of NS-398 and ibuprofen on selected cellular targets in another human prostate carcinoma cell line, DU-145, which did not express COX-2 protein.
This comprehensive microarray study revealed that 24h treatment with low concentrations of NS-398, ibuprofen and COX-2 RNAi altered very few genes in PC3 cells. However, treatment with high concentrations of NS-398 and ibuprofen resulted in differential expression of several COX-2-independent targets of NSAIDs. This study highlights the need for preclinical drug fingerprinting to compare drugs, their dosages and schedules to understand the similarities and differences of agents even within the same class, with the ultimate goal of potentially selecting the drug and schedule appropriate for personalized medicine.
Materials and Methods
Cells
PC3 and DU-145 human prostate carcinoma cells were obtained from American Type Culture Collection (Rockville, MD) and maintained in RPMI 1640 supplemented with 10% fetal bovine serum, glutamine, and antibiotics. Tissue culture reagents were purchased from Life Technologies, Inc. (Grand Island, NY). Microarray analysis was done on PC3 cells. In addition, we examined the NSAID-induced changes in selected genes and proteins in DU-145 cells.
Treatment
NS-398 was purchased from Cayman chemicals (Ann Arbor, MI), dissolved in dimethyl sulfoxide (DMSO) and stored at -20°C. Ibuprofen (I1892; Sigma chemicals) was prepared fresh as a 100mmols/L stock in distilled water and filter sterilized before adding to cells. Cells were cultured to 60-70% confluence and treated with 0.01 and 0.1mmols/L NS-398 and 0.1 and 1.5mmols/L ibuprofen for 24h. Control dishes were treated with equivalent amounts of DMSO or H2O.
Transfection of COX-2 small interfering RNA
PC3 cells were transfected with 100nmol/L siRNA targeting COX-2 (SMART pool, M-004557-00, Dharmacon, Lafayette, CO) using oligofectamine reagent (Invitrogen, Carlsbad, CA) as described previously (30).
Measurement of PGE2 Production
To measure the effect of NS-398 and ibuprofen on PGE2 production PC3 cells were treated with NS-398 (1-100 μmol/L) and ibuprofen (5-100μmol/L) for 24h. At the end of 24h cells were stimulated with 30μmol/L arachidonic acid (AA). After 15min culture supernatants were collected, centrifuged to remove debris and stored at -70°C. To measure the PGE2 production in cells after COX-2 RNAi cells were transfected with vehicle (oligofectamine) or COX-2 siRNA. At 24h, 48h and 72h after transfection cells were stimulated with 30μmol/L arachidonic acid and culture supernatants were collected after 15min. Prostaglandin levels were determined by a competitive enzyme immunoassay using PGE2 Monoclonal EIA Kit (Cayman chemicals, Ann Arbor, MI) according to the manufacturer's instructions.
Clonogenic Cell Survival
Cells were treated with NS-398, ibuprofen or siCOX-2. After 24h cells were trypsinized, counted, and 50-200 cells were plated in triplicates in six-well plates for clonogenic assay. Colonies were stained with crystal violet after 12days and colonies of >50 cells were counted.
Cell Cycle Study
Cells were treated with NS-398, ibuprofen or siCOX-2 for 24h and fixed in 70% ethanol. Cells were processed for cell cycle analysis using Guava Cell Cycle Reagent according to the protocol provided by the manufacturer (Cat. No. 4700-0160). Data were collected on Guava cytosoft (Hayward, CA) and changes in cell cycle distribution were analyzed by Modfit program.
Microarray analysis
PC3 cells were used for microarray analysis. Total RNA was extracted from cells treated with NS-398, ibuprofen or siCOX-2 for 24h from 3 separate biological replicates, using QIAshredder spin column (Cat. No. 79654, Qiagen, USA). All extracted RNAs were purified with an RNeasy mini kit (Qiagen, USA). The concentration of total RNA was measured by spectrophotometry at OD260/280 and the quality of the total RNA sample was assessed using an Agilent Bioanalyzer with the RNA6000 Nano Lab Chip (Agilent Technologies). Biotin-labeled cRNA was prepared by linear amplification of the Poly (A) + RNA population within the total RNA sample. Briefly, 2 μg of total RNA was reverse transcribed after priming with a DNA oligonucleotide containing the T7 RNA polymerase promoter 5′ to a d(T)24 sequence. After second-strand cDNA synthesis and purification of double-stranded cDNA, in vitro transcription was performed using T7 RNA polymerase in the presence of biotinylated UTP. The quantity and quality of the cRNA was assayed by spectrophotometry and on the Agilent Bioanalyzer as indicated for total RNA analysis.
Ten μg of purified cRNA was fragmented to uniform size and applied to CodeLink Human Whole Genome Bioarrays (Applied microarrays Inc.) in hybridization buffer. CodeLink Human Whole Genome arrays are comprised of approximately 55,000 30-mer probes designed to conserved exons across the transcripts of targeted genes. These probes represent well-annotated, full length, and partial human gene sequences from major public databases. All fragmented samples were visualized on the Agilent Bioanlyzer to verify complete fragmentation to ∼0.1 kb size before the sample was applied to the array. Arrays were hybridized at 37° C for 18 hrs in a shaking incubator, washed in 0.75× TNT at 46° C for 1 hr, and stained with Cy5-Streptavidin dye conjugate for 30 min. Rinsed and dried arrays were scanned with a GenePix™ 4000B scanner (Axon Instruments) at 5μm resolution.
Statistical Analysis
CodeLink Expression Analysis software (GE Healthcare) was used to process the scanned images from arrays (gridding and feature intensity) and the data generated for each feature on the array was analyzed with GeneSpring software (Agilent Technologies). Raw intensity data for each gene on every array was normalized to the median intensity of the raw values from that array. Data for all arrays were filtered for intensity values that were above background in at least two of any set of three replicates for any condition within each drug treatment. To ensure that genes were reliably measured, ANOVA was used to compare the means of each condition (n=3). Cut off ratios greater than 2.0 and less than 0.5 and a p value <0.05 relative to the respective control group were selected for this study.
Ingenuity pathway analysis (IPA)
The functional significance of differentially expressed genes perturbed by NS 398, ibuprofen and COX-2 RNAi was evaluated using Ingenuity Pathway analysis (IPA) software (Ingenuity Systems Version 6.3-1402, Redwood City, CA,). Genes with a minimal 2-fold change and a p value <0.05 were selected for network generation and pathway analyses implemented in IPA tools. GenBank IDs of the selected genes were uploaded into the IPA, which were next mapped to the functional networks available in the Ingenuity Pathway Knowledge Base. Networks are comprised of biological functions assigned to networks using significant p values for focus gene functions compared with the whole Ingenuity Pathway Knowledge Base. Focus genes were identified as the subset having modeled interaction(s) with the other molecules in the database. A maximum of 35 molecules comprised a network. Each network was given a score reflecting the negative logarithm of the p value, based on the chance of the significant molecules falling in to the network by random. A score of 2 implies that there is a 1 in 100 chance that the focus genes are together in a network because of random chance. Therefore, scores of 2 or higher have at least a 99% confidence of not being generated by random chance alone.
Western blotting
Cell extracts were prepared and proteins were separated as described previously (13). Membranes were processed by enhanced chemiluminescence method (Santa Cruz Biotechnology, Santa Cruz, CA). Protein bands were captured by digital CCD camera (Fuji, LAS 3000). The membranes were stripped and reprobed for actin. Signal intensities were quantified using Image Quant (5.2 version) software (Molecular Dynamics, Sunnyvale, CA), normalized to their loading control actin and expressed as fold change compared with vehicle treated controls.
Antibodies used for immunoblotting were purchased from the following sources: COX-2 goat polyclonal antibody was purchased from Cayman Chemicals (Ann Arbor, MI). NAG-1 rabbit polyclonal antibody was purchased from Upstate cell signaling solutions (New York). Cyclin A1 and E2F2 were purchased from BD Pharmingen (San Diego, CA). Cdk2 and ATF3 were purchased from Santa Cruz Biotechnology and Cyclin E2 was from Cell signaling (Danvers, MA). Horseradish peroxidase-conjugated goat, mouse and rabbit antibodies were purchased from Santa Cruz Biotechnology and anti-actin antibody was purchased from Chemicon (Temecula, CA).
Real-Time RT-PCR
The expression of angiopoietin-like 4 (ANGPTL4) gene was validated by real-time PCR using Taqman gene expression assays and the ABI PRISM 7500 Sequence Detection System instrument equipped with the SDS version 1.4.0 software (Applied Biosystems, Foster City, CA). Forward and reverse primers and probes were designed and produced by Applied Biosystems for ANGPTL4 (Hs01101127_m1). PCR was carried out in a 50-μl reaction volume that contained 100 ng of RNA using TaqMan One-Step RT-PCR Master Mix (Applied Biosystems). Each sample was analyzed in duplicate, for 3 different biological sets of RNA and 18S ribosomal RNA was used as endogenous control (Hs9999901_s1). Negative controls were processed under the same conditions without RNA template. The threshold cycle (CT) of the endogenous control was used to normalize target gene expression (ΔCt) to correct for experimental variation. The relative change in gene expression (ΔΔCT) was used for comparison of the gene expression in drug treated samples versus vehicle control by use of a paired t test.
In addition to ANGPTL4, the mRNA expression of COX-2 (Hs00153133 m1), NAG-1 (also known as GDF-15 and MIC-1) (Hs00171132_m1), MMP3 (Hs00153133m1) and E2F2 (Hs00918091_m1) was analyzed in DU-145 cells. Gene expression levels were normalized to GAPDH (Hs99999905_m1) expression and data are presented as the fold change in the target genes in treated cell normalized to the internal control gene (GAPDH) and relative to untreated cells. The baseline mRNA levels were compared between control PC3 and DU-145 expression and the fold differences calculated using the ΔΔCT method as described by Livak and Schmittgen (31).
Immunoassay
The Quantitative MMP3 immunoassay kit (R&D systems) was used for the determination of active and pro-Matrix Metalloproteinase 3 (total MMP3) in cell culture supernatants. Cells were treated with NS-398, ibuprofen or siCOX-2 in six-well plates in duplicate for each condition. At 24 hours, media were collected, centrifuged to remove debris, and stored at -70°C. Cells from each well were trypsinized and counted. MMP3 concentration was determined by ELISA according to the manufacturer's instructions and expressed as ng of MMP3 / 106 cells.
Data analysis
Each data point represents average ± SEM of 3 experiments. Differences between the groups were statistically evaluated by two-tailed paired t test. A p value of <0.05 was considered statistically significant.
Results
Inhibition of PGE2 synthesis by NS-398, ibuprofen and COX-2 RNAi
PGE2 synthesis was inhibited by both NSAIDs over the concentration range studied (NS-398 1-100μmol/L, ibuprofen 5-100μmol/L). Treatment with 0.01 mmol/L NS-398 and 0.1 mM ibuprofen for 24h reduced AA stimulated PGE2 secretion by 70% and 60% respectively, compared to the vehicle treated controls. Transfection of cells with COX-2 siRNA reduced PGE2 secretion by 40% at 24h and by 60% at 48h compared to the vehicle (oligofectamine) treated cells.
Clonogenic survival
The plating efficiencies of DMSO control, H2O control and oligofectamine control were 0.69 ± 0.12, 0.65 ± 0.07 and 0.67, respectively. The surviving fractions of PC3 cells treated with 0.01 and 0.1mM NS-398 for 24h were 1.09 ± 0.04 and 1.06 ± 0.09 respectively compared to the DMSO control (n=3). The surviving fractions of 0.1 and 1.5mM ibuprofen treated cells were 1.03 ± 0.3 and 0.46 ± 0.12 respectively, compared to the control (n=3). The surviving fractions of cells transfected with COX-2 siRNA was 0.93 compared to its vehicle control (n=2). Thus, only the high concentration of ibuprofen reduced the clonogenic survival and by 50%.
Microarray analysis of PC3 cells
Out of the total 55,000 genes represented in the Code-link Human Whole Genome array 3,362 genes were differentially expressed by the NSAID treatments and COX-2 RNAi with high confidence (>2 fold change, p<0.05). Less than 3% genes were altered by low concentrations of NSAIDs or COX-2 RNAi (supplementary table S1) while high concentrations of NS-398 and ibuprofen altered 17% and 80% genes respectively (supplementary tables S2 and S3).
A comparison of differentially expressed genes in cells treated with low concentrations of NS-398, ibuprofen and COX-2 RNAi showed that not a single gene was commonly up regulated (Fig. 1A) or down regulated (Fig. 1B) among these treatments. High concentration of both NSAIDs showed much greater effect on gene expression patterns compared to the low concentration. Global gene expression changes were much greater with ibuprofen than for NS-398. Venn diagrams in Fig. 1C and D (lower panel) show the comparison of differentially expressed genes by high concentrations of NS-398, ibuprofen and COX-2 RNAi. The number of commonly expressed genes with high concentration of the two NSAIDs was 264, out of which 97 were up regulated (Fig. 1C) and 167 were down regulated (Fig. 1D). MMP3 was the only one gene that was commonly up regulated by high concentrations of the two NSAIDs and COX-2 RNAi (Fig. 1C).
Fig. 1.
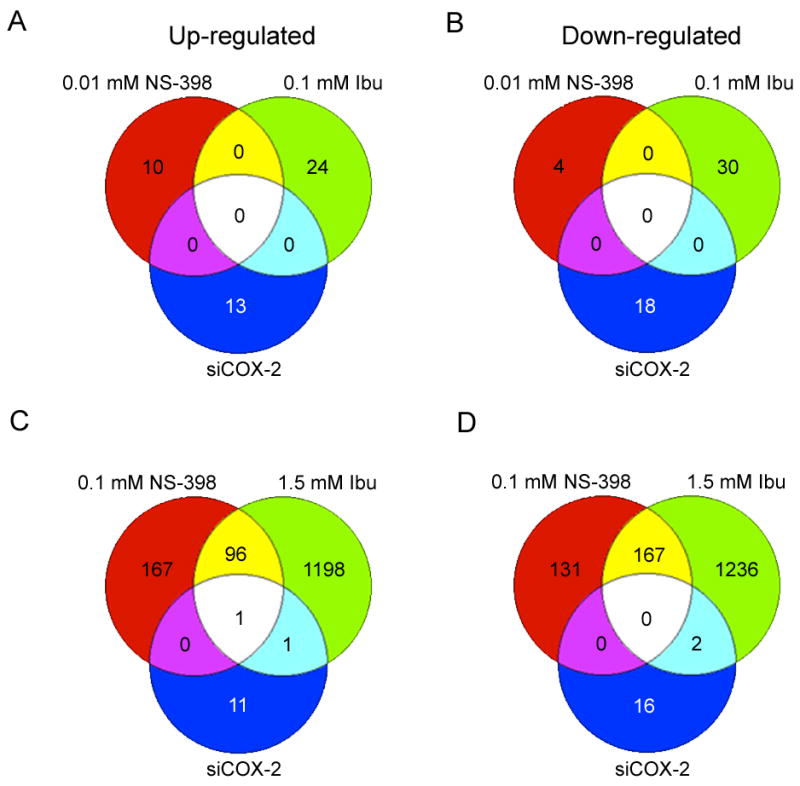
Venn diagrams showing the number of overlapping genes in comparison of 0.01mM NS-398, 0.1mM ibuprofen and siCOX-2 (A, B); and 0.1mM NS-398, 1.5mM ibuprofen and siCOX-2 (C, D). A and C: - up regulated genes, B and D: - down regulated genes. The number of differentially expressed genes with more than 2-fold changes for each treatment was 0.01mM NS-398 (14 genes), 0.1mM NS-398 (562 genes), 0.1mM ibuprofen (54 genes) and 1.5mM ibuprofen (2701 genes) and siCOX-2 (31 genes).
Ingenuity Pathway Analysis
Genes significantly affected by NSAIDs and COX-2 RNAi (> 2 fold change and p value <0.05) were mapped to the functional networks in the IPA data base and ranked by score. Table 1 and 2 show up to the top 10 networks affected by NSAIDs and COX-2 RNAi. Treatment with 0.01mM NS-398, 0.1mM ibuprofen and COX-2 RNAi perturbed very few genes (see Venn diagram- Fig.1A, B) and there were less than 10 functional IPA networks to classify these genes (Table 1). However, treatment with 0.1mM NS-398 and 1.5mM ibuprofen resulted in differential expression of 562 and 2701 genes, respectively (Fig. 1C, D). IPA showed that for 0.1mM NS-398 there were 18 networks that had more than 10 focus molecules whereas for 1.5mM ibuprofen there were 65 networks that had more than 10 focus molecules. Table 2 describes the top 10 networks in which the genes affected by high concentration of NSAIDs were mapped. DNA replication, recombination and repair, gastrointestinal disease and immune response were the functional categories included in the top 3 networks for 0.1mM NS-398. The functional categories in the top 3 networks for 1.5mM ibuprofen included DNA replication, recombination and repair, cell cycle, cellular movement, cell growth and proliferation. Significantly, IPA analysis also revealed renal and urological disease and gastrointestinal disease categories in the top 3 networks of high ibuprofen.
Table 1. Functions associated with the top 10 networks for genes whose expression was affected by low dose NSAIDs or SiCOX2 treatment in PC3 Cells.
Genes significantly affected in PC3 cells by NSAIDs and COX-2 RNAi (>2-fold change, p<0.05) were classified in to functional networks using IPA software. Table 1 shows the networks and the associated functional categories identified by IPA for NS-398 (0.01mM), ibuprofen (0.1mM) and COX-2 siRNA. Score refers to the statistical significance and focus molecules indicate the number of genes that could be mapped to molecules, out of a possible 35 molecules in each network.
| Networks | Score | Focus Molecules | Top Functions |
|---|---|---|---|
| 0.01 mM NS-398 | |||
| 1 | 14 | 5 | Protein Synthesis, Cellular Growth and Proliferation, Cell Cycle |
| 2 | 3 | 1 | Cellular Development, Developmental Disorder, Digestive System Development and Function |
| 0.1 mM IBU | |||
| 1 | 44 | 18 | Cellular Development, Cellular Growth and Proliferation, Cancer |
| 2 | 26 | 12 | Cell Cycle, Hepatic System Development and Function, Gene Expression |
| 3 | 3 | 1 | Hematological Disease, Infectious Disease, Cardiovascular Disease |
| 4 | 3 | 1 | Cell Morphology, Cell-To-Cell Signaling and Interaction, Nervous System Development and Function |
| 5 | 3 | 1 | |
| 6 | 3 | 1 | |
| 7 | 2 | 1 | Cardiovascular Disease, Organismal Injury & Abnormalities, Reproductive System Development and Function |
| SiCOX2 | |||
| 1 | 38 | 14 | Reproductive System Disease, Cellular Movement, Cardiovascular System Development and Function |
| 2 | 3 | 1 | Molecular Transport, Small Molecule Biochemistry, Amino Acid Metabolism |
| 3 | 3 | 1 | Gene Expression |
| 4 | 3 | 1 | Small Molecule Biochemistry |
Table 2. Functions associated with the top 10 networks for genes whose expression was affected by high dose NSAIDs in PC3 Cells.
Top 10 networks and the associated functional categories of the genes significantly altered by high NS-398 (0.1mM) and high ibuprofen (1.5mM), identified by IPA.
| Networks | Score | Focus Molecules | Top Functions |
|---|---|---|---|
| 0.1 mM NS-398 | |||
| 1 | 57 | 32 | DNA Replication, Recombination, and Repair, Cancer, Gastrointestinal Disease |
| 2 | 52 | 30 | DNA Replication, Recombination, and Repair, Viral Function, Cancer |
| 3 | 42 | 26 | Cancer, Immune Response, Connective Tissue Disorders |
| 4 | 35 | 23 | DNA Replication, Recombination, and Repair, Cell Cycle, Cellular Assembly and Organization |
| 5 | 27 | 19 | Cancer, Cellular Growth and Proliferation, Hematological Disease |
| 6 | 25 | 18 | Neurological Disease, Tissue Morphology, Cancer |
| 7 | 23 | 17 | Gene Expression, Viral Function, RNA Post-Transcriptional Modification |
| 8 | 23 | 17 | Gene Expression, Cellular Growth and Proliferation, Cell Death |
| 9 | 21 | 16 | Cellular Assembly and Organization, Cellular Function and Maintenance, Cancer |
| 10 | 21 | 16 | Carbohydrate Metabolism, Lipid Metabolism, Small Molecule Biochemistry |
| 1.5 mM IBU | |||
| 1 | 44 | 35 | DNA Replication, Recombination, and Repair, Cell Cycle, Cellular Movement |
| 2 | 42 | 34 | Cancer, Cellular Growth and Proliferation, Renal and Urological Disease |
| 3 | 42 | 34 | Cancer, Cell Cycle, Gastrointestinal Disease |
| 4 | 39 | 33 | Cell-To-Cell Signaling and Interaction, Cellular Movement, Cellular Assembly and Organization |
| 5 | 39 | 33 | Connective Tissue Disorders, Immunological Disease, Inflammatory Disease |
| 6 | 37 | 32 | Cell Cycle, DNA Replication, Recombination, and Repair, Cellular Assembly and Organization |
| 7 | 35 | 31 | Lipid Metabolism, Small Molecule Biochemistry, Hematological Disease |
| 8 | 33 | 30 | DNA Replication, Recombination, and Repair, Cell Cycle, Cancer |
| 9 | 33 | 30 | Cancer, Cell Death, Cellular Movement |
| 10 | 33 | 30 | Embryonic Development, Nervous System Development and Function, Organ Development |
Heat maps
Fig. 2A shows heat maps of the selected top functions cell cycle, cell proliferation, DNA replication and DNA repair identified by IPA. The fold changes in individual genes within a functional category for all five experimental conditions were color coded to represent the expression patterns in the heat maps. Almost all genes included in the functional networks DNA replication and DNA repair were down regulated by high concentrations of NS-398 and ibuprofen. Genes included in cell cycle, cell proliferation and DNA repair networks were more affected by high concentration of ibuprofen. These heat maps clearly show that low concentrations of NSAIDs and COX-2 RNAi had very little effect on these genes.
Fig. 2.
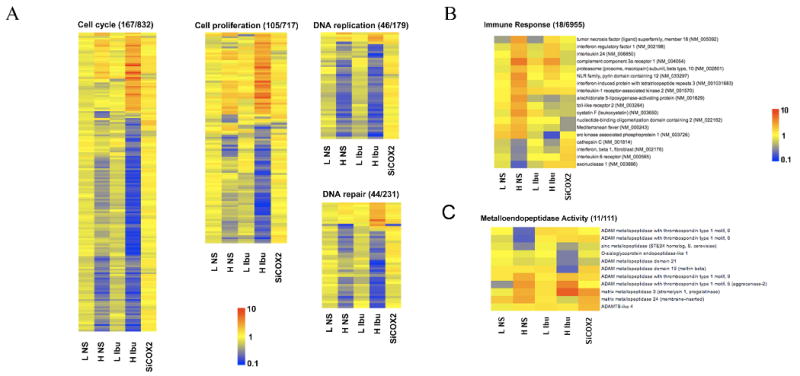
Differentially expressed genes exhibiting greater than 2-fold changes and p<0.05 were classified in to functional networks using IPA software (Table 1 and 2). (A) Heat maps of Cell cycle, Cell proliferation, DNA replication and DNA repair categories which were among the top 3 networks for the high concentration of NS-398 and ibuprofen. Heat maps represent the fold change in each individual gene in these categories for each experimental condition- 0.01mol/L NS-398 (L NS), 0.1mM NS-398 (H NS), 0.1mM ibuprofen (L IBU), 1.5mM ibuprofen (H IBU), and siCOX-2. Yellow in to dark orange indicates up-regulated genes; Blue indicates down-regulated genes. (B) Immune response function was identified as one of the top function for high NS-398 by IPA analysis. Heat map shows the expression patterns of 18 immune response genes differentially expressed (>2-fold, p<0.05) by high concentration NS-398 and by other experimental conditions. (C) Heat map of 11 metallopeptidases genes (>2-fold, p<0.05) altered by NSAIDs and COX-2 RNAi in PC3 cells.
One of the top 3 networks for high concentration of NS-398 included immune response as a functional category. Fig. 2B shows the fold changes in 18 immune response genes that were differentially expressed by NS-398 and by other treatments. MMP3 was the only gene commonly up regulated by high concentration of NS-398, ibuprofen and COX-2 RNAi. Fig. 2C shows the changes in other metallopeptidases in response to NSAIDs and COX-2 RNAi.
Confirmation of microarray data
NSAID-induced changes in gene expression of selected genes detected by microarray analysis were confirmed by western blot analysis, ELISA or real-time RT-PCR (Fig. 3A, B, C and D). Microarray analysis revealed ∼9-fold increase in COX-2 by 0.1mM NS-398 and 1.5mM Ibuprofen, and >4 fold reduction in COX-2 by COX-2 RNAi, which was confirmed by western blot analysis (Fig. 3A). COX-2 gene expression and protein were not altered by lower concentrations of the NSAIDs. NAG-1, (NAG-1 is NSAID-activated gene, also called GDF-15 and MIC-1) was significantly up regulated with higher concentrations of both NSAIDs. Increase in NAG-1 was confirmed by western blot analysis (Fig. 3A). ATF3 (activating transcription factor 3) gene upregulation was seen only in cells treated with ibuprofen; however, the protein was found to be up regulated by both NSAIDs. Microarray analysis showed that cell cycle-related genes Cyclin E2, E2F2 transcription factor and cyclin-dependent kinase 2 were down regulated and Cyclin A1 was up regulated by higher concentrations of NSAIDs and this was confirmed at protein level by western blot analysis (Fig. 3B). None of these genes, except COX-2, was altered by COX-2 RNAi. Angiopoietin-like 4 (ANGPTL 4) gene was significantly up regulated in microarray analysis by both low (49-fold) and high concentrations (112-fold) of ibuprofen. Real-Time RT-PCR analysis confirmed the increase at message level. Although not statistically significant, ANGPTL 4 gene expression was also up regulated by 0.1 mM NS-398 (9-fold). The increase in ANGPTL4 by NS-398 at the message level was confirmed by Real-time RT-PCR (Fig. 3C). MMP3 gene expression was up regulated by 0.1mM NS-398, 1.5mM ibuprofen, and also by COX-2 RNAi. Analysis of culture supernatants by ELISA confirmed the increase in MMP3 at protein level (Fig. 3D).
Fig. 3.


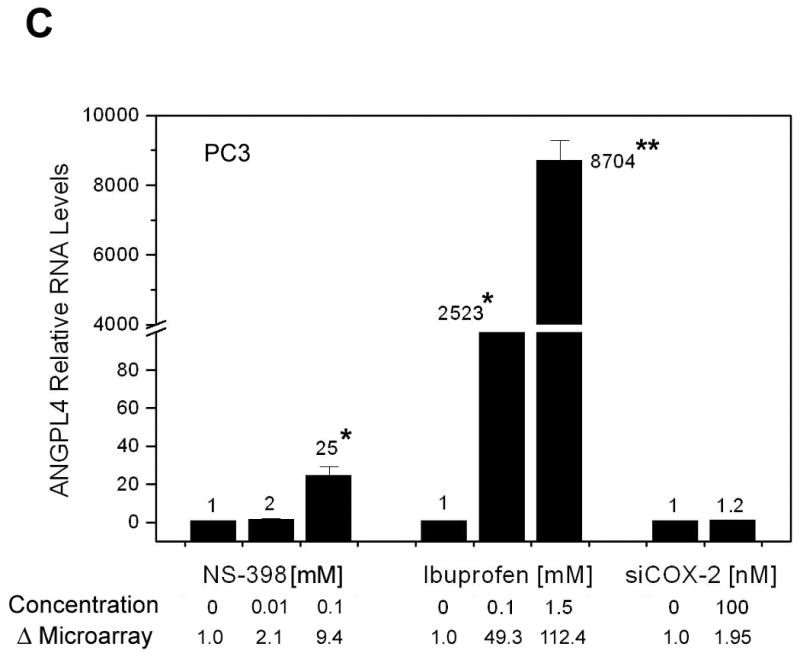
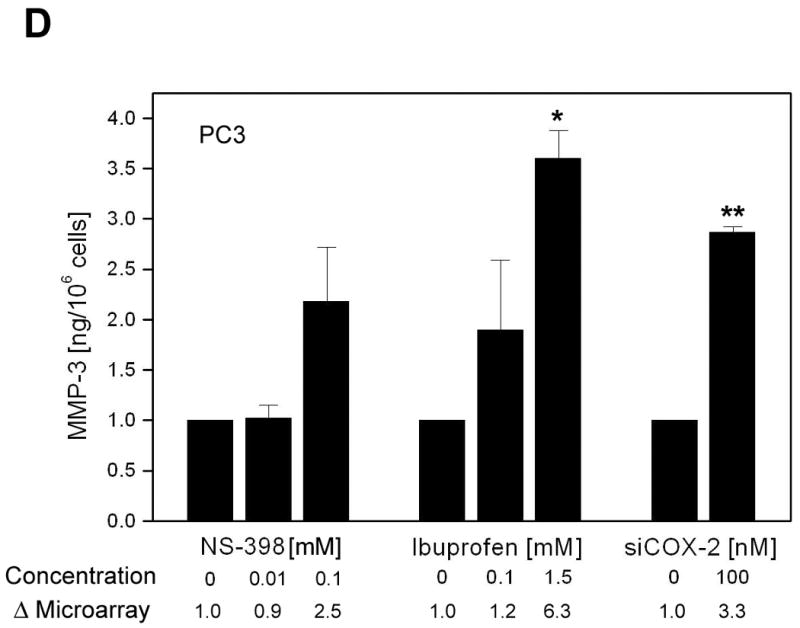
Validation of selected genes. (A) Western blot analysis confirming changes in COX-2, NAG-1 and ATF3 protein levels in PC3 cells after 24h treatment with NS-398 (0.01mM and 0.1mM), ibuprofen (0.1mM and 1.5mM) and COX-2 siRNA. Δ= fold change normalized to control or vehicle. Fold change by microarray and fold change by densitometry are indicated. (B) Western blot analysis demonstrating alterations in cell cycle regulatory proteins Cyclin E2, E2F2, Cdk-2 and Cyclin A1 in PC3 cells treated with NS-398 (0.01mM and 0.1mM), ibuprofen (0.1mM and 1.5mM) and COX-2 siRNA. Δ= fold change normalized to control or vehicle. Fold change by microarray and fold change by densitometry are indicated. (C) Real-Time RT- PCR analysis of angiopoietin-like4 (ANGPTL4) mRNA in PC3 cells treated with NS-398 (0.01mM and 0.1mM), ibuprofen (0.1mM and 1.5mM) and COX-2 siRNA. Each data point represents Average ± SEM of 3 separate experiments. * p<0.05, ** p<0.01. (D) MMP3 levels in culture supernatants from PC3 cells after 24h treatment with NS-398 (0.01mM and 0.1mM), ibuprofen (0.1mM and 1.5mM) and COX-2 siRNA were determined by ELISA and normalized to control or vehicle. Fold change is expressed as nanogram MMP3/106 cells. Each data point represents Average ± SEM of 3 separate experiments. * p<0.01, ** p<0.001
Effect of NSAIDs on cell cycle progression
Microarray data and western blot analysis showed that several genes regulating cell cycle progression were altered by high concentrations of both NSAIDs at 24h. Lower concentrations of NSAIDs or COX-2 RNAi had no effect on these genes or proteins (Fig. 3B). Therefore, we evaluated the effect of all 5 treatments on the DNA distribution by flow cytometry (Table 3). Treatment of cells with high concentration of NS-398 and ibuprofen resulted in significant G1 arrest and reduction in the percentage of cells in S phase (p<0.05). At lower concentrations, NSAIDs showed no effect on the cell cycle distribution. Silencing COX-2 by siRNA resulted in some increase in G2M but had no effect on G1 and S phase as compared to the vehicle-treated cells.
Table 3. Cell cycle distribution of PC3 cells following treatment.
Cell cycle distribution of PC3 cells following treatment with NS-398, ibuprofen and COX-2 siRNA. PC3 cells were treated for 24h and percentages of cells in different cell cycle compartments were determined by flow cytometry. The data represents Average ± SEM of 3 separate experiments.
| Treatment | G1 | S | G2M |
|---|---|---|---|
| DMSO control | 39.9 ± 0.6 | 43.5 ± 0.5 | 16.6 ± 0.3 |
| 0.01mM NS-398 | 38.6 ± 0.4 | 43.4 ± 1.3 | 18.0 ± 0.9 |
| 0.1mM NS-398 | 61.2 ± 3.1** | 21.9 ± 1.8** | 17.0 ± 4.1 |
| Control | 36.8 ± 3.6 | 45.6 ± 3.9 | 17.6 ± 2.4 |
| 0.1mM ibuprofen | 40.2 ± 1.1 | 43.6 ± 1.8 | 16.2 ± 1.7 |
| 1.5mM ibuprofen | 78.9 ± 2.1** | 14.0 ± 2.9*** | 7.1 ± 0.9* |
| Vehicle control | 35.8 ± 3.5 | 44.7 ± 2.1 | 19.5 ± 1.6 |
| siCOX-2 | 34.0 ± 3.6 | 42.1 ± 0.8 | 23.9 ± 2.9* |
p <0.05,
p <0.01 and
p <0.001
Effect of NS-398 and ibuprofen on DU-145 cells
We evaluated the effects of NS-398 and ibuprofen on cell survival, cell cycle and selected molecular targets that were validated in PC3 cells, in a second human prostate carcinoma cell line, the DU-145 cells.
Clonogenic cell Survival
The plating efficiencies of DMSO control and H2O control were 0.44 ± 0.01, 0.50 ± 0.01 respectively. The surviving fractions of DU-145 cells treated with 0.01 and 0.1mM NS-398 for 24h were 1.10 ± 0.07 and 1.01 ± 0.04 respectively compared to the DMSO control (n=3). The surviving fractions of 0.1 and 1.5mM ibuprofen treated cells were 1.00 ± 0.02 and 0.67 ± 0.02 respectively compared to the H2O control (n=3). Thus, as in the case of PC3 cells, 24h exposure to NS-398 was not cytotoxic to DU-145 cells also. However, Ibuprofen was less cytotoxic to DU-145 cells as compared to the PC3 cells.
Cell Cycle
Effect of NSAIDs on cell cycle in DU-145 cells is shown in Table 4. Although higher concentration of NS-398 resulted in some increase in cells in G1, it was not statistically significant. Significant increase in cells in G1 accompanied by reduction in cells in S and G2M compartments was seen after treatment with high concentration of ibuprofen (Table 4).
Table 4. Cell cycle distribution of DU-145 cells following treatment.
Cell cycle distribution of DU-145 cells following treatment with NS-398 and ibuprofen. DU-145 cells were treated for 24h and percentages of cells in different cell cycle compartments were determined by flow cytometry. The data represents Average ± SEM of 3 separate experiments.
| Treatment | G1 | S | G2M |
|---|---|---|---|
| DMSO control | 53.3 + 0.9 | 24.2 + 0.1 | 22.2 + 0.9 |
| 0.01mM NS-398 | 51.9 + 0.6 | 25.5 + 0.4 | 22.4 + 0.5 |
| 0.1mM NS-398 | 58.7 + 1.7 | 22.2 + 1.4 | 18.6 + 1.0* |
| Control | 51.9 + 1.7 | 24.3 + 1.5 | 23.5 + 1.1 |
| 0.1mM ibuprofen | 50.4 + 1.3 | 25.0 + 1.4* | 24.5 + 0.4 |
| 1.5mM ibuprofen | 60.4 + 0.9* | 20.5 + 0.8* | 19.1 + 1.3* |
p <0.05
Effect of NSAIDs on COX-2, NAG-1, and ATF3 in DU-145 cells
COX-2, NAG-1 and ATF3, some of the known targets of NSAIDs, were significantly up regulated by high concentrations of NS-398 and ibuprofen in PC3 cells. COX-2 mRNA was very low in DU-145 cells compared to the levels in PC3 (< 42 fold). DU-145 cells did not express COX-2 protein nor was it induced by high concentrations of NS-398 or ibuprofen (data not shown), although mRNA expression increased by 25 fold with 1.5 mM ibuprofen (Table 5). Similarly, NAG-1 message was very low in DU-145 cells compared to the PC3 (< 76 fold). NAG-1 protein was not detected in DU-145 cells, however Real Time RT PCR analysis revealed significant increases in NAG-1 at message level by both, NS-398 and ibuprofen (Table 5). ATF3 protein was significantly up regulated by ibuprofen but not by NS-398 (Fig. 4). Fig. 4 shows the effect of NS-398 and ibuprofen on the selected cell cycle regulatory proteins in DU-145 cells. Cyclin A1 was up regulated by ibuprofen and not by NS-398. Cyclin E2 and CDK2 were inhibited by ibuprofen but only to a lesser extent by NS-398. Real Time RT PCR data showed that NS-398 had no significant effect on E2F2 message, however, ibuprofen significantly down regulated E2F2 at message level (Table 5).
Table 5. Changes in gene expression by NSAIDs in DU-145 cells.
Real-Time RT- PCR analysis of GDF 15, angiopoietin-like4 (ANGPTL4) and E2F2 mRNA in DU-145 cells treated with NS-398 (0.01mM and 0.1mM) and ibuprofen (0.1mM and 1.5mM). Each data point represents Average ± SEM of 3 separate experiments.
| Treatment | COX-2 | NAG-1 | ANGPTL4 | E2 F2 | MMP 3 |
|---|---|---|---|---|---|
| DMSO Control | 1 | 1 | 1 | 1 | 1 |
| 0.01mM NS-398 | 1.1 ± 0.0** | 1.0 ± 0.1 | 1.2 ± 0.1 | 1.3 ± 0.1* | 1.1 ± 0.1 |
| 0.1mM NS-398 | 2.5 ± 0.2* | 6.7± 0.4** | 2.0 ± 0.3 | 0.9 ± 0.1 | 1.9 ± 0.2* |
| Control | 1 | 1 | 1 | 1 | 1 |
| 0.1mM ibuprofen | 1.3 ± 0.1* | 0.7 ± 0.2 | 6.2 ± 0.9* | 1.0 ± 0.3 | 1.9 ± 0.2 |
| 1.5mM ibuprofen | 25.0 ± 0.9** | 106.1 ± 10.1* | 39.5 ± 3.7* | 0.4 ± 0.1* | 149.4 ± 0.1* |
p<0.05 and
p<0.005
Fig. 4.

Western blot analysis demonstrating alterations in ATF3, and cell cycle regulatory proteins Cyclin E2, Cdk-2 and Cyclin A1 in DU-145 cells treated with NS-398 (0.01mM and 0.1mM) and ibuprofen (0.1mM and 1.5mM). Δ= fold change normalized to control. Data shown are representative of 3 separate experiments.
ANGPTL4
Effect of NSAIDs on Angiopoietin-like 4 (ANGPTL4) in DU-145 cells was analyzed by Real Time RT PCR (Table 5). There was no significant increase in ANGPTL4 by NS-398 in DU-145 cells. ANGPTL4 was significantly up regulated by low and high concentration of ibuprofen in DU-145 cells, although to a much smaller extent compared to the PC3 cells.
MMP3
Whereas MMP3 was up regulated in PC3 cells by high concentrations of NSAIDs and COX-2 RNAi, MMP3 was not detectable in DU-145 cell culture supernatants following NSAID treatment (data not shown). Real Time PCR data revealed significant lower baseline expression level of MMP3 mRNA in DU-145 (<400 fold) compared to PC3 cells, however 1.5 mM ibuprofen significantly upregulated MMP3 mRNA expression (Table 5).
Discussion
The primary purpose of this study was to better understand the similarities and differences among drug treatments from the same general class of drugs, the COX inhibitors using drug concentrations commonly used in preclinical studies in comparison to the drug concentration achievable in the clinic. That the published literature includes a wide range of drugs, doses and schedules and identifies a wide range of drug targets, makes it an challenge to reconcile the differences and thereby to understand how a drug may work in the clinic and also to determine potential biomarkers of drug effect. The striking observation of this study is that the 5 treatments, low and high concentration of the COX-2 specific NSAID NS-398, the non-specific NSAID ibuprofen, and COX-2 RNAi produce very different gene expression profiles. siRNA is considered to be a useful means of understanding the impact of turning off a particular gene. COX-2 siRNA produced very few genes in common with any of the drug conditions.
In preclinical studies the antitumor effects of NSAIDs are mainly seen at higher concentrations and at these concentrations NSAIDs target a wide variety of cellular processes (5). In the present study, treatment of PC3 cells with higher concentrations of NS-398 and ibuprofen resulted in an accumulation of cells in G1 with a decrease in the number of cells in S phase. These data are in agreement with earlier studies showing similar cell cycle perturbations in cells treated with NS-398 (50-200μM) (16, 32) and ibuprofen (1mM) (18). Interestingly, inhibition of COX-2 in OVCAR-3 cells by treatment with COX-2 siRNA did not affect cell cycle progression (32) and this is confirmed in PC3 cells. The cell cycle perturbations by NSAIDs correlated with alterations in cell cycle regulatory genes. The microarray data revealed that high concentrations of NS-398 and ibuprofen down regulated several cell cycle regulatory genes including Cyclin E2, cdk2 and the transcription factor E2F2. Some of the other cell cycle regulatory genes targeted by NSAIDs included MCM family members, P18, geminin, aurora kinase B, E2F8 and BRCA1. In addition p21, Mdm2, CHK2, E2F5 and GADD45 were up regulated and cdc2, cdc25, E2F1, Cyclins (A2, B1, B2, K and F), and Wee1 homolog were down regulated specifically by high concentration of ibuprofen. Significantly, low concentrations of the both NSAIDs and COX-2 RNAi had no influence on cell cycle distribution and also did not alter any of these cell cycle regulatory genes. Although some of the previous microarray studies have reported changes in a few cell cycle regulatory genes by NSAIDs, the present global microarray analysis is more comprehensive and has revealed a large number of cell cycle regulatory genes that are differential expressed by NSAIDs.
The cell cycle response of DU-145 cells to the NSAIDs was different than the response of PC3 cells. Whereas NS-398 and ibuprofen both induced significant G1 accumulation in PC3 cells, only ibuprofen treatment resulted in significant G1 accumulation in DU-145 cells. As in case of PC3 cells, the cell cycle perturbations by NSAIDs in DU-145 cells correlated with alterations in cell cycle regulatory proteins. The inhibition of Cyclin E2, CDK2 and E2F2, and up regulation of Cyclin A1 was more pronounced in cells treated with ibuprofen than those treated with NS-398. We recently observed a difference in cell cycle response of PC3 and DU-145 cells to another compound, PX-478, an inhibitor of hypoxia-inducible factor-1α (33)
In addition to cell cycle regulatory genes, our PC3 microarray data revealed significant upregulation of genes with antitumorigenic and proapoptotic activities, including NAG-1 (GDF-15, MIC-1) and ATF3, mainly at higher concentrations of the both NSAIDs. We have previously shown that ibuprofen induced apoptotic DNA fragmentation in PC3 cells but not in DU-145 cells (12). NAG-1 which belongs to TGF-β superfamily, is up regulated by several NSAIDs as well as by other antitumorogenic or dietary compounds and the induction of NAG-1 has been reported to be independent of COX-2 and p53 status (34-36). It has been shown that the increase in NAG-1 mRNA by a panel of NSAIDs correlated with the induction of apoptosis (34). NAG-1 is also implicated in cell growth arrest (35). Increase in NAG-1 by sulindac sulfide in ovarian carcinoma cells was associated with suppression of cell growth, and transfection with NAG-1 siRNA reversed the suppression of cell growth (35). The basal expression of NAG-1 appears to be cell type dependent. It has been reported that of the 3 established human prostate carcinoma cell lines PC3 and LNCaP cells secreted NAG-1 (MIC-1) protein at high levels whereas DU-145 cells produced no NAG-1 (MIC-1) protein (37). Beak et al studied NAG-1 mRNA induction by nonspecific NSAIDs and COX-2 specific NSAIDs in COX-2 deficient HCT-16 colon carcinoma cells and found that, in general, the nonspecific NSAIDs increased NAG-1 expression by 2- to 5-fold whereas the COX-2 specific inhibitors did not (32). NS-398 failed to induce NAG-1 mRNA or protein in COX-2 deficient SKOV3 ovarian and HCT-16 colon carcinoma cells (34, 35). Our data showed an increase in NAG-1 message and protein in COX-2 expressing PC3 cells treated with 0.1mmolar NS-398 whereas in COX-2 deficient DU-145 cells the induction of NAG-1 mRNA by NS-398 was much smaller compared to the induction by ibuprofen. Interestingly, DU-145 cells did not express COX-2 and NAG-1 proteins, nor they were up regulated in DU-145 cells by high concentrations of NS-398 or ibuprofen. Thus the NSAID-induced up regulation of COX-2 and NAG-1 appears to be cell type dependent and appears to differ for COX-2 specific NSAIDs and nonspecific NSAIDs.
Antitumor and proapoptotic gene ATF3 is also reportedly activated by a wide variety of NSAIDs, including the traditional NSAIDs sulindac sulfide and indomethacin (22). ATF3 is known to regulate several downstream genes related to cell growth (38), and invasion (39). In PC3 cells, although the increase in ATF3 gene expression was specific to ibuprofen, ATF3 protein was up regulated by high concentration of NS-398 as well. In DU-145 cells the up regulation in ATF3 protein was much greater with ibuprofen as compared to the increase with NS-398. It appears that ATF3 is involved in the induction of NAG-1. In HCT-116 cells polyphenolic compound epigallocatechin (ECG) induces the transcription factor ATF3 which binds to NAG-1 promoter and transactivates NAG-1 expression(40). In the present study the NSAIDs-induced NAG-1 protein pattern, in general, paralleled the ATF3 protein pattern. Increase in NAG-1 and ATF3 may contribute to the antitumorigenic effects of ibuprofen and NS-398, as reported for other NSAIDs (22, 34-36).
Up regulation in COX-2 protein by high concentrations of NSAIDs has been reported in several earlier studies (13, 30, 37, 38). The NSAIDs-induced COX-2 mRNA in chick embryo fibroblast cells was reportedly truncated and non-functional (41). Simmons et al. showed that non-specific NSAID Diclofenac-induced COX-2 has distinct COX active site and also is more sensitive to acetaminophen than COX-2 induced by Lipopolysaccharide (LPS) (42). In the present study NS-398 and ibuprofen both induced ∼9-fold increase in COX-2 gene and 10-fold increase in the protein in PC3 cells, as seen in our previous studies (13, 30). Low concentrations of NSAIDs did not induce COX-2, and COX-2 RNAi as expected, reduced COX-2 expression. The functional role of the NSAIDs-induced COX-2 is unclear. Overall, the change in COX-2, ATF3 and NAG-1 in PC3 cells as well as in DU-145 cells appears to occur in tandem suggesting the possibility that these targets may be interrelated. We are currently pursuing this observation in further details.
In addition to confirming the changes in NAG-1, ATF3 and COX-2, the known targets of NSAIDs, the present microarray data identified several new targets of NSAIDs including angiopoietin like-4 (ANGPTL4). There was a concentration dependent significant upregulation of ANGPTL4 gene in PC3 cells treated with NS-398 and ibuprofen and ibuprofen was much more effective than NS-398 in inducing ANGPTL4. ANGPTL4 protein is a circulating plasma protein, expressed in the liver, adipose tissue, and placenta (43, 44). It is implicated in regulation of angiogenesis and metastasis (45). ANGPTL4 expression is regulated by hypoxia both in endothelial cells and in tumor cells (46, 47). It has been reported that ANGPTL4 reduces endothelial cell adhesion, and decreases cell migration and sprouting (48). Recent studies indicate that ANGPL4, through its action on both vascular and tumor compartments, prevents the metastatic process by inhibiting vascular activity as well as tumor cell motility and invasiveness (49). ANGPTL4 is also an important regulator of glucose homeostasis, insulin sensitivity and lipid metabolism (50). Interestingly, one of the top 10 networks identified by IPA for high concentration of ibuprofen included lipid metabolism as a functional category. PC3 Microarray data also showed changes in many genes associated with lipid metabolism. In DU-145 cells also ibuprofen significantly increased ANGPTL4 mRNA expression, although to a much smaller extent, compared to the increase by ibuprofen in PC3 cells. Increase in ANGPL4 by NSAIDs is a novel observation of this study and further work is needed to determine the significance of this finding.
Several members of extracellular matrix class were altered by NSAIDs. Of particular interest is MMP3, which was the only common gene up regulated with high concentrations of NS-398, ibuprofen and COX-2 RNAi. Many cytokines and growth factors increase MMP3 expression and several signaling molecules including COX-2 derived PGE2 are implicated in the regulation of MMP3. The reports on the effect of NSAIDs on MMP3 expression appear to be contradictory. While some studies have shown an inhibition in cytokine-induced MMP3 by NS-398 (51), others have reported an increase in IL-1α induced MMP3 by indomethacin and NS-398, even though both drugs inhibited IL-1α induced PGE2 (52). In patients with osteoarthritis given 1200mg/day ibuprofen for 28 days MMP3 serum concentration was found to be increased (53). Similarly, in a clinical model of acute inflammation, treatment with rofecoxib (50 mg daily) or ibuprofen (400 mg 4 times per day) increased MMP3 in the oral mucosa biopsy specimens of volunteers (54). Although the precise mechanism of the overexpression of MMP3 by NSAIDs and COX-2 RNAi in the present study is not clear, inhibition of PGE2 could be one of the factors for this increase as reported by others (52, 54).
The present microarray data elucidates some of the differences in the cellular effects of NS-398 and ibuprofen that are seen in high concentration experiments. Ibuprofen was more cytotoxic compared to NS-398 and 24h drug treatment reduced the plating efficiency of ibuprofen-treated cells to ∼50% whereas NS-398 was not toxic. At high concentrations both drugs induced cell cycle perturbations although ibuprofen was more effective. The microarray data showed that at 24h, treatment with high concentration of NS-398 and ibuprofen resulted in alterations in 17% and 80% of the 3,362 genes. Ibuprofen was much more effective in down regulating genes related to cell cycle, proliferation and DNA repair. In addition the microarray data and the IPA analysis showed that ibuprofen affected genes regulating signal transduction, gene expression, cellular function and maintenance, drug metabolism, molecular transport, lipid and carbohydrate metabolism, more effectively compared to NS-398. This may account for the higher ibuprofen toxicity.
The use of expression profiling has been proposed to (a) profile the molecular pathway of a tumor to guide drug choice (55), (b) to guide the use of chemotherapeutic drugs and combinations by predicting sensitivity and resistance (56), and (c) to predict how one drug will behave compared to others (57). This present study adds an additional dimension in that it is important to profile and thereby “fingerprint” drugs and drug regimens even within drugs of the same class and, if gene silencing is used to predict how a drug is working, this must also be fingerprinted to compare it to pharmacologic inhibition. The choice of cell lines to use for fingerprinting could be tumor type specific or a few “standard” cell lines could be employed with the purpose of providing an comparison of drug effect with the wide range of concentrations used in mechanistic studies, some of which may have minimal relevance to the clinical application.
While more work is required, drug fingerprinting could facilitate matching the drug, drug dose and schedule with the tumor profile, optimizing the drug prior to clinical application and facilitating understanding the drug effect in the patient. Further work in our laboratory is “fingerprinting” radiation therapy doses and schedules. As analytical tools are more fully developed and validated, we hypothesize that it might be possible to compare expression profile of the tumor, the drug induced changes and the radiation induced changes to pre-select effective combined modality treatment combinations, including potentially predicting normal tissue injury. While the in vitro and in vivo expression profiles differ (58, 59), it may be that the in vitro data using well defined cells and tissue arrays may allow such predictions to be made on a limited set of genes, as is now being used to predict clinical outcome in the clinic.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge and thank Dr. David Goldstein and the CCR for support, and Dr. Joanna Shih, Biomedical Research Branch, NCI, for consultation and help with statistical analysis. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
ABBREVIETIONS
- IPA
Ingenuity Pathway Analysis
- PGE2
Prostaglandin Endoperoxidase2
- MMP3
Matrix Metallo Protease-3
- ANGPTL4
Angiopoeitin-like 4
- NAG-1
NSAID Activated Gene-1
- ATF3
Activating Transcription Factor-3
Bibliography
- 1.Teicher BA, Bump EA, Palayoor ST, Northey D, Coleman CN. Signal transduction inhibitors as modifiers of radiation therapy in human prostate carcinoma xenografts. Radiation Oncology Investigations. 1996;4:221–40. [Google Scholar]
- 2.Choy H, Milas L. Enhancing radiotherapy with cyclooxygenase-2 enzyme inhibitors: a rational advance? J Natl Cancer Inst. 2003;95:1440–52. doi: 10.1093/jnci/djg058. [DOI] [PubMed] [Google Scholar]
- 3.Rao CV, Reddy BS. NSAIDs and chemoprevention. Curr Cancer Drug Targets. 2004;4:29–42. doi: 10.2174/1568009043481632. [DOI] [PubMed] [Google Scholar]
- 4.DuBois RN. NSAIDs and prostate cancer risk. Cancer J. 2006;12:108–9. [PubMed] [Google Scholar]
- 5.de Groot DJ, de Vries EG, Groen HJ, de Jong S. Non-steroidal anti-inflammatory drugs to potentiate chemotherapy effects: from lab to clinic. Crit Rev Oncol Hematol. 2007;61:52–69. doi: 10.1016/j.critrevonc.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 7.Meric JB, Rottey S, Olaussen K, et al. Cyclooxygenase-2 as a target for anticancer drug development. Crit Rev Oncol Hematol. 2006;59:51–64. doi: 10.1016/j.critrevonc.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Rostom A, Dube C, Lewin G, et al. Nonsteroidal anti-inflammatory drugs and cyclooxygenase-2 inhibitors for primary prevention of colorectal cancer: a systematic review prepared for the U.S. Preventive Services Task Force. Ann Intern Med. 2007;146:376–89. doi: 10.7326/0003-4819-146-5-200703060-00010. [DOI] [PubMed] [Google Scholar]
- 9.Fung HB, Kirschenbaum HL. Selective cyclooxygenase-2 inhibitors for the treatment of arthritis. Clin Ther. 1999;21:1131–57. doi: 10.1016/S0149-2918(00)80018-1. [DOI] [PubMed] [Google Scholar]
- 10.Radi ZA, Khan NK. Effects of cyclooxygenase inhibition on the gastrointestinal tract. Exp Toxicol Pathol. 2006 doi: 10.1016/j.etp.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Levesque LE, Brophy JM, Zhang B. The risk for myocardial infarction with cyclooxygenase-2 inhibitors: a population study of elderly adults. Ann Intern Med. 2005;142:481–9. doi: 10.7326/0003-4819-142-7-200504050-00113. [DOI] [PubMed] [Google Scholar]
- 12.Palayoor ST, Bump EA, Calderwood SK, Bartol S, Coleman CN. Combined antitumor effect of radiation and ibuprofen in human prostate carcinoma cells. Clin Cancer Res. 1998;4:763–71. [PubMed] [Google Scholar]
- 13.Palayoor ST, Tofilon PJ, Coleman CN. Ibuprofen-mediated reduction of hypoxia-inducible factors HIF-1alpha and HIF-2alpha in prostate cancer cells. Clin Cancer Res. 2003;9:3150–7. [PubMed] [Google Scholar]
- 14.Coleman CN, Kelly L, Riese Daly N, et al. Phase III study of ibuprofen versus placebo for radiation-induced genitourinary side effects. Int J Radiat Oncol Biol Phys. 2002;54:191–4. doi: 10.1016/s0360-3016(02)02907-3. [DOI] [PubMed] [Google Scholar]
- 15.Raju U, Nakata E, Yang P, Newman RA, Ang KK, Milas L. In vitro enhancement of tumor cell radiosensitivity by a selective inhibitor of cyclooxygenase-2 enzyme: mechanistic considerations. Int J Radiat Oncol Biol Phys. 2002;54:886–94. doi: 10.1016/s0360-3016(02)03023-7. [DOI] [PubMed] [Google Scholar]
- 16.Minter HA, Eveson JW, Huntley S, Elder DJ, Hague A. The cyclooxygenase 2-selective inhibitor NS398 inhibits proliferation of oral carcinoma cell lines by mechanisms dependent and independent of reduced prostaglandin E2 synthesis. Clin Cancer Res. 2003;9:1885–97. [PubMed] [Google Scholar]
- 17.Palayoor ST, Youmell MY, Calderwood SK, Coleman CN, Price BD. Constitutive activation of IkappaB kinase alpha and NF-kappaB in prostate cancer cells is inhibited by ibuprofen. Oncogene. 1999;18:7389–94. doi: 10.1038/sj.onc.1203160. [DOI] [PubMed] [Google Scholar]
- 18.Andrews J, Djakiew D, Krygier S, Andrews P. Superior effectiveness of ibuprofen compared with other NSAIDs for reducing the survival of human prostate cancer cells. Cancer Chemother Pharmacol. 2002;50:277–84. doi: 10.1007/s00280-002-0485-8. [DOI] [PubMed] [Google Scholar]
- 19.Tegeder I, Pfeilschifter J, Geisslinger G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. Faseb J. 2001;15:2057–72. doi: 10.1096/fj.01-0390rev. [DOI] [PubMed] [Google Scholar]
- 20.Masferrer JL, Leahy KM, Koki AT, et al. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res. 2000;60:1306–11. [PubMed] [Google Scholar]
- 21.Gao XQ, Han JX, Huang HY, Song B, Zhu B, Song CZ. Effect of NS398 on metastasis-associated gene expression in a human colon cancer cell line. World J Gastroenterol. 2005;11:4337–43. doi: 10.3748/wjg.v11.i28.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bottone FG, Jr, Martinez JM, Collins JB, Afshari CA, Eling TE. Gene modulation by the cyclooxygenase inhibitor, sulindac sulfide, in human colorectal carcinoma cells: possible link to apoptosis. J Biol Chem. 2003;278:25790–801. doi: 10.1074/jbc.M301002200. [DOI] [PubMed] [Google Scholar]
- 23.Yin H, Xu H, Zhao Y, Yang W, Cheng J, Zhou Y. Cyclooxygenase-independent effects of aspirin on HT-29 human colon cancer cells, revealed by oligonucleotide microarrays. Biotechnol Lett. 2006;28:1263–70. doi: 10.1007/s10529-006-9084-9. [DOI] [PubMed] [Google Scholar]
- 24.Tseng WW, Deganutti A, Chen MN, Saxton RE, Liu CD. Selective cyclooxygenase-2 inhibitor rofecoxib (Vioxx) induces expression of cell cycle arrest genes and slows tumor growth in human pancreatic cancer. J Gastrointest Surg. 2002;6:838–43. doi: 10.1016/s1091-255x(02)00061-6. discussion 44. [DOI] [PubMed] [Google Scholar]
- 25.Hardwick JC, van Santen M, van den Brink GR, van Deventer SJ, Peppelenbosch MP. DNA array analysis of the effects of aspirin on colon cancer cells: involvement of Rac1. Carcinogenesis. 2004;25:1293–8. doi: 10.1093/carcin/bgh118. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, DuBois RN. Detection of differentially expressed genes in human colon carcinoma cells treated with a selective COX-2 inhibitor. Oncogene. 2001;20:4450–6. doi: 10.1038/sj.onc.1204588. [DOI] [PubMed] [Google Scholar]
- 27.Lou J, Fatima N, Xiao Z, et al. Proteomic profiling identifies cyclooxygenase-2-independent global proteomic changes by celecoxib in colorectal cancer cells. Cancer Epidemiol Biomarkers Prev. 2006;15:1598–606. doi: 10.1158/1055-9965.EPI-06-0216. [DOI] [PubMed] [Google Scholar]
- 28.Konstan MW, Byard PJ, Hoppel CL, Davis PB. Effect of high-dose ibuprofen in patients with cystic fibrosis. N Engl J Med. 1995;332:848–54. doi: 10.1056/NEJM199503303321303. [DOI] [PubMed] [Google Scholar]
- 29.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999;96:272–7. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palayoor ST, Arayankalayil MJ, Shoaibi A, Coleman CN. Radiation sensitivity of human carcinoma cells transfected with small interfering RNA targeted against cyclooxygenase-2. Clin Cancer Res. 2005;11:6980–6. doi: 10.1158/1078-0432.CCR-05-0326. [DOI] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Denkert C, Furstenberg A, Daniel PT, et al. Induction of G0/G1 cell cycle arrest in ovarian carcinoma cells by the anti-inflammatory drug NS-398, but not by COX-2-specific RNA interference. Oncogene. 2003;22:8653–61. doi: 10.1038/sj.onc.1206920. [DOI] [PubMed] [Google Scholar]
- 33.Palayoor ST, Mitchell JB, Cerna D, Degraff W, John-Aryankalayil M, Coleman CN. PX-478, an inhibitor of hypoxia-inducible factor-1alpha, enhances radiosensitivity of prostate carcinoma cells. Int J Cancer. 2008;123:2430–7. doi: 10.1002/ijc.23807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baek SJ, Kim KS, Nixon JB, Wilson LC, Eling TE. Cyclooxygenase inhibitors regulate the expression of a TGF-beta superfamily member that has proapoptotic and antitumorigenic activities. Mol Pharmacol. 2001;59:901–8. [PubMed] [Google Scholar]
- 35.Kim JS, Baek SJ, Sali T, Eling TE. The conventional nonsteroidal anti-inflammatory drug sulindac sulfide arrests ovarian cancer cell growth via the expression of NAG-1/MIC-1/GDF-15. Mol Cancer Ther. 2005;4:487–93. doi: 10.1158/1535-7163.MCT-04-0201. [DOI] [PubMed] [Google Scholar]
- 36.Pang RP, Zhou JG, Zeng ZR, et al. Celecoxib induces apoptosis in COX-2 deficient human gastric cancer cells through Akt/GSK3beta/NAG-1 pathway. Cancer Lett. 2007;251:268–77. doi: 10.1016/j.canlet.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 37.Liu T, Bauskin AR, Zaunders J, et al. Macrophage inhibitory cytokine 1 reduces cell adhesion and induces apoptosis in prostate cancer cells. Cancer Res. 2003;63:5034–40. [PubMed] [Google Scholar]
- 38.Fan F, Jin S, Amundson SA, et al. ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene. 2002;21:7488–96. doi: 10.1038/sj.onc.1205896. [DOI] [PubMed] [Google Scholar]
- 39.Yan C, Wang H, Boyd DD. ATF3 represses 72-kDa type IV collagenase (MMP-2) expression by antagonizing p53-dependent trans-activation of the collagenase promoter. J Biol Chem. 2002;277:10804–12. doi: 10.1074/jbc.M112069200. [DOI] [PubMed] [Google Scholar]
- 40.Baek SJ, Kim JS, Jackson FR, Eling TE, McEntee MF, Lee SH. Epicatechin gallate-induced expression of NAG-1 is associated with growth inhibition and apoptosis in colon cancer cells. Carcinogenesis. 2004;25:2425–32. doi: 10.1093/carcin/bgh255. [DOI] [PubMed] [Google Scholar]
- 41.Lu X, Xie W, Reed D, Bradshaw WS, Simmons DL. Nonsteroidal antiinflammatory drugs cause apoptosis and induce cyclooxygenases in chicken embryo fibroblasts. Proc Natl Acad Sci U S A. 1995;92:7961–5. doi: 10.1073/pnas.92.17.7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simmons DL, Botting RM, Robertson PM, Madsen ML, Vane JR. Induction of an acetaminophen-sensitive cyclooxygenase with reduced sensitivity to nonsteroid antiinflammatory drugs. Proc Natl Acad Sci U S A. 1999;96:3275–80. doi: 10.1073/pnas.96.6.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Lam KS, Lam JB, et al. Overexpression of Angiopoietin-like Protein 4 Alters Mitochondria Activities and Modulates Methionine Metabolic Cycle in the Liver Tissues of db/db Diabetic Mice. Mol Endocrinol. 2007 doi: 10.1210/me.2006-0249. [DOI] [PubMed] [Google Scholar]
- 44.Yoon JC, Chickering TW, Rosen ED, et al. Peroxisome proliferator-activated receptor gamma target gene encoding a novel angiopoietin-related protein associated with adipose differentiation. Mol Cell Biol. 2000;20:5343–9. doi: 10.1128/mcb.20.14.5343-5349.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang YH, Wang Y, Lam KS, et al. Suppression of the Raf/MEK/ERK signaling cascade and inhibition of angiogenesis by the carboxyl terminus of angiopoietin-like protein 4. Arterioscler Thromb Vasc Biol. 2008;28:835–40. doi: 10.1161/ATVBAHA.107.157776. [DOI] [PubMed] [Google Scholar]
- 46.Le Jan S, Amy C, Cazes A, et al. Angiopoietin-like 4 is a proangiogenic factor produced during ischemia and in conventional renal cell carcinoma. Am J Pathol. 2003;162:1521–8. doi: 10.1016/S0002-9440(10)64285-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lal A, Peters H, St Croix B, et al. Transcriptional response to hypoxia in human tumors. J Natl Cancer Inst. 2001;93:1337–43. doi: 10.1093/jnci/93.17.1337. [DOI] [PubMed] [Google Scholar]
- 48.Cazes A, Galaup A, Chomel C, et al. Extracellular matrix-bound angiopoietin-like 4 inhibits endothelial cell adhesion, migration, and sprouting and alters actin cytoskeleton. Circ Res. 2006;99:1207–15. doi: 10.1161/01.RES.0000250758.63358.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galaup A, Cazes A, Le Jan S, et al. Angiopoietin-like 4 prevents metastasis through inhibition of vascular permeability and tumor cell motility and invasiveness. Proc Natl Acad Sci U S A. 2006;103:18721–6. doi: 10.1073/pnas.0609025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu A, Lam MC, Chan KW, et al. Angiopoietin-like protein 4 decreases blood glucose and improves glucose tolerance but induces hyperlipidemia and hepatic steatosis in mice. Proc Natl Acad Sci U S A. 2005;102:6086–91. doi: 10.1073/pnas.0408452102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Domeij H, Yucel-Lindberg T, Modeer T. Signal pathways involved in the production of MMP-1 and MMP-3 in human gingival fibroblasts. Eur J Oral Sci. 2002;110:302–6. doi: 10.1034/j.1600-0722.2002.21247.x. [DOI] [PubMed] [Google Scholar]
- 52.Yan M, Noguchi K, Ruwanpura SM, Ishikawa I. Cyclooxygenase-2-dependent prostaglandin (PG) E2 downregulates matrix metalloproteinase-3 production via EP2/EP4 subtypes of PGE2 receptors in human periodontal ligament cells stimulated with interleukin-1alpha. J Periodontol. 2005;76:929–35. doi: 10.1902/jop.2005.76.6.929. [DOI] [PubMed] [Google Scholar]
- 53.Bevilacqua M, Devogelaer JP, Righini V, Famaey JP, Manicourt DH. Effect of nimesulide on the serum levels of hyaluronan and stromelysin-1 in patients with osteoarthritis: a pilot study. Int J Clin Pract Suppl. 2004:13–9. [PubMed] [Google Scholar]
- 54.Wang XM, Wu TX, Lee YS, Dionne RA. Rofecoxib regulates the expression of genes related to the matrix metalloproteinase pathway in humans: implication for the adverse effects of cyclooxygenase-2 inhibitors. Clin Pharmacol Ther. 2006;79:303–15. doi: 10.1016/j.clpt.2005.12.306. [DOI] [PubMed] [Google Scholar]
- 55.Bild AH, Yao G, Chang JT, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 56.Potti A, Dressman HK, Bild A, et al. Genomic signatures to guide the use of chemotherapeutics. Nat Med. 2006;12:1294–300. doi: 10.1038/nm1491. [DOI] [PubMed] [Google Scholar]
- 57.Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–35. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 58.Tsai MH, Cook JA, Chandramouli GV, et al. Gene expression profiling of breast, prostate, and glioma cells following single versus fractionated doses of radiation. Cancer Res. 2007;67:3845–52. doi: 10.1158/0008-5472.CAN-06-4250. [DOI] [PubMed] [Google Scholar]
- 59.Camphausen K, Purow B, Sproull M, et al. Orthotopic growth of human glioma cells quantitatively and qualitatively influences radiation-induced changes in gene expression. Cancer Res. 2005;65:10389–93. doi: 10.1158/0008-5472.CAN-05-1904. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.