

Deoxysugars are prevalent structural components of many antibiotics and often contribute substantially to their biological properties.[1-3] Methylation of hydroxyl group(s) on the deoxysugar ring is relatively common (Scheme 1) since O-methylation not only protects the reactive hydroxyl group from undesired modifications such as oxidation or dehydration, but also alters the solubility and pharmacokinetic properties of the resulting molecule.[4] Biosynthetically, these O-methylation reactions are mainly catalysed by a variety of S-adenosyl-L-methionine (SAM or AdoMet) dependent methyltransferases in a site-specific manner. For example, the two SAM-dependent O-methyltransferases TylE and TylF in the tylosin biosynthetic pathway of Streptomyces fradiae sequentially methylate individual hydroxyl groups (C2‴OH and C3‴-OH) in the 6-deoxyallose moiety of demethylmacrocin to generate the macrolide antibiotic tylosin.[5-8] ElmMI, ElmMII, and ElmMIII are responsible for the consecutive methylation of three hydroxyl groups of L-rhamnose in the antitumor polyketide antibiotic elloramycin.[9] Moreover, a growing number of O-methyltransferases involved in various deoxysugar methylation reactions such as EryG,[10] OleY,[11] SpinH, SpinI, SpinK[12] have been reported in diverse antibiotic biosynthetic systems.
Scheme 1.
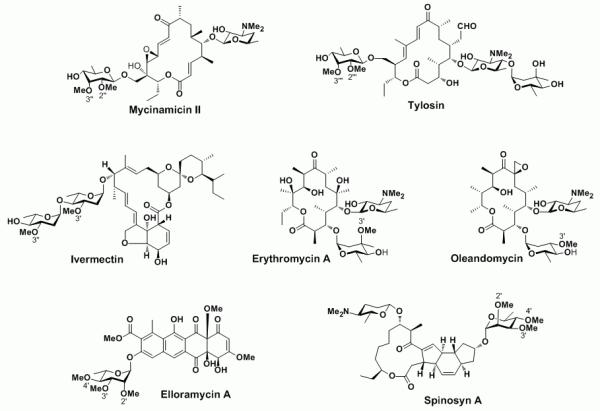
Antibiotics containing various O-methylated deoxysugars. The methyl groups installed by O-methyltransferases are numbered.
Mycinamicins represent a family of macrolide antibiotics with more than twenty members produced by the rare actinomycete Micromonospora griseorubida.[13-15] The antibacterial activities of some mycinamicin products against Staphylococcus aureus are higher than those of clinical macrolide antibiotics erythromycin and leucomycin. More importantly, mycinamicins have shown strong activity against a number of antibiotic-resistant human pathogens.[13] Structurally, the major mycinamicin products of wild type strain M. griseorubida A11725 including mycinamicin I, II, IV and V are composed of a 16-membered ring macrolactone core, an N,N-dimethylated deoxysugar desosamine and a di-O-methylated deoxyhexose mycinose (Scheme 1). During the past two decades, the biosynthesis of mycinamicin has been elucidated through strain mutagenesis, bioconversion studies[16,17] and sequence analysis of the complete mycinamicin gene cluster,[18] wherein two putative O-methyltransferases genes mycE and mycF were tentatively assigned.
Initial bioinformatics analysis of the corresponding genes revealed that MycE and MycF show high amino acid sequence similarities to TylE (demethylmacrocin O-methyltransferase) and TylF (macrocin O-methyltransferase), respectively, in the tylosin biosynthetic pathway.[18] Accordingly, the function of MycE was proposed to methylate the C2′-OH group of 6-deoxyallose in mycinamicin VI, leading to mycinamicin III, whereas MycF was presumed to transfer a methyl group to the C3′′-OH group of javose (i. e., C2′′-methylated 6-deoxyallose) in mycinamicin III to generate mycinamicin IV (Scheme 2). The proposed functions of MycE and MycF were supported by in vivo precursor feeding studies.[16,19] Herein, we report the expression of mycE and mycF in Escherichia coli, and purification of MycE and MycF to establish their biochemical function for regiospecific deoxysugar O-methylation in mycinamicin macrolide antibiotics.
Scheme 2.

Physiological reactions catalysed by MycE and MycF.
Comparative analysis revealed that MycE (399 amino acids) is significantly larger than MycF (222 amino acids). Alignment of these two O-methyltransferases exhibits low sequence identity (11.3%), suggesting they might have evolved from distinct ancestors. A protein BLAST search revealed a number of O-methyltransferases when either MycE or MycF were used as the query protein. Interestingly, all candidates with high sequence similarities are O-methyltransferases involved in deoxysugar biosynthesis. In the phylogenetic tree (Figure 1A) of selected O-methyltransferases with high similarities to MycE and MycF, it is evident that they are located in distinct branches, indicating their potentially disparate evolutionary origins. In the sequence alignment of MycE with corresponding close relatives (Figure 1B and 1C), three conserved motifs (Motif I-III)[9,20] were identified that are predicted to contribute to SAM binding. In contrast, there are only two conserved SAM-binding motifs (Motif I and III) found in MycF.
Figure 1.

Amino acid sequence analysis of MycE and MycF. A. Phylogenetic tree of selected O-methyltransferases generated by MegAlign (DNASTAR) with Clustal W method. MycE and MycF are highlighted in bold and italic. The selected O-methyltransferases include MycE and MycF (mycinamicin pathway), TylE and TylF (tylosin pathway), ChmMI and ChmMII (chalcomycin pathway), SpnH, SpnI, and SpnK (spinosad pathway), BusH, BusI, and BusK (butenyl spinosyn pathway), SnogY (nogalamycin pathway), OleY (oleandomycin pathway), ElmMI, ElmMII, and ElmMIII (elloramycin pathway), NovP (novobiocin pathway), CumN (coumermycin pathway), and MtfB (mycobacterial serovar-specific glycopeptidolipid pathway); B. SAM binding motifs of MycE and its homologs; C. SAM binding motifs of MycF and its homologs.
To confirm the proposed function of MycE and MycF, we cloned mycE and mycF genes into pET28b for overexpression in E. coli BL21 (DE3). The recombinant N-terminal His6-tagged proteins were purified to homogeneity by one-step Ni-NTA agarose chromatography (Figure 2). With the purified enzymes in hand, we initially tested the activities of MycE and MycF in 50 mM sodium phosphate buffer (pH 7.4) at 30 °C using SAM as methyl donor, and mycinamicin VI and mycinamicin III as substrates (Scheme 2). MycE was unreactive toward both substrates, whereas MycF was able to moderately methylate mycinamicin III (but not VI), forming mycinamicin IV (data not shown). Since the dependence of this class of methyltransferases on a metal co-factor is not uncommon, we next investigated the effect of different divalent metal ion on the activities of MycE and MycF. Both enzymes achieved optimal activity in the presence of 10 mM MgCl2 (Figure 3A). A number of alternative divalent ions including Co2+, Fe2+, Mn2+, and Zn2+ are capable of supporting sub-optimal activities. Interestingly, the Mg2+-dependence of MycE appears to be more pronounced than MycF, as MycF remained moderately active in the absence of Mg2+. The metal dependence of MycF was further assessed by addition of 2 mM of EDTA, however, this treatment failed to abrogate activity, suggesting that a metal ion might be dispensible in the O-methyltransferase reaction catalysed by MycF. In contrast, EDTA significantly lowered the activity of MycE even in the presence of 10 mM Mg2+. The optimal pH and temperature range for MycE and MycF assays were determined through comparison of enzymatic activities under different reaction conditions. The optimal reaction condition of the two O-methyltransferases is pH 9.0 (Figure 3B), significantly higher than the corresponding homologs TylE (optimal pH: 7.5-8.5) and TylF (7.5-8.0).[7] At pH 9.0, the maximal activities of MycE and MycF were observed at 50 °C and 37 °C (Figure 3C), respectively, higher than those of TylE (42 °C) and TylF (31 °C).[7]
Figure 2.

SDS-PAGE analysis of purified MycE and MycF
Figure 3.

Optimisation of methylation reactions catalysed MycE and MycF. A. Effect of divalent metal ion; B. pH optimisation; C. Temperature optimisation.
Under optimal conditions, the in vitro activities of MycE and MycF were analysed to reveal that MycE methylated the C2′′-OH of 6-deoxyallose, converting a majority of mycinamicin VI to mycinamicin III (Figure 4B). MycE was incapable of double methylation to generate mycinamicin IV. The second C3′′-OH methylation of javose was catalysed by MycF with a higher conversion than MycE toward mycinamicin VI (Figure 4F). Co-incubation of MycE and MycF with starting substrate mycinamicin VI resulted in accumulation of both mycinamicin III and IV (Figure 4D). It is noteworthy that mycinamicin IV cannot be further methylated by these two methytransferases although there remains a hydroxyl group at C4′′ position in mycinose. Collectively, it is evident that both MycE and MycF possess high substrate specificity.
Figure 4.

LC-MS analysis (UV: 280 nm) of in vitro conversions catalysed by MycE and MycF. A. mycinamicin VI (M-VI) standard; B. M-VI + MycE; C. M-VI + MycF; D. M-VI + MycE +MycF; E. mycinamicin III (M-III) standard; F. M-III + MycF; G. M-III + MycE; H. mycinamicin IV (M-IV) standard. The identity of compound was confirmed by mass spectrometry, comparison to standard compound regarding retention time and co-injection.
Finally, we determined the steady-state kinetic parameters (Table 1) for MycE and MycF based on substrate consumption monitored by HPLC. MycE converted M-VI to M-III with a Km of 26.4 ± 7.0 μM and a kcat of 5.0 ± 0.5 min−1. In contrast, MycF methylated M-III approximately two fold more efficient (with respect to kcat/Km value) than MycE did toward M-VI. Notably, these kinetic data of MycE and MycF are comparable to those of TylE and TylF in tylosin pathway as previously reported.[7]
Table 1.
Steady-state kinetic parameters of MycE and MycF
| Km (μM) | kcat (min−1) | kcat/Km (μM−1·min−1) | |
|---|---|---|---|
| MycE | 26.4 ± 7.0 | 5.0 ± 0.5 | 0.19 |
| MycF | 30.7 ± 6.9 | 13.5 ± 1.1 | 0.44 |
The mycinamicin post-PKS (polyketide synthase) tailoring pathway includes two glycosylation steps mediated by two glycotransferases (MycB and MycD), four oxidation steps mediated by two cytochrome P450 monooxygenases (MycCI and MycG), and two methylations catalysed by MycE and MycF.[16] These post-PKS modifications not only lead to structural diversification, but also confer biologically active properties on the resulting metabolites. Recently, we confirmed all oxidative tailoring steps in vitro through functional analysis of two P450 enzymes.[21] This work revealed the importance of both methylation steps in 6-deoxyallose for substrate recognition by the MycG monooxygenase. In this report, we have advanced the knowledge about this pathway by analysing MycE and MycF O-methyltransferases in vitro. Taking advantage of the reconstituted optimal in vitro assay, the substrate specificity of MycE and MycF and hence the order of sugar modification (mycinamicin VI → III → IV) in this pathway was unambiguously determined. This new information will help facilitate future efforts to manipulate deoxysugar biosynthesis for generation of novel macrolide antibiotics.
Experimental Section
MycE and MycF Gene Cloning
Using cosmid pMR01[18] as template, mycE and mycF genes were amplified by PCR under standard conditions with primers as follow: forward, 5′-GGAGTTCCATATGACCGCACAGACCGAA-3 ′ for mycE and 5′-GGAGTTCCATATGAGCCCGTCGACCGGA-3′ for mycF (the italic letters represent the NdeI cutting site); reverse, 5′-ACATCAAGCTTTCATGTCGCGCCTCCGGA-3′ for mycE and 5′-ACATCAAGCTTTCAGGCCGAGCGACGCCA-3′ for mycF. (the underlined bases indicate the HindIII restriction site). The gel cleaned cDNAs were double digested by NdeI and HindIII (New England Biolabs), followed by the ligation of fragments containing mycE and mycF genes into NdeI/HindIII treated pET28b (Novagen) to generate recombinant plasmids pET28b-mycE and pET28b-mycF for expression of N-terminal His6-tagged MycE and MycF, respectively. The accuracy of the inserted gene was confirmed by nucleotide sequencing.
Protein Expression and Purification
Recombinant plasmids pET28b-mycE and pET28b-mycF were utilized to transform Escherichia coli BL21(DE3) cells with Z-Competent™ Kit (Zymo Research). The resulting transformants were grown at 37 °C in 1 liter of LB broth containing kanamycin (50 μg/ml) for 2~3 h until OD600 reached 0.6~0.8. Then, isopropyl-β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 0.1 mM to induce the gene expressions, and the cells were cultured at 18 °C overnight. The culture was centrifuged at 5000 × g for 10 min to collect cells. The freeze-thaw cell pellet was resuspended in 30 ml of lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 10% glycerol, pH8.0) and applied to sonication. Cell debris was removed by centrifugation at 35,000 × g for 30 min, and the supernantant was mixed with 1 ml of Ni-NTA agarose (Qiagen) for 1 h at 4 °C. The slurry was loaded onto an empty column, and the column was washed stepwise with 10 ml of lysis buffer and 40~60 ml of wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, 10% glycerol, pH8.0). The bound His6-tagged proteins were eluted with elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, 10% glycerol, pH 8.0). The MycE (~ 45 kDa) and MycF (~ 30 kDa) proteins were further purified and concentrated with 30K and 10K size exclusion filters (Amicon), respectively. The final desalting step was attained by buffer exchange into storage buffer (50 mM NaH2PO4, 10% glycerol, pH7.4) with a PD-10 column (GE Healthcare).
Enzymatic Assays
The optimised enzyme assay was carried out in 100 μl of 50 mM Tris-buffer (pH 9.0) containing 2 μM MycE or MycF, 250 μM substrate (mycinamicin VI for MycE or mycinamicin III for MycF), 10 mM MgCl2, and 500 μM SAM at 50 °C (for MycE) or 37 °C (for MycE) for 1 h. The reactions were quenched by extraction with 2 × 200 μl of CHCl3. The resulting organic extracts were dried and redissolved in 120 μl of methanol. The LC-MS analysis of reaction extract was performed on LCMS-2010 EV (Shimadzu) by using ×Bridge™ (Waters) C18; 3.5 μm; 150 mm reverse-phase HPLC column under following conditions: 20~100% solvent B over 18 min (solvent A = deionized water + 0.1% formic acid, B = acetonitrile + 0.1% formic acid); flow rate: 0.2 ml/min; UV wavelength: 280 nm.
Steady-state Kinetics
The standard reaction buffered with 50 mM Tris-HCl (pH 9.0) contains 0.3 μM MycE or MycF, 500 μM MgCl2, 2~100 μM mycinamicin VI for MycE or 3~100 μM mycinamicin III in a total volume of 396 μl. After pre-incubation at optimal temperature for 5 min, the reaction with different substrate concentration was initiated by adding 4 μl of SAM (50 mM) and three aliquots (100 μl) were taken at three time points (0, 1, 2 min and 0, 2, 4 min for reactions with substrate concentrations under 40 μM and above 60 μM, respectively) within the linear range to thoroughly mix with 100 μl of methanol for reaction termination. The proteins were removed by centrifugation at 16,000 × g for 15 min. The supernantant was subject to HPLC analysis to monitor the substrate consumption within the linear range, thereby deducing the initial velocity of the O-methylation reaction. The HPLC conditions were: ×Bridge™ (Waters) C18; 5 μm; 250 mm reverse-phase HPLC column, 20~100% solvent B over 20 min (solvent A = deionized water + 0.1% trifluoroacetic acid, B = acetonitrile + 0.1% trifluoroacetic acid), flow rate: 1.0 ml/min, UV wavelength: 280 nm. All measurements were performed in duplicate, and velocities determined under different substrate concentrations were fit into the Michaelis-Menten equation to calculate the kinetic parameters.
Acknowledgements
This work was supported by NIH grant GM078553 and the Hans W. Vahlteich Professorship to D. H. S.
References
- [1].Thibodeaux CJ, Melancon CE, H.-w. Liu. Nature. 2007;446:1008–1016. doi: 10.1038/nature05814. [DOI] [PubMed] [Google Scholar]
- [2].Trefzer A, Salas JA, Bechthold A. Nat. Prod. Rep. 1999;16:283–299. doi: 10.1039/a804431g. [DOI] [PubMed] [Google Scholar]
- [3].Weymouth-Wilson AC. Nat. Prod. Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- [4].Zubieta C, He X, Dixon RA, Noel JP. Nat. Str. Biol. 2001;8:271–279. doi: 10.1038/85029. [DOI] [PubMed] [Google Scholar]
- [5].Fouces R, Mellado E, Diez B, Barredo JL. Microbiology. 1999;145:855–868. doi: 10.1099/13500872-145-4-855. [DOI] [PubMed] [Google Scholar]
- [6].Bauer NJ, Kreuzman AJ, Dotzlaf JE, Yeh W. J. Biol. Chem. 1988;263:15619–15625. [PubMed] [Google Scholar]
- [7].Kreuzman AJ, Turner JR, Yeh W. J. Biol. Chem. 1988;263:15626–15633. [PubMed] [Google Scholar]
- [8].Bate N, Cundliffe E. J. Ind. Microbiol. Biotech. 1999;23:118–122. doi: 10.1038/sj.jim.2900707. [DOI] [PubMed] [Google Scholar]
- [9].Patallo EP, Blanco G, Fischer C, Brana AF, Rohr J, Mendez C, Salas JA. J. Biol. Chem. 2001;276:18765–18774. doi: 10.1074/jbc.M101225200. [DOI] [PubMed] [Google Scholar]
- [10].Paulus TJ, Tuan JS, Luebke VE, Maine GT, DeWitt JP, Katz L. J. Bacteriol. 1990;172:2541–2546. doi: 10.1128/jb.172.5.2541-2546.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Aguirrezabalaga I, Olano C, Allende N, Rodriguez L, Brana AF, Mendez C, Salas JA. Antimicrob. Agents Chemother. 2000;44:1266–1275. doi: 10.1128/aac.44.5.1266-1275.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Waldron C, Matsushiba P, Rosteck PR, Broughton MC, Turner J, Madduri K, Crawford KP, Merlo DJ, Baltz RH. Chem. Biol. 2001;8:487–499. doi: 10.1016/s1074-5521(01)00029-1. [DOI] [PubMed] [Google Scholar]
- [13].Satoi I, Muto N, Hayashi M, Fujii T, Otani M. J. Antibiot. 1980;33:364–376. doi: 10.7164/antibiotics.33.364. [DOI] [PubMed] [Google Scholar]
- [14].Kinoshita K, Takenaka S, Suzuki H, Morohoshi T, Hayashi M. J. Antibiot. 1992;45:1–9. doi: 10.7164/antibiotics.45.1. [DOI] [PubMed] [Google Scholar]
- [15].Kinoshita K, Takenaka S, Hayashi M. J. Antibiot. 1991;44:1270–1273. doi: 10.7164/antibiotics.44.1270. [DOI] [PubMed] [Google Scholar]
- [16].Suzuki H, Takenaka S, Kinoshita K, Morohoshi T. J. Antibiot. 1990;43:1508–1511. doi: 10.7164/antibiotics.43.1508. [DOI] [PubMed] [Google Scholar]
- [17].Anzai Y, Ishii Y, Yoda Y, Kinoshita K, Kato F. FEMS Microbiol. Lett. 2004;238:315–320. doi: 10.1016/j.femsle.2004.07.050. [DOI] [PubMed] [Google Scholar]
- [18].Anzai Y, Saito N, Tanaka M, Kinoshita K, Koyama Y, Kato F. FEMS Microbiol. Lett. 2003;218:135–141. doi: 10.1111/j.1574-6968.2003.tb11509.x. [DOI] [PubMed] [Google Scholar]
- [19].Inouye M, Suzuki H, Takada Y, Muto N, Horinouchi S, Beppu T. Gene. 1994;141:121–124. doi: 10.1016/0378-1119(94)90138-4. [DOI] [PubMed] [Google Scholar]
- [20].Kagan RM, Clarke S. Arch. Biochem. Biophy. 1994;310:417–427. doi: 10.1006/abbi.1994.1187. [DOI] [PubMed] [Google Scholar]
- [21].Anzai Y, Li S, Chaulagain MR, Kinoshita K, Kato F, Montgomery J, Sherman DH. Chem. Biol. 2008;15:950–959. doi: 10.1016/j.chembiol.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]