

Over the past half century, numerous protocols for carbonyl propargylation using allenylmetal reagents have been developed.[1] Allenic Grignard reagents were used by Prévost et al.[2a] in carbonyl additions to furnish mixtures of β-acetylenic and α-allenic carbinols, which led to them to coin the term “propargylic transposition.”[2a,b] Subsequent studies by Chodkiewicz and co-workers[2c] demonstrated relative stereocontrol in such additions. Shortly thereafter, Lequam and Guillerm[2d] reported that isolable allenic stannanes provide products of carbonyl propargylation upon exposure to chloral. Later, Mukaiyama and Harada[2e] demonstrated that stannanes generated in situ from propargyl iodides and stannous chloride reacted with aldehydes to provide mixtures of β-acetylenic and α-allenic carbinols. Related propargylations employing allenylboron reagents were first reported by Favre and Gaudemar,[2f] and propargylations employing allenylsilicon reagents were first reported by Danheiser and Carini.[2g] Asymmetric variants followed (Scheme 1). Allenylboron reagents chirally modified at the boron center engage in asymmetric propargylation, as was first reported by Yamamoto and co-workers[2h] and Corey et al.[2i] Allenylstannanes chirally modified at the tin center also induce asymmetric carbonyl propargylation, as was first reported by Minowa and Mukaiyama.[2j] Axially chiral allenylstannanes, allenylsilanes, and allenylboron reagents propargylate aldehydes enantiospecifically, as was first described by Marshall et al.,[2k,l] and Hayashi and coworkers,[2m] respectively. Finally, asymmetric aldehyde propargylation using allenylmetal reagents may be catalyzed by chiral Lewis acids or chiral Lewis bases, as was first reported by Keck et al.,[2n] and Denmark and Wynn,[2o] respectively.
Scheme 1.

Chirally modified allenylmetal reagents for carbonyl propargylation. Tf =trifluoromethanesulfonyl, Ts =para-toluenesulfonyl.
Here, we report a new approach to carbonyl propargylation based on ruthenium-catalyzed C–C bond-forming transfer hydrogenation.[3–5] Specifically, upon exposure of 1,3-enynes 1a–1g to alcohols 2a–2o in the presence of [RuHCl(CO)(PPh3)3]/dppf (dppf =1,1′-bis(diphenylphosphino)ferrocene), hydrogen shuffling between reactants occurs to generate nucleophile–electrophile pairs that regioselectively combine to furnish products of carbonyl propargylation.[6] Under related transfer hydrogenation conditions and employing isopropanol as the terminal reductant, 1,3-enynes couple to aldehydes to furnish identical products of carbonyl propargylation. The observed regiochemistry is unique with respect to related enyne–carbonyl reductive coupling reactions that are catalyzed by rhodium[5,7] and nickel complexes, [8,9,10] which favor coupling at the acetylenic terminus of the enyne. Significantly, this protocol enables carbonyl propargylation from the alcohol or aldehyde oxidation level in the absence of preformed allenylmetal reagents (Scheme 2).
Scheme 2.

Divergent regioselectivity observed in metal-catalyzed enyne–carbonyl coupling.
In connection with our efforts to exploit catalytic hydrogenation in C–C coupling reactions beyond hydroformylation,[5] we recently demonstrated that C–C bond formation may be achieved under the conditions of iridium- and ruthenium-catalyzed transfer hydrogenation.[11] These processes enable direct carbonyl allylation from the alcohol or aldehyde oxidation level by using commercially available allenes or dienes as allyl donors. Seeking to develop corresponding carbonyl propargylations, diverse iridium and ruthenium complexes were assayed for their ability to catalyze the coupling of enyne 1a and alcohol 2a. Gratifyingly, both [{Ir(cod)Cl}2]/biphep (biphep =diphenylphosphine, cod =cycloocta-l,5-diene) and [RuHCl(CO)(PPh3)3]/dppf catalyze the desired coupling. The ruthenium-based catalyst was most effective and, under optimized conditions, enyne 1a coupled to benzylic, allylic, and aliphatic alcohols 2a–2o to form homopropargyl alcohols 3a–3o in good to excellent yields (Table 1). To probe the scope of the enyne coupling partner, enynes 1b–1g were coupled to benzyl alcohol 2b under standard reaction conditions. Good to excellent yields of propargylation products 3p–3u were observed (Table 2). Substitution at the olefinic terminus of the enyne was found to diminish conversion to product. Finally, carbonyl allylation can also be achieved from the aldehyde oxidation level by employing isopropanol as the terminal reductant. Under standard reaction conditions, aldehydes 4a–4c couple to enyne 1a to provide the products of carbonyl propargylation 3a–3c, respectively, in good to excellent yield. Thus, carbonyl propargylation may be achieved from either the alcohol or aldehyde oxidation level (Table 3). The coupling products 3a–3u are remarkably resistant to over-oxidation to form the corresponding β,γ-acetylenic ketones. However, such over-oxidation is observed if cationic ruthenium complexes are employed as catalysts. This result suggests that, for the neutral ruthenium complexes employed in this study, the alkyne moiety of the coupling product blocks a coordination site required for β hydride elimination of the carbinol C–H bond. Other aspects of the catalytic mechanism, including determination of the structural and interactive features of the ruthenium complex that influence relative and absolute stereocontrol, are currently under investigation.
Table 1.
Carbonyl propargylation from the alcohol oxidation level by ruthenium-catalyzed transfer hydrogenation.a
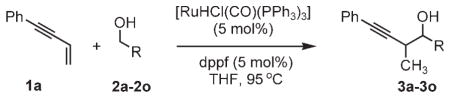 | ||
| 2a, R =p-NO2Ph | 2f, R =p-BrPh | 2k, R =geranyl |
| 2b, R =phenyl | 2g, R =2-furyl | 2l, R =crotyl |
| 2c, R =p-MeOPh | 2h, R =3-indolyl | 2m, R =cyclopropyl |
| 2d, R =o-MeOPh | 2i, R =2-(6-BrPy) | 2n, R =benzyl |
| 2e, R =5-piperonyl | 2j, R =cinnamyl | 2o, R =n-pentyl |
| Coupling to benzylic alcohols | ||
 |
 |
 |
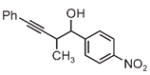
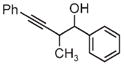
| 3a | 3b | 3c |
| 65% yield | 81% yield | 81% yield |
| 1:1 d.r. | 1:1 d.r. | 1:1 d.r. |
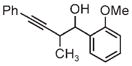 |
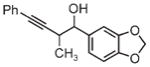 |
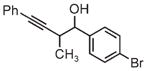 |
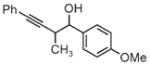
| 3d | 3e | 3f |
| 91% yield | 83% yield | 73% yield |
| 2:1 d.r. | 2:1 d.r. | 1:1 d.r. |
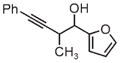 |
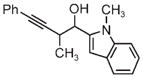 |
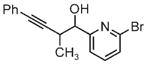 |
| 3g | 3h | 3i |
| 71% yield | 94% yield | 42% yield |
| 1.5:1 d.r. | 1:1 d.r. | 1.3:1 d.r. |
| Coupling to allylic alcohols | ||
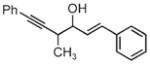 |
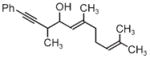 |
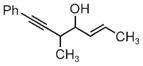 |
| 3j | 3k | 3l |
| 68% yield | 63% yield | 72% yield |
| 1:1 d.r. | 1.5:1 d.r. | 2:1 d.r. |
| Coupling to aliphatic alcohols | ||
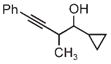 |
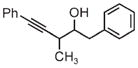 |
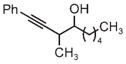 |
| 3m | 3n | 3o |
| 75% yield | 70% yield | 72% yield |
| 2:1 d.r. | 1:1 d.r. | 2:1 d.r. |
Yields of isolated material. Standard reaction conditions employed 1 equivalent of alcohol/aldehyde and 2 equivalents of enyne. See the Supporting Information for detailed experimental procedures. Py =pyridine.
Table 2.
Coupling of enynes 1b–1g to benzyl alcohol 2b by ruthenium-catalyzed transfer hydrogenation.a
 | ||
| 1b, R =2-thienyl | 1d, R =TBSO(CH2)4 | 1f, R =TBSOC(CH3)2 |
| 1c, R =BocNH(CH2)2 | 1e, R =TBSOCH2 | 1g, R =cyclohexyl |
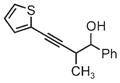 |
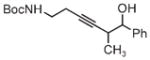 |
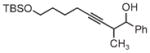 |
| 3pb | 3q | 3r |
| 71% yield | 54% yield | 63% yield |
| 1:1 d.r. | 1:1 d.r. | 1:1 d.r. |
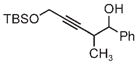 |
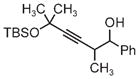 |
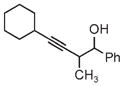 |
| 3s | 3tb | 3ub |
| 78% yield | 56% yield | 70% yield |
| 1.5:1 d.r. | 1:1 d.r. | 1:1 d.r. |
See the footnotes of Table 1 for details.
m-NO2BzOH (5 mol%) was employed as a cocatalyst. Boc =tert-butyloxycarbonyl, Bz =benzyl, TBS =tert-butyldimethylsilyl.
Table 3.
Carbonyl propargylation from the aldehyde oxidation level by ruthenium-catalyzed transfer hydrogenation.a
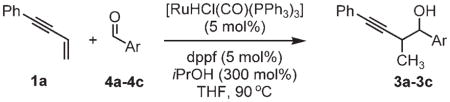 | ||
| 4a, Ar =p-NO2Ph | 4b, Ar =Ph | 4c, Ar =p-MeOPh |
| 3a | 3b | 3c |
| 61% yield | 74% yield | 91% yield |
| 1:1 d.r. | 1:1 d.r. | 1:1 d.r. |
Formation of 3a and 3b were accompanied by about 10% alkyne reduction. See the Supporting Information for detailed experimental procedures.
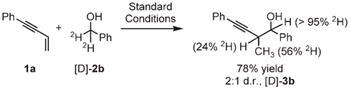
A general catalytic mechanism is likely to involve the following steps:[11] a) alcohol dehydrogenation to generate a ruthenium hydride is followed by b) enyne hydrometalation to generate an allenyl metal–aldehyde/nucleophile–electrophile pair, which undergoes c) carbonyl addition with propargylic transposition. Consistent with this interpretation, the coupling of enyne 1a to [D]-2b under standard reaction conditions provides [D]-3b, in which deuterium is incorporated at the benzylic position (>95%), the allylic methyl group (56%), and the allylic methine position (24%), thus suggesting reversible olefin-hydrometalation [Eq. (1)].
 |
(1) |
Our collective studies on hydrogenative and transfer hydrogenative C–C coupling define a departure from the use of preformed organometallic reagents in carbonyl addition chemistry.[5,11] For such transfer hydrogenative coupling reactions, hydrogen embedded within isopropanol or an alcohol substrate is redistributed among reactants to generate nucleophile–electrophile pairs, thus enabling carbonyl addition from the aldehyde or alcohol oxidation level. In this way, carbonyl additions that transcend the boundaries of oxidation level are devised. In the present study, we have demonstrated that 1,3-enynes serve as allenylmetal equivalents under the conditions of transfer hydrogenative coupling, thus also enabling carbonyl propargylation from the alcohol or aldehyde oxidation level. These studies contribute to a growing body of catalytic methods for the direct functionalization of carbinol C–H bonds.[11,12] Future studies will focus on the development of related alcohol–unsaturate C–C coupling processes.
Supplementary Material
Footnotes
Acknowledgements are made to Merck, the Robert A. Welch Foundation, the American Chemical Society Green Chemistry Institute Pharmaceutical Roundtable, and the NIH (Grant No. RO1-GM069445) for partial support of this research.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200801359.
References
- 1.For reviews that encompass carbonyl propargylation employing allenylmetal reagents, see: Moreau J-L. In: The Chemistry of Ketenes, Allenes and Related Compounds. Patai S, editor. Wiley; New York: 1980. pp. 363–413.Marshall JA. Chem Rev. 1996;96:31–48. doi: 10.1021/cr950037f.Gung BW. Org React. 2004;64:1–113.Marshall JA, Gung BW, Grachan ML. In: Modern Allene Chemistry. Krause N, Hashmi ASK, editors. Wiley-VCH; Weinheim: 2004. pp. 493–592.Marshall JA. J Org Chem. 2007;72:8153–8166. doi: 10.1021/jo070787c.
- 2.For selected milestones in carbonyl propargylation, see: Prévost C, Gaudemar M, Honigberg J. C R Hebd Seances Acad Sci. 1950;230:1186–1188.Wotiz JH. J Am Chem Soc. 1950;72:1639–1642.Karila M, Capmau ML, Chodkie-wicz W. C R Hebd Seances Acad Sci. 1969;269:342–345.Lequan M, Guillerm G. J Organomet Chem. 1973;54:153–164.Mukaiyama T, Harada T. Chem Lett. 1981:621–624.Favre E, Gaudemar M. C R Hebd Seances Acad Sci. 1966;263:1543–1545.Danheiser RL, Carini DJ. J Org Chem. 1980;45:3925–3927.Haruta R, Ishiguro M, Ikeda N, Yamamoto H. J Am Chem Soc. 1982;104:7667–7669.Corey EJ, Yu CM, Lee DH. J Am Chem Soc. 1990;112:878–879.Minowa N, Mukaiyama T. Bull Chem Soc Jpn. 1987;60:3697–3704.Marshall JA, Wang XJ. J Org Chem. 1991;56:3211–3213.Marshall JA, Maxson K. J Org Chem. 2000;65:630–633. doi: 10.1021/jo991543y.Matsumoto Y, Naito M, Uozumi Y, Hayashi T. J Chem Soc Chem Commun. 1993:1468–1469.Keck GE, Krishnamurthy D, Chen X. Tetrahedron Lett. 1994;35:8323–8324.Denmark SE, Wynn T. J Am Chem Soc. 2001;123:6199–6200. doi: 10.1021/ja016017e.
- 3.For selected reviews on ruthenium-catalyzed transfer hydrogenation, see: Zassinovich G, Mestroni G, Gladiali S. Chem Rev. 1992;92:1051–1069.Noyori R, Hashiguchi S. Acc Chem Res. 1997;30:97–102.Noyori R, Yamakawa M, Hashiguchi S. J Org Chem. 2001;66:7931–7944. doi: 10.1021/jo010721w.Noyori R, Ohkuma T. Angew Chem. 2001;113:40–75.Angew Chem Int Ed. 2001;40:40–73.Noyori R. Angew Chem. 2002;114:2108–2123.Angew Chem Int Ed. 2002;41:2008–2022.Noyori R. Adv Synth Catal. 2003;345:15–32.Muñiz K. Angew Chem. 2005;117:6780–6785.Angew Chem Int Ed. 2005;44:6622–6627. doi: 10.1002/anie.200501787.Noyori R. Chem Commun. 2005:1807–1811. doi: 10.1039/b502713f.Gladiali S, Alberico E. Chem Soc Rev. 2006;35:226–236. doi: 10.1039/b513396c.Ikariya T, Blacker AJ. Acc Chem Res. 2007;40:1300–1308. doi: 10.1021/ar700134q.
- 4.For selected reviews of ruthenium-catalyzed C–C coupling, see: Trost BM, Toste FD, Pinkerton AB. Chem Rev. 2001;101:2067–2096. doi: 10.1021/cr000666b.Kondo T, Mitsudo T-a. Curr Org Chem. 2002;6:1163–1179.Derien S, Monnier F, Dixneuf PH. Top Organomet Chem. 2004;11:1–44.
- 5.For selected reviews on hydrogenative C–C coupling, see: Ngai MY, Kong JR, Krische MJ. J Org Chem. 2007;72:1063–1072. doi: 10.1021/jo061895m.Iida H, Krische MJ. Top Curr Chem. 2007;279:77–104.Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394–1401. doi: 10.1021/ar7001123.
- 6.The transformations may be considered as hydrogen autotransfer processes; for reviews, see: Guillena G, Ram+n DJ, Yus M. Angew Chem. 2007;119:2410–2416.Angew Chem Int Ed. 2007;46:2358–2364. doi: 10.1002/anie.200603794.Hamid MHSA, Slatford PA, Williams JMJ. Adv Synth Catal. 2007;349:1555–1575.
- 7.For rhodium-catalyzed reductive coupling of 1,3-enynes to carbonyl compounds and imines, see: Jang HY, Huddleston RR, Krische MJ. J Am Chem Soc. 2004;126:4664–4668. doi: 10.1021/ja0316566.Kong JR, Cho CW, Krische MJ. J Am Chem Soc. 2005;127:11269–11276. doi: 10.1021/ja051104i.Kong JR, Ngai MY, Krische MJ. J Am Chem Soc. 2006;128:718–719. doi: 10.1021/ja056474l.Koman-duri V, Krische MJ. J Am Chem Soc. 2006;128:16448–16449. doi: 10.1021/ja0673027.Hong YT, Cho CW, Skucas E, Krische MJ. Org Lett. 2007;9:3745–3748. doi: 10.1021/ol7015548.
- 8.For nickel-catalyzed reductive coupling of 1,3-enynes to car-bonyl compounds, see: Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J Am Chem Soc. 2004;126:4130–4131. doi: 10.1021/ja0491735.Miller KM, Jamison TF. Org Lett. 2005;7:3077–3080. doi: 10.1021/ol051075g.Miller KM, Colby EA, Woodin KS, Jamison TF. Adv Synth Catal. 2005;347:1533–1536.
- 9.For seminal contributions to nickel-catalyzed alkyne/carbonyl reductive coupling, see: Oblinger E, Montgomery J. J Am Chem Soc. 1997;119:9065–9066.Huang WS, Chan J, Jamison TF. Org Lett. 2000;2:4221–4223. doi: 10.1021/ol006781q.
- 10.For reviews encompassing nickel-catalyzed alkyne/carbonyl reductive coupling, see: Montgomery J. Angew Chem. 2004;116:3980–3998.Angew Chem Int Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634.Montgomery J, Sormunen GJ. Top Curr Chem. 2007;279:1–23.Moslin RM, Miller-Moslin K, Jamison TF. Chem Commun. 2007:4441–4449. doi: 10.1039/b707737h.
- 11.a) Bower JF, Skucas E, Patman RL, Krische MJ. J Am Chem Soc. 2007;129:15134–15135. doi: 10.1021/ja077389b. [DOI] [PubMed] [Google Scholar]; b) Bower JF, Patman RL, Krische MJ. Org Lett. 2008;10:1033–1035. doi: 10.1021/ol800159w. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:6338–6339. doi: 10.1021/ja801213x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Lin Y, Ma D, Lu X. Tetrahedron Lett. 1987;28:3249–3252. [Google Scholar]; b) Shi L, Tu YQ, Wang M, Zhang FM, Fan CA, Zhao YM, Xia WJ. J Am Chem Soc. 2005;127:10836–10837. doi: 10.1021/ja0528331. [DOI] [PubMed] [Google Scholar]; c) Pastine SJ, McQuaid KM, Sames D. J Am Chem Soc. 2005;127:12180–12181. doi: 10.1021/ja053337f. [DOI] [PubMed] [Google Scholar]; d) Jiang YJ, Tu YQ, Zhang E, Zhang SY, Cao K, Shi L. Adv Synth Catal. 2008;350:552–556. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.