

Abstract
Tumor immune escape is a major obstacle in cancer immunotherapy but the mechanisms involved remain poorly understood. We have previously developed an immune evasion tumor model using an in vivo immune selection strategy and revealed Akt-mediated immune resistance to anti-tumor immunity induced by various cancer immunotherapeutic agents. In the current study, we employed microarray gene analysis to identify an Akt-activating candidate molecule overexpressed in immune resistant tumors compared to parental tumors. X-linked lymphocyte-regulated protein pM1 (XLR) gene was the most upregulated in immune-resistant tumors compared to parental tumor cells. Furthermore, the retroviral transduction of XLR in parental tumor cells led to activation of Akt, resulting in upregulation of anti-apoptotic proteins and the induction of immune resistance phenotype in parental tumor cells. In addition, we found that transduction of parental tumor cells with other homologous genes from the mouse XLR family, such as synaptonemal complex protein 3 (SCP3) and XLR-related, meiosis regulated protein (XMR) and its human counterpart of SCP3 (hSCP3) also led to activation of Akt, resulting in the upregulation of anti-apoptotic proteins and induction of immune resistance phenotype. Importantly, characterization of a panel of human cervical cancers revealed relatively higher expression levels of hSCP3 in human cervical cancer tissue compared to normal cervical tissue. Thus, our data indicate that ectopic expression of XLR and its homologs in tumor cells represents a potentially important mechanism for tumor immune evasion and serves as a promising molecular target for cancer immunotherapy.
Keywords: XLR, Akt, immune resistance, human papillomavirus (HPV), E7
Introduction
Cancer immunotherapy has been reasonably successful in generating tumor-specific immune responses. However, in some cases, it is observed that the generation of tumor-specific immune responses does not translate into tumor regression in cancer patients (1). One potential explanation is that tumor cells with their aberrant gene expression, can possibly influence and impair the immune system in many ways. A better understanding of the mechanisms of tumor immune evasion will serve as an important objective in the field of cancer immunotherapy.
To elucidate mechanisms of immune escape associated with immune resistance, we employed an in vivo selection strategy using a previously developed HPV-16 E7-expressing cancer cell line, called TC-1/ P0, which has served as a preclinical tumor model for testing various E7-specific cancer immunotherapies (2, 3) including an HPV-16 E7-expressing vaccinia virus vaccine termed Vac-Sig/E7/LAMP-1. The vaccine encodes a fusion protein consisting of an endoplasmic reticulum (ER) signal sequence, HPV-16 E7 gene, and the transmembrane and cytoplasmic domains of lysosome-associated membrane protein-1 (LAMP-1) (4). Using immune selection, we generated an immune resistant clone which has high expression of MHC class I, called TC-1 P3 (A17) (5, 6). The TC-1 P3 (A17) cells demonstrated immune resistance to cytotoxic T lymphocyte (CTL)-induced apoptosis in vitro and in vivo as compared to parental TC-1 cells (5). In addition, the resistance of A17 tumor cells was shown to depend on prosurvival Akt signaling pathway, which was confirmed by a pharmacological approach using various kinase inhibitors and using siRNA targeting Akt (5). Thus, we have successfully generated an immune-resistant tumor cell model (TC-1 P3 (A17)), thereby developing a system that would allow us to identify genes that may contribute to the Akt-mediated immune resistance to anti-tumor immunity induced by various cancer immunotherapeutic agents.
In the current study, we have employed microarray gene analysis to identify candidate molecules that are overexpressed in A17 tumors compared to P0 tumors. Our data indicate that the expression of XLR and its homologs in tumor cells represents a new mechanism of immune resistance via activation of the Akt pathway and has important implications for the development of a novel therapeutic strategy against immune-resistant tumor cells.
Materials and methods
Mice
Six- to 8-week-old female C57BL/6 mice and nude mice were purchased from Daehan Biolink (Chungbuk, Korea). All animal procedures were done in accordance with recommendations for the proper use and care of laboratory animals.
Microarray data analysis
For the microarray analysis, the RNA samples from TC-1 and TC-1 P3 (A17) cell lines were prepared and analyzed with Affymetrix GeneChip Mouse Genome 430 2.0 arrays. To estimate the gene expression signals, image analysis was done for the CEL files of the chips using the statistical technique and package guanine-cytosine content–robust multiarray analysis as described previously (6, 7). The normalized signal intensities were analyzed using parametric empirical Bayes method to estimate the posterior probabilities of differential expression of genes between TC-1 and TC-1 P3 (A17) cell lines (8, 9).
DNA constructs and siRNA transfection
For the generation of the pMSCV construct encoding XLR, SCP3, XMR or hSCP3, the DNA fragments encoding the XLR and SCP3 were amplified from A17 tumor cell cDNA, DNA fragments encoding XMR were amplified from mouse testis cDNA and human SCP3 were amplified from a human testis cDNA library (Clontech, USA) with PCR using a set of primers. The amplified DNAs were subsequently cloned into pMSCV retroviral vector (Clontech, CA). Plasmid constructs were confirmed by DNA sequencing.
Synthetic small interfering RNA (siRNA) specific for Akt were purchased from Dharmacon (Lafayette, CO). XLR and GFP siRNAs were synthesized using 20-O-ACE-RNA phosphoramides (Dharmacon, Lafayette, CO) (See Supplementary Table 1).
Chemical reagents
LY294002 (Calbiochem Corp, San Diego, CA) for PI3K inhibition, API-2 (Calbiochem Corp, San Diego, CA) for Akt inhibition, SB203580 (Calbiochem corp, San Diego, CA) for p38 inhibition, PD98059 (Stressgen, Michigan, USA) for ERK inhibition were used for inhibition of the individual kinase pathway.
Cells
Six HPV-16 E7-expressing cell lines, TC-1, TC-1 P3 (A17), TC-1/No insert, TC-1/XLR, TC-1/XMR, and TC-1/SCP3 were used as murine tumor models and 293Db/No insert and 293Db/hSCP3 cell lines were used as human tumor models (10). The production and maintenance of TC-1 and TC-1 P3 (A17) cells has been described previously (2). B16, B16F10, EL4, CT26, CMT93, WEHI164, Neuro-2a and NIH3T3 cell line purchased from American Type Culture Collection (ATCC, USA). TC-1/No insert, TC-1/XLR, TC-1/XMR, TC-1/SCP3, 293Db/No insert and 293Db/hSCP3 cell lines were generated using the constructed pMSCV encoding No insert, XLR, XMR, SCP3, hSCP3 or XLR by methods described previously (5).
Flow cytometry analysis
For in vitro E7-specific CD8+ T cell activation, TC-1/no insert and TC-1/XLR cells were incubated with an E7-specific CD8+ T cell line at 0.01:1, 0.1:1, and 1:1 ratio of tumor:T cells for 16 hours. Cell surface marker staining of CD8 and intracellular cytokine staining for IFN-γ as well as FACScan analysis was performed using conditions described previously (11-14). To detect MHC class I expression, PE-labeled anti-mouse H-2Kb or H-2Db (BD Biosciences) monoclonal antibody was used. Analysis was done on a Becton Dickinson FACScan with CELLQuest software (BD Biosciences). The in vitro CTL assay was performed using methods described previously (5, 11-14) and analyzed by flow cytometry analysis.
In vivo tumor treatment experiments
The in vivo tumor treatment experiment using vaccinia virus vaccination or adoptive T cell transfer using chitosan hydrogel containing 5ug API-2 (5, 15) was performed as described previously (5).
Western blot
A total of 5×105 cells were used as described previously (5). The primary antibodies phospho Akt (Ser473), Akt, phospho Erk (T202/Y204), Erk, p38 MAPK, Bcl-w, Bid, Bim, Bad, phospho Bad (Ser 136), cIAP-1, XIAP (1:1000, Cell signaling), Dual phosphor p38 MAPK (1:1000, Stressgen, Victoria, Canada), Mcl-1, Bcl-2, cIAP-2, Bcl-xL, Bax, HA-Probe, survivin (1:1000, Santa Cruz Biotechnology), Bak, SCP3 (1:1000, BD Biosciences), E7 (provided by Dr. Ju-hong Jun, Seoul National University, Korea) and XLR (provided by Dr. Henri-Jean Garchon, Institut National de la Santé et de la Recherche Médicale) were used for Western blotting followed by the appropriate secondary antibodies. Immunoreactive bands were visualized by enhanced chemiluminescence (ECL, Elpis Biotech, Korea) reaction.
Statistical analysis
All data are representative of at least two separate experiments. Results for intracellular cytokine staining with flow cytometry analysis, and tumor treatment experiments were evaluated by ANOVA. Comparisons between individual data points were made using Student’s t-test. All p-values < 0.05 were considered statistically significant.
Results
Overexpression of XLR in TC-1 P3(A17) tumor cells leads to an immune resistance to apoptotic cell death induced by E7-specific CD8+ T cells
In our previous study, we have characterized the immunoresistant A17 tumors and the parental TC-1/P0 tumors and found that A17 tumor cells demonstrated comparable levels of E7 and MHC class I expression and the ability to activate E7-specific CD8+ T cells compared to TC-1 tumors (5). We also sequenced the HPV-16 E7 gene within the A17 tumor cells and found no mutations (data not shown), thus precluding the alteration of the immunodominant epitope of E7. In order to identify candidate genes that are overexpressed in the immune resistant TC-1 P3 tumor cell clone, A17, we performed a microarray analysis and observed that the X-linked lymphocyte regulated protein pM1 (XLR) demonstrated the highest fold increase in expression in the TC-1 P3 (A17) cells compared to TC-1 P0 cells (Supplementary Table 2). To confirm this, we also performed Western blot analysis to compare the expression of XLR in TC-1 P3 (A17) cells and TC-1 P0 cells. As shown in Figure 1A, XLR was found to be upregulated in TC-1 P3 (A17) cells compared to TC-1 P0 cells. Furthermore, we have demonstrated that several murine tumor cells express significant levels of XLR (See Supplementary Figure 1).
Figure 1. Characterization of the expression and anti-apoptotic function of XLR in TC-1 P0 and A17 tumor cells.
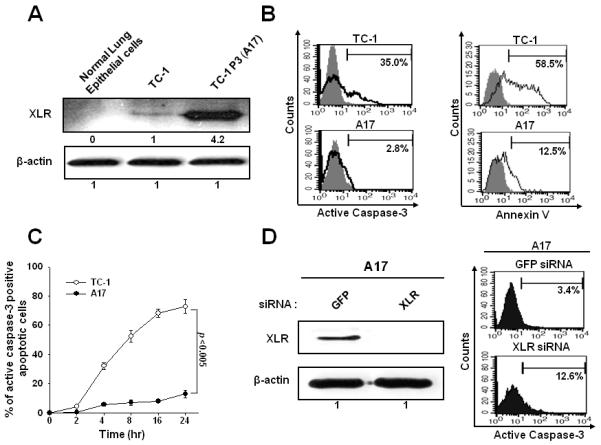
A. Western blot analysis characterizing the XLR protein expression in TC-1 and A17 cells. XLR-specific antibody was used and incubated with goat anti-mouse IgG conjugated to horseradish peroxidase for visualization of XLR protein using Hyperfilm-enhanced chemiluminescence. β-actin was used as a loading control. B. Flow cytometry analysis of apoptotic cell death induced by E7-specific CD8+ T cells at 1:1 ratio for 4 hours. Apoptosis was measured by quantification of active caspase-3 expression and annexin-V staining. C. Kinetics of caspase-3 positive apoptosis after CTL assay described in Fig 1B. D. Western blot analysis to characterize the XLR expression in GFP or XLR siRNA transfected A17 cells and flow cytometry analysis of caspase-3 positive apoptosis induced by E7-specific CD8+ T cells in A17 cells transfected with GFP or XLR siRNA. A17 cells were transfected with 0.5uM GFP or XLR siRNA and then cultured for 48 hours and Western blot analysis was performed. Data shown are representative of three independent experiments.
Previously, we demonstrated that the immunoresistance of TC-1 P3 (A17) was associated with its resistance against E7-specific CD8+ T cell mediated apoptosis and the observed resistance against apoptosis was found to be associated with the upregulation of anti-apoptotic molecules caused by AKT activation (5). Thus, we first compared the apoptotic cell death induced by E7-specific CD8+ T cells in A17 and TC-1 cells. Consistently, we found that A17 tumor cells demonstrated significant resistance to tumor cell killing compared to the TC-1 tumor cells (Figure 1B). We also found that the percentage of apoptotic cells increased with time (Figure 1C). We intended to further confirm the link between XLR expression level and resistance to cytotoxic T lymphocyte (CTL) killing by employing siRNA technology. For this, A17 cells were transfected with GFP siRNA or XLR siRNA and in turn characterized for expression of XLR by Western blot analysis. XLR expression was completely abolished in XLR siRNA transfected A17 cells and this led to reduction in the immune resistance to apoptotic cell death mediated by E7-specific CD8+ T cells compared to GFP siRNA transfected cells (Figure 1D). Thus, our data indicate that overexpression of XLR in TC-1 P3(A17) cells contributes to immune resistance to apoptotic cell death mediated by E7-specific CD8+ T cells.
TC-1/XLR tumor cells demonstrate comparable levels of E7 expression and the ability to activate E7-specific CD8+ T cells compared to TC-1 tumors but demonstrate immune resistance phenotype
In order to determine if the expression of XLR in TC-1/P0 cells results in an immune resistance phenotype, we introduced DNA construct encoding XLR into TC-1/P0 cells via a retroviral system. TC-1/P0 cells transduced with no insert (pMSCV) were used as a control (TC-1/no insert). We then performed Western blot analysis to determine the expression of XLR and E7 in TC-1/XLR and TC-1/no insert cells. We observed that TC-1/XLR demonstrated significantly higher expression of XLR compared to TC-1/no insert cells (Figure 2A). The TC-1/XLR and TC-1/no insert cells also showed comparable expression levels of E7 (Figure 2A). We further evaluated the phenotypic and functional characteristics of the TC-1/XLR cells. As shown in Figure 2B, we observed that TC-1/XLR cells demonstrated comparable expression levels of MHC class I compared to the TC-1/no insert cells. Furthermore, in order to determine the ability of TC-1/XLR cells to process and present the E7 peptide through the MHC class I pathway, we characterized the ability of TC-1/XLR cells to activate E7-specific CD8+ T cells compared to TC-1/no insert tumors. We observed that TC-1/XLR cells demonstrated a similar ability to activate E7-specific CD8+ T cells in vitro compared to TC-1/no insert tumors (Figure 2C). This result suggests that the antigen processing and presentation of E7 through the MHC class I was not impaired in TC-1/XLR cells relative to TC-1/P0 cells.
Figure 2. Functional characterization of TC-1/XLR tumor cell line.

A. Western blot analysis to characterize the expression of XLR and E7 in TC-1/no insert and TC-1/XLR cells. B. Flow cytometry analysis to characterize MHC class I expression on TC-1/no insert and TC-1/XLR cells. PE-conjugated anti-mouse H-2Db monoclonal antibody was used to detect MHC class I expression. The isotype antibody was used as the negative control (black profile). C. Intracellular cytokine staining and flow cytometry analysis to determine the number of IFN-γ secreting E7-specific CD8+ T cells induced by TC-1/no insert and TC-1/XLR cells. TC-1/no insert and TC-1/XLR cells were incubated with E7-specific CD8+ T cells at different E:T rations (0.01:1, 0.1:1, and 1:1) for 16 hours. After incubation, cells were stained for CD8 and IFN-γ, and analyzed by flow cytometry analysis. D. Graphical representation of the tumor volume in mice challenged with TC-1/No insert and TC-1/XLR cells with or without vaccination with Vac-Sig/E7/LAMP-1. Data shown are representative of three independent experiments.
We further determined if the TC-1/XLR tumor cells maintained the immune resistance phenotype similar to the A17 tumors in tumor-bearing mice. As shown in Figure 2D, TC-1/XLR tumor-challenged mice vaccinated with Vac-Sig/E7/LAMP-1 showed significantly higher tumor volumes over time compared to TC-1/no insert tumor-challenged mice, thus demonstrating immune resistance. We also determined if the ectopic expression of XLR in tumor cells leads to a resistance to apoptotic cell death induced by CTL killing. We found a significant reduction in the apoptotic cell death in TC-1/XLR cells compared to apoptotic TC-1/no insert tumor cells at different E:T ratios (Supplementary Figure 2A). We further assessed the immune resistance of the tumor cell lines to the therapeutic effect induced by adoptively transferred E7-specific CD8+ T cells in C57BL/6 mice. We found that TC-1/no insert tumor challenged mice treated with E7-specific CD8+ T cells demonstrated significantly lower tumor volume over time compared to TC-1/XLR tumor challenged mice treated with E7-specific CD8+ T cells (Supplementary Figure 2B). Thus, our data indicate that TC-1/XLR tumor cells are highly resistant to the therapeutic effect induced by tumor antigen specific CD8+ T cells both in vitro and in vivo.
Akt activation plays a crucial role in the resistance of apoptotic tumor cell death of TC-1/XLR cells induced by E7-specific CD8+ T cells
To determine the expression of the various signaling molecules that may play a role in the global control of apoptosis, we performed Western blot analysis to compare the expression of total Akt, Ser 473 phosphorylated pAkt, total Erk, Thr 202/Tyr 204 phosphorylated pErk, total p38 MAP kinase and Thr 180/Tyr 182 phosphorylated pp38 MAP kinase in TC-1/XLR cells and TC-1/no insert cells. We observed that the active form of Akt (pAkt) was the only molecule that demonstrated significantly increased expression in the TC-1/XLR cells compared to TC-1/no insert cells among all of the signaling molecules characterized (Figure 3A). Additionally, downregulation of XLR using XLR siRNA in A17 led to decrease of pAKT level (Supplementary Figure 3). Taken together, our data indicate that expression of XLR in TC-1 tumor cells led to activation of Akt.
Figure 3. Characterization of the signal pathways that may play a role in increased immune resistance in TC-1/XLR tumor cells.

A. Western blot analysis to characterize the expression of total Akt, Ser 473 pAkt, total Erk, Thr 202/Tyr 204 pErk, total p38 MAP kinase and Thr 180/Tyr 182 pp38 MAP kinase in the TC-1/no insert and TC-1/XLR cells. B. Western blot analysis to characterize the expression of total Akt, Ser 473 pAkt, total Erk, Thr 202/Tyr 204 pErk, total p38 MAP kinase and Thr 180/Tyr 182 pp38 MAP kinase in TC-1/XLR cells treated with the various inhibitors. TC-1/XLR cells were incubated with DMSO, PI3K inhibitor LY294002, p38 MAP kinase inhibitor SB203580 or Erk inhibitor PD98059 for 18 hours prior to lysate preparation. C. Representative flow cytometry data for detection of apoptotic TC-1/XLR cells in the presence or absence of E7-specific CD8+ T cells after treatment of DMSO or each inhibitor. TC-1/XLR cells were pretreated with the various inhibitors and incubated with E7-specific CD8+ T cells at 1:1 ratio. After 4 hour incubation, apoptosis was evaluated The data are representative of three separate experiments. D. Bar graph depicting the percentage of apoptotic TC-1/XLR cells.
Since Akt is a key signaling molecule in the PI3K pathway, we sought to confirm our results by incubating TC-1/XLR cells with the PI3K inhibitor LY294002. The effectiveness of each of the inhibitors is demonstrated by Western blot analysis in Figure 3B. We observed that TC-1/XLR cells pretreated with the PI3K inhibitor demonstrated significant decrease in the resistance to tumor killing by E7-specific CD8+ T cells compared to cells pretreated with DMSO or any of the other inhibitors (Figure 3C and D). Thus, our data indicate that PI3K/Akt signal pathway plays a crucial role in the resistance of apoptotic tumor cell death of TC-1/XLR cells induced by E7-specific CD8+ T cells.
To determine if the expression of XLR in TC-1 cells results in upregulation of antiapoptotic proteins, we characterized the expression of key pro- and anti-apoptotic proteins in TC-1/no insert and TC-1/XLR cells. We found that the expression of anti-apoptotic proteins (Bcl-2, Bcl-xL, p-Bad, survivin, cIAP2) was significantly increased in the TC-1/XLR cells compared to the TC-1/no insert cells while the expression of key pro-apoptotic proteins (Bak, Bax, Bim, Bad) showed no significant difference between TC-1/XLR cells and TC-1/no insert cells (Supplementary Figure 4A).
Treatment of TC-1/XLR tumor cells with the Akt inhibitor, API-2, reduces the expression of anti-apoptotic proteins resulting in the abolishment of immune resistance phenotype
To confirm the role of Akt in the immune resistance of TC-1/XLR apoptotic tumor cell death induced by E7-specific CD8+ T cells, we employed a pharmacological inhibitor of Akt, Akt/protein kinase B signaling inhibitor-2 (API-2) which has been shown to suppress the kinase activity and phosphorylation level of Akt leading to inhibition of cell growth and induction of apoptosis (16). As shown in Figure 4A, the expression of the key anti-apoptotic proteins (Bcl-xL, Bcl-2) in the TC-1/XLR cells was significantly reduced in the presence of API-2. Consistently, similar phenotypes were observed after siAKT treatment (Supplementary Figure 4B).
Figure 4. Characterization of anti-tumor effects generated by E7 specific CD8+ T cell adoptive transfer combined with AKT inhibitor API-2 treatment.

A. Western blot analysis to characterize the expression of p-AKT, AKT, Bcl-XL and Bcl-2 in DMSO or Akt inhibitor API-2 treated TC-1/XLR cells. B. Bar graph representing tumor volumes from tumor challenged mice treated with or without API-2 in the presence or absence of E7-specific CD8+ T cells on 10 days after tumor challenge. Tumor volumes from TC-1/XLR tumor were recorded twice per week for 10 days following adoptive transfer.
We then characterized the in vivo tumor growth in TC-1/XLR tumor-bearing mice treated with or without the inhibitor. For this, chitosan hydrogel (CH) containing 5ug API-2 was injected intra-tumorally (5). The CH was completely degraded and disappeared 7-9 days after injection. Thus, the experiment was terminated, and tumor volumes were measured 10 days following T cell adoptive transfer. As shown in Figure 4B, tumor challenged mice treated with adoptive transfer E7-specific CD8+ T cells in the presence of API-2 demonstrated significantly lower tumor volume compared to tumor challenged mice treated with E7-specific CD8+ T cells without API-2 treatment. Thus, our data indicate that inhibition of Akt in TC-1/XLR tumor reduces the expression of anti-apoptotic proteins, resulting in the abolishment of immune resistance phenotype.
TC-1/P0 cells transduced with homologous genes from the XLR family lead to activation of Akt, upregulation of antiapoptotic proteins and induction of the immune resistance phenotype
To explore whether other genes from the XLR family which share significant homology with XLR could induce immune resistance phenotype on TC-1/P0 cells, we aligned the amino acid sequence of XLR gene with other XLR family members, XMR, SCP3 and the human homolog of the SCP3 gene (hSCP3). We observed more than 60% homology of these genes compared to XLR gene (See Supplementary Table 3) (17, 18-20). We then characterized the expression and function of SCP3 or XMR in transduced TC-1/P0 cells. As shown in Figure 5A, TC-1 cells transduced with XMR or SCP3 also demonstrated significant activation of Akt and upregulation of Mcl-1, Bcl-XL and Bcl-2 compared to the control TC-1/no insert cells.
Figure 5. Characterization of expression of XLR family genes in the TC-1/No insert and TC-1/XLR tumor cells.

A. Western blot analysis to characterize the expression of SCP3 in TC-1/no insert and TC-1/SCP3 and HA expression in the TC-1/no insert and TC-1/XMR/HA cells and p-AKT, total Akt, Mcl-1, Bcl-xL and Bcl-2 expression in TC-1 cells expressing the XLR homologs. B. Flow cytometry analysis to characterize MHC class I expression on TC-1 cells expressing the XLR homologs compared to TC-1/no insert cells and in vitro T cell activation to determine the number of IFN-γ secreting E7-specific CD8+ T cells induced by TC-1 cells expressing the XLR homologs compared to TC-1/no insert cells. MFI; mean fluorescence intensity. C. Bar graph depicting percentage of apoptotic TC-1 cells expressing the XLR homologs compared to TC-1/no insert cells incubated with E7-specific CD8+ T cells at 1:1 ratio for 4 hours. D. Graphical representation of the tumor volume in mice challenged with TC-1/XLR, TC-1/SCP3 or TC-1/XMRtumor cells with or without adoptive transfer of E7-specific CD8+ T cells. Data shown are representative of three independent experiments.
We also determined if TC-1/SCP3 and TC-1/XMR cells demonstrate resistance to the apoptotic cell death induced by E7-specific cytotoxic T lymphocytes. As shown in Figure 5C, there was a significant reduction of apoptotic cell death in TC-1/XLR, TC-1/SCP3 and TC-1/XMR cells compared to TC-1/no insert cells while there was no significant change in MHC class I expression and ability to activate E7-specific CD8+ T cells in vitro (Figure 5B).
We further assessed whether these tumor cell lines are resistant to the therapeutic effect of adoptive transfer of E7-specific CD8+ T cells in vivo. As shown in Figure 5D, TC-1/no insert tumor challenged mice treated with E7-specific CD8+ T cells demonstrated significantly lower tumor volume over time compared to TC-1/XLR, TC-1/SCP3 or TC-1/XMR tumor-bearing mice treated with E7-specific CD8+ T cells. Taken together, our data suggest that TC-1/P0 cells transduced with homologous genes from the XLR family lead to activation of Akt, upregulation of antiapoptotic proteins and induction of the immune resistance phenotype.
Human 293Db cells transduced with hSCP3 are highly resistant to apoptotic cell death induced by CTL killing
Our data indicate that SCP3 plays an important role in the immune resistance phenotype in murine tumor cells. Thus, we characterized the expression of hSCP3 gene in cervical tissue from various stages of human cervical cancer by immunohistochemistry staining analysis using antibody against hSCP3. hSCP3 expression was observed in human cervical cancer cells but not in the surrounding stromal tissue (Supplementary Figure 5A). In general, the higher stages of cervical cancer demonstrated higher level of hSCP3 expression compared to lower stage cervical cancers or normal cervical tissue (Supplementary Figure 5B). Thus, our data indicate the upregulation of hSCP3 in human cervical cancers.
In order to determine if human tumor cells ectopically expressing SCP3 were resistant to CTL-induced apoptosis, we characterized the function of human 293Db cells retrovirally transduced with hSCP3. The expression of hSCP3 in the transduced 293Db cells is shown in Figure 6A. As shown in Figure 6B, 293Db/hSCP3 cells demonstrated significant expression of pAkt compared to the control 293Db/no insert cells. Furthermore, 293Db/hSCP3 cells showed a significant upregulation in key antiapoptotic proteins such as Bcl-XL and Bcl-2 compared to the control (Figure 6B). We also determined if the immune resistance of 293Db/hSCP3 cells is related to the apoptotic cell death induced by cytotoxic T lymphocyte (CTL) killing. As shown in Figure 6C and D, there was a significant reduction of apoptotic cell death in 293Db/hSCP3 cells compared to 293Db/no insert cells. Thus, our data indicate that human tumor cells transduced with hSCP3 are highly resistant to apoptotic cell death induced by cytotoxic T cells.
Figure 6. Characterization of the apoptotic cell death of 293Db/no insert and 293Db/SCP3 induced by E7 CTL killing in vitro.

A. Western blot analysis to characterize the expression of SCP3 in 293Db/no insert and 293Db/hSCP3 cells. B. Western blot analysis to characterize the expression of p-AKT, Bcl-2 and Bcl-xL in 293Db/no insert and 293Db/hSCP3 cells. β-actin was used as a loading control. C. Representative flow cytometry data demonstrating the percentage of apoptotic 293Db/no insert and 293Db/hSCP3 cells. 293Db/no insert and 293Db/hSCP3 cells were incubated with E7-specific CD8+ T cells at different E:T ratios (1:1 or 5:1) for 4 hours. After incubation, the percentage of apoptotic cells was analyzed using intracellular flow cytometry analysis for active caspase-3 expression. D. Graphical representation of the percentage of apoptotic cells among 293Db/no insert and 293Db/hSCP3 cells at different E:T ratios.
Discussion
In the current study, we identified XLR as a candidate molecule that is overexpressed in immune resistant TC-1 P3 (A17) tumors compared to P0 tumors. We found that the expression of XLR in TC-1/P0 cells could induce an immune resistance phenotype. Thus, we have identified a novel immune resistance mechanism through characterization of the overexpressed protein in immune resistant tumors by microarray analysis.
We report that XLR and its homologs render tumor cells resistant to apoptosis induced by CTL killing possibly through the activation of Akt, leading to the upregulation of antiapoptotic proteins, thus resulting in the immune resistant phenotype. The Akt dependent resistance to CTL killing was also observed in other tumor cell line models including EL4 and LLC cell lines (data not shown). This is consistent with our previous study wherein we demonstrated activation of PI3K/Akt pathway represents a new mechanism of immune escape (5). XLR has been shown to be a transcriptional regulatory protein. It is conceivable that overexpression of XLR in tumor cells may result in modulation of proteins important for the Akt activation pathway, such as phosphoinositide 3-kinase (PI3K) and PI3K-dependent kinase 1 (PDK1) (for reviews see (21, 22)). However, there was no difference was observed in expression level of phosphatase and tensin homologue (PTEN), which have been known as a central negative regulator of PI3K/AKT signal transduction cascade, in TC-1/XLR and TC-1/no insert cells. Interestingly, we observed that rapamycin treatment led to reverse the immune-resistance to CTL killing in TC-1/XLR cells, which had increase of phophorylated mammalian target of rapamycin (mTOR) compared to TC-1/no insert (unpublished data). These data suggest that mTOR could be one of downstream signaling pathways which are activated by XLR-mediated AKT activation. Thus, it will be interesting to explore the mTOR could form a positive feedback loop of AKT signaling provoked by ectopic expression of XLR. However, we do not exclude other possibility that XLR and its homolog may interact with and thus, modulate molecules that are associated with Akt signaling.
In our study, we observed that several cervical cancers demonstrate significant expression of the XLR homolog, hSCP3. The murine XLR gene does not have a human counterpart, but the murine SCP3 gene belong to the XLR family and has 62% homology with XLR (See Supplementary Table 3) (18). The human counterpart of SCP3 (hSCP3) shares 63% homology with XLR. We also demonstrated that human tumor cells transduced with hSCP3 could lead to activation of Akt, upregulation of antiapoptotic proteins and induction of the immune resistance phenotype. Thus, it will be important to further determine if overexpression of hSCP3 is a common phenomenon in advanced stage cancers. This could potentially serve as an important mechanism for tumor immune evasion.
In conclusion, this is the first report, to our knowledge, to show that XLR and its human homolog, typical cancer testis antigens, are expressed in immune resistant mouse tumor cells and human cervical cancer cells, respectively and play a role in the resistance of tumor cells to CTL killing. On the basis of these findings, further expression profile studies are planned in clinical specimens from patients with HPV-16 associated high-grade cervical intraepithelial neoplasia (CIN) lesions who were vaccinated with an HPV-16 E7 therapeutic DNA vaccine but failed to show lesion regression. Our study has important implications for future development of a novel strategy for enhancing cancer immunotherapy against immune-resistant tumor cells.
Supplementary Material
Acknowledgements
This work was supported by the NCI SPORE P50 CA098252, P50 CA96784-06, 1RO1 CA114425-01 in the United States, NRF grant funded by MEST (R11-2005-017-03003-0), KOSEF grant funded by MEST (2009-0076972), a grant of National R&D Program for Cancer Control (070355) and a grant of the Korea Healthcare technology R&D Project (A062260), Ministry of Health & Welfare, in Republic of Korea.
References
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin KY, Guarnieri FG, Staveley-O’Carroll KF, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21–6. [PubMed] [Google Scholar]
- 3.Wu TC. The role of vascular cell adhesion molecule-1 in tumor immune evasion. Cancer Res. 2007;67:6003–6. doi: 10.1158/0008-5472.CAN-07-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu TC, Guarnieri FG, Staveley-O’Carroll KF, et al. Engineering an intracellular pathway for major histocompatibility complex class II presentation of antigens. Proc Natl Acad Sci U S A. 1995;92:11671–5. doi: 10.1073/pnas.92.25.11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noh KH, Kang TH, Kim JH, et al. Activation of Akt as a Mechanism for Tumor Immune Evasion. Mol Ther. 2008;17:439–47. doi: 10.1038/mt.2008.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin KY, Lu D, Hung CF, et al. Ectopic expression of vascular cell adhesion molecule-1 as a new mechanism for tumor immune evasion. Cancer Res. 2007;67:1832–41. doi: 10.1158/0008-5472.CAN-06-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 8.Kendziorski CM, Newton MA, Lan H, Gould MN. On parametric empirical Bayes methods for comparing multiple groups using replicated gene expression profiles. Stat Med. 2003;22:3899–914. doi: 10.1002/sim.1548. [DOI] [PubMed] [Google Scholar]
- 9.Newton MA, Kendziorski CM, Richmond CS, Blattner FR, Tsui KW. On differential variability of expression ratios: improving statistical inference about gene expression changes from microarray data. J Comput Biol. 2001;8:37–52. doi: 10.1089/106652701300099074. [DOI] [PubMed] [Google Scholar]
- 10.Bloom MB, Perry-Lalley D, Robbins PF, et al. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J Exp Med. 1997;185:453–9. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hung CF, Cheng WF, He L, et al. Enhancing major histocompatibility complex class I antigen presentation by targeting antigen to centrosomes. Cancer Res. 2003;63:2393–8. [PubMed] [Google Scholar]
- 12.Cheng WF, Hung CF, Chai CY, et al. Tumor-specific immunity and antiangiogenesis generated by a DNA vaccine encoding calreticulin linked to a tumor antigen. J Clin Invest. 2001;108:669–78. doi: 10.1172/JCI12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen CH, Wang TL, Hung CF, et al. Enhancement of DNA vaccine potency by linkage of antigen gene to an HSP70 gene. Cancer Res. 2000;60:1035–42. [PubMed] [Google Scholar]
- 14.Hung CF, Cheng WF, Hsu KF, et al. Cancer immunotherapy using a DNA vaccine encoding the translocation domain of a bacterial toxin linked to a tumor antigen. Cancer Res. 2001;61:3698–703. [PubMed] [Google Scholar]
- 15.Han HD, Song CK, Park YS, et al. A chitosan hydrogel-based cancer drug delivery system exhibits synergistic antitumor effects by combining with a vaccinia viral vaccine. Int J Pharm. 2008;350:27–34. doi: 10.1016/j.ijpharm.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Yang L, Dan HC, Sun M, et al. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64:4394–9. doi: 10.1158/0008-5472.CAN-04-0343. [DOI] [PubMed] [Google Scholar]
- 17.Calenda A, Allenet B, Escalier D, Bach JF, Garchon HJ. The meiosis-specific Xmr gene product is homologous to the lymphocyte Xlr protein and is a component of the XY body. Embo J. 1994;13:100–9. doi: 10.1002/j.1460-2075.1994.tb06239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lammers JH, Offenberg HH, van Aalderen M, Vink AC, Dietrich AJ, Heyting C. The gene encoding a major component of the lateral elements of synaptonemal complexes of the rat is related to X-linked lymphocyte-regulated genes. Mol Cell Biol. 1994;14:1137–46. doi: 10.1128/mcb.14.2.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garchon HJ. The Xlr (X-linked lymphocyte regulated) gene family (a candidate locus for an X-linked primary immune deficiency) Immunodefic Rev. 1991;2:283–302. [PubMed] [Google Scholar]
- 20.Siegel JN, Turner CA, Klinman DM, et al. Sequence analysis and expression of an X-linked, lymphocyte-regulated gene family (XLR) J Exp Med. 1987;166:1702–15. doi: 10.1084/jem.166.6.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27:6473–88. doi: 10.1038/onc.2008.313. [DOI] [PubMed] [Google Scholar]
- 22.Du K, Tsichlis PN. Regulation of the Akt kinase by interacting proteins. Oncogene. 2005;24:7401–9. doi: 10.1038/sj.onc.1209099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.