

Abstract
Phase I of the hypoxic pulmonary vasoconstriction (HPV) response begins upon transition to hypoxia and involves an increase in cytosolic calcium ([Ca2+]i). Phase II develops during prolonged hypoxia and involves increases in constriction without further increases in [Ca2+]i, suggesting an increase in Ca2+ sensitivity. Prolonged hypoxia activates RhoA and RhoA kinase, which may increase Ca2+ sensitivity, but the mechanism is unknown. We previously found that reactive oxygen species (ROS) trigger Phase I. We therefore asked whether ROS generation during prolonged hypoxia activates RhoA in PA smooth muscle cells (PASMCs) and endothelial cells (PAECs) during Phase II. By using a cytosolic redox sensor, RoGFP, we detected increased oxidant signaling in prolonged hypoxia in PASMCs (29.8 ± 1.3% to 39.8 ± 1.4%) and PAECs (25.9 ± 2.1% to 43.7.9 ± 3.5%), which was reversed on the return to normoxia and was attenuated with EUK-134 in both cell types. RhoA activity increased in PASMCs and PAECs during prolonged hypoxia (6.4 ± 1.2-fold and 5.8 ± 1.6-fold) and with exogenous H2O2 (4.1- and 2.3-fold, respectively). However, abrogation of the ROS signal in PASMCs or PAECs with EUK-134 or anoxia failed to attenuate the increased RhoA activity. Thus, the ROS signal is sustained during prolonged hypoxia in PASMCs and PAECs, and this is sufficient but not required for RhoA activation. Antioxid. Redox Signal. 12, 603–610.
Introduction
Initially described in 1946 by von Euler and colleagues (20), hypoxic pulmonary vasoconstriction (HPV) is a physiologic response to low alveolar oxygen tension that improves lung gas exchange by redistributing blood flow away from poorly ventilated areas. HPV is a biphasic response in which Phase I (acute hypoxia) begins within seconds of a hypoxic challenge, lasts for several minutes, and involves an initial transient constriction due to a hypoxia-induced increase in cytosolic calcium concentration. Phase II occurs during prolonged hypoxia and lasts for hours to days if the hypoxic challenge is maintained. Phase II also involves hypoxia-induced increases in [Ca2+]i, but progressive increases in contraction occur without further increases in [Ca2+]i, suggesting that calcium sensitivity is augmented.
Recent studies indicate that RhoA, a small GTPase, and its downstream effector, RhoA kinase, play an important role in calcium sensitization and sustained contraction of vascular smooth muscle cells (5, 15, 19). The RhoA switch operates by alternating between an active, GTP-bound state and an inactive, GDP-bound state. The GTP-bound RhoA state translocates to the plasma membrane, where it activates RhoA kinase. RhoA kinase appears to increase calcium sensitivity by inactivating myosin light chain phosphatase through phosphorylation of its myosin-binding subunit. Because vascular smooth muscle tone is regulated by the balance between myosin light-chain kinase and myosin light-chain phosphatase activity, inactivation of myosin light-chain phosphatase by RhoA kinase could augment pulmonary artery constriction without a significant increase in [Ca2+]i (14, 15, 19).
Prolonged hypoxia has been shown to activate RhoA and subsequently RhoA kinase activity (1, 13), but the mechanisms by which RhoA is activated during hypoxia are not established. Endogenous endothelin-1 (ET-1) and serotonin have been shown to contribute to sustained activation of RhoA kinase in chronically hypoxic hypertensive rat pulmonary arteries (8). In distal rat pulmonary arteries, evidence indicates that Src-family kinases maybe involved in the activation of RhoA kinase during hypoxia (10). Additionally, superoxide has been shown to mediate the ET-1–mediated increases in RhoA activity in pulmonary arteries from chronically hypoxic rats (9). However, the role of ROS production during prolonged hypoxia on the activation of RhoA activity has not been established.
Although ROS signaling during acute hypoxia has been the subject of numerous investigations, little investigation has been undertaken into the nature and action of the ROS signal during prolonged hypoxia. We previously showed that an increased ROS signal is required for Phase I of the HPV response (22–24). In this study, we investigate the nature of the ROS signal during prolonged hypoxia in pulmonary artery smooth muscle cells (PASMCs) and pulmonary artery endothelial cells (PAECs) by using RoGFP, a novel ratiometric protein sensor of redox dynamics. We hypothesize that a continued increase in ROS signaling during prolonged hypoxia subsequently activates RhoA, thereby initiating increases in Ca2+ sensitivity during Phase II HPV.
Materials and Methods
Pharmacologic agents
EUK-134, a chemical antioxidant, was obtained from Cayman Chemicals Co. (Ann Arbor, MI) and dissolved in DMSO (final concentration of DMSO,<0.1%) to make 1 M stock solution. EUK-134 is a salen-manganese complex that has been modified to increase its catalase activity while retaining superoxide dismutase (SOD) activity. dl-Dithiothreitol (DTT), tert-butyl hydroperoxide (tBH), thrombin, and bovine heparin were obtained from Sigma-Aldrich (St. Louis, MO). Stock solutions of DTT and tBH (1 M) were stored at −20°C. All stock solutions were thawed on the day of the experiment.
RoGFP redox sensor
RoGFP is a ratiometric protein sensor of thiol redox status. The RoGFP probe was created by the substitution of cysteines at surface-exposed residues of a green fluorescent protein at positions enabling disulfide bond formation and thus conferring redox sensitivity (3, 6). RoGFP exhibits two emission maxima whose relative amplitudes depend on the redox status of a pair of cysteine thiols, providing a ratiometric measurement of the redox status that is independent of protein concentration, photobleaching, and variations in excitation intensity. The RoGFP cDNA was generated from the eGFP-N1 expression vector (Invitrogen, Carlsbad, CA), and the resulting construct was used to generate a recombinant adenoviral vector to facilitate transduction of primary cells (ViraQuest, North Liberty, IA). PASMCs were infected with five plaque-forming units (PFU) per cell, whereas PAECs were infected with 10 PFU per cell. Experiments were carried out ∼36 h after initial infection to allow RoGFP expression.
RoGFP fluorescence images were obtained with a 40× 1.3 n.a. oil-immersion objective. RoGFP protein was excited at 400 and 450 nm, whereas fluorescence images were acquired at 517 nm by using a cooled CCD camera (Cascade 650, Photometrics, Tucson, AZ) controlled by Metamorph or Metafluor software (Molecular Devices, Downingtown, PA). RoGFP images at the two excitation wavelengths were divided to yield a ratiometric image, and responses of individual cells were monitored by defining regions of interest. Oxidation/reduction of the regulatory thiol groups in RoGFP is fully reversible, allowing calibration of the ratios obtained during the experiment. Calibration was performed at the end of the experiment by treatment with DTT (1 mM, to reduce dithiols fully) followed by tBH (1 mM, to oxidize thiols fully). Oxidation of the protein during the experiment was then determined from the following equation:
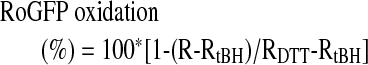 |
where R is the experimentally measured ratio, RtBH is the ratio obtained on complete oxidation by using tBH, and RDTT is the ratio obtained on complete reduction of the sensor by using DTT.
Pulmonary microvessel smooth muscle and endothelial cell isolation
All animal studies were approved by the Northwestern University Institutional Animal Care and Use Committee. PASMCs were isolated from adult Sprague–Dawley rats (Harlan Sprague Dawley, Indianapolis, IN) by using a modification of the method of Marshall et al. (11). Freshly excised rat heart and lungs were rinsed with PBS containing penicillin and streptomycin (1%). The right ventricle was cannulated, and the pulmonary vasculature was flushed with the PBS solution (30hairsp;ml). By use of the pulmonary artery cannula, growth medium 199 (M199, 30 ml) containing HEPES (25 mM) along with penicillin and streptomycin (1%) plus low-melting-point agarose (0.5%) and iron particles (0.5%) was flushed through the pulmonary vasculature. The iron particles were too large to pass through the capillaries; therefore, only the arteries were filled with the agarose and iron particles. The airways were filled via the trachea with M199 (15 ml) containing low-melting-point agarose (1%) without iron. The lungs were placed in cold PBS to cause the agarose to solidify. After 10 min, the lobes were dissected free and finely minced in a Petri dish. Lung fragments were then resuspended and washed (3 times) with PBS by use of a magnet to retain the iron-containing fragments. The iron-containing pieces were resuspended in M199 (25 ml) containing collagenase (80 U/ml) and incubated at 37°C for 30–60 min. To remove extravascular tissue, fragments were first drawn through a 15-gauge needle and subsequently through an 18-gauge needle. The iron-containing fragments then were washed (3 times) with M199 containing FBS (5%) and drained. The resulting fragments were placed in a T-75 flask and resuspended in M199 containing FBS (5%). The T-75 flasks were incubated at 37°C with CO2 (5%) in air for 4 or 5 days, during which time the myocytes were observed to migrate and adhere.
After 4 or 5 days, the media and iron-containing particles were transferred to a new flask containing fresh media. The adherent myocytes continued to propagate until the cells were 50% confluent. Cells were confirmed to be pulmonary artery myocytes through fluorescent staining for smooth muscle actin.
PAECs were isolated in a fashion similar to the PASMC isolation. After lung isolation from adult Sprague–Dawley rats (described earlier), lobes were finely chopped in a Petri dish, and lung fragments were resuspended and washed (3 times) with PBS by using a magnet to retain the iron-containing fragments. The iron-containing pieces were resuspended in F-12 Kaighn's modification (25 ml) containing collagenase (80 U/ml) and incubated at 37°C for 30–60 min. To remove extravascular tissue, fragments were first drawn through a 15-gauge needle and subsequently through an 18-gauge needle. The iron-containing fragments then were washed (3 times) with F-12 Kaighn's modification containing FBS (5%) and drained. The resulting fragments were placed in a T-75 flask and resuspended in F-12 Kaighn's modification containing FBS (5%), 50 mg heparin, and 15 mg endothelial cell growth supplement (Calbiochem, Gibbstown, NJ). The T-75 flasks were incubated at 37°C with CO2 (5%) for 4 or 5 days, during which time the endothelial cells (distinguished from smooth muscle cells by morphology) were observed to migrate and adhere. After 4 or 5 days, the clusters of endothelial cells were removed by using a 1,000-μl pipette and placed in a new T-75 flask with new media. The adherent endothelial cells continued to propagate. Cells were confirmed to be PAECs through immunofluorescence staining for N-cadherin and by their distinct morphology.
Measurement of ROS signal during prolonged hypoxia
Primary culture rat PASMCs (passage 2–8) and PAECs (passage 2–4) were seeded on collagen-coated glass coverslips (Fisher 25CIR-2, Fisher Scientific, Pittsburgh, PA) at a density of 150,000 cells per coverslip. Cells were given 24 h to adhere and then were infected with recombinant adenovirus expressing cytosolic targeted RoGFP. The following day, coverslips were placed in an environmentally controlled incubator (Coy Laboratory Products, Inc., Grass Lake, MI) and exposed to hypoxia (1.5% O2, 5% CO2, balance N2) for 24 h. After 24 h of hypoxia, the coverslips containing cells were transferred to a flow-through chamber that permitted study of the cells on an inverted microscope under controlled O2 concentrations. The flow-through chamber was then mounted on a heated platform of an inverted microscope (Nikon, Inc., Melville, NY), and cells were superfused with a buffered salt solution (NaCl 117 mM, KCl 4.0 mM, NaHCO3 22 mM, MgSO4 0.76 mM, NaH2PO4 1.0 mM, CaCl2 1.21 mM, and glucose 5.6 mM) that was previously equilibrated with a hypoxic gas mixture (1.5% O2, 5% CO2, balance N2) in a water-jacketed glass equilibration column (Radnotti Glass Technology, Inc., Monrovia, CA) mounted above the microscope stage. Cells were superfused with buffered solutions at 1 ml/min, as ratiometric images of RoGFP were obtained to assess oxidant stress. After ∼10 min of hypoxic superfusion, the gas bubbling the perfusate was changed to normoxia (21% O2, 5% CO2, balance N2) as the data collection continued. Changes in the redox status of the cell with reversal of hypoxia were then evaluated for 20 min. Finally, the RoGFP sensor was calibrated by reducing the protein with DTT and then oxidizing it with tBH.
In some experiments, ROS were scavenged by treating PASMCs with EUK-134 (7 μM) during hypoxia 1 h before initiation of RoGFP redox measurements. This concentration of EUK-134 was determined from initial studies exploring the dose dependence of its effects. In PAECs, EUK-134 (10 μM, also determined from dose-dependence studies) was added during the hypoxia treatment while the RoGFP oxidation status was being measured. Cells were then returned to normoxia as RoGFP images were acquired in the continued presence of EUK-134.
RhoA activity
Primary culture rat PASMCs (from passage 2 through 8) and PAECs (from passage 2 through 4) were grown to ∼80% confluence on 10-cm tissue-culture dishes (∼750,000 cells per dish). Cells were serum starved for 24 h before the initiation of the experiment. Cells were placed either in the hypoxia incubator (1.5% O2, 5% CO2, balance N2) or in an anoxic chamber using an O2-scavenging system (Anoxic Bugbox, 0% O2, 5% CO2, 5% H2, balance N2; Ruskinn Technology Ltd., Bridgend, U.K.) for 24 h. The anoxic chamber was used to deprive the cells completely of O2, a necessary substrate for superoxide formation, to prevent any ROS generation. Thrombin (100 U, administered 20 min before RhoA assay) served as a positive control for RhoA activity. Hydrogen peroxide (H2O2, 25 μM for PASMCs, 50 μM for PAECs, administered as a bolus every 15 min for 1 h) served as acute oxidant treatment. In experiments to attenuate RhoA activity with antioxidant EUK-134, cells were treated with EUK-134 (7 μM every 3 or 6 h for 24 h before RhoA assay). Cell medium was replaced during each EUK-134 treatment with new media previously equilibrated to the experimental oxygen environment.
RhoA activity was determined by using the RhoA Activation Assay Biochem Kit (Cytoskeleton Inc., Denver, Co). The assay uses the Rho-binding domain (RBD) of the Rho effector protein, Rhotekin. The RBD motif has been shown to bind specifically to the active GTP-bound form of RhoA. The Rhotekin-RBD protein supplied in the kit contains Rhotekin residues 7 to 89 and is in the form of a GST fusion protein, which allows “pull-down” of Rhotekin-RBD/Rho-GTP complex with glutathione affinity beads. Active RhoA is normalized to the total RhoA by Western blot analysis (RhoA monoclonal antibody, Cytoskeleton, anti-mouse secondary antibody; Cell Signaling Technology, Danvers, MA). For cells under hypoxic or anoxic conditions, collection of cell lysates was performed in the respective environmental boxes without exposure to alterations in O2 concentration.
Statistical analysis
Analysis of variance (ANOVA) was used to establish statistical significance, and the Newman–Keuls or Student's t test post hoc tests were used to explore specific differences. To control for differences in the hypoxic responses of cultured pulmonary smooth muscle and endothelial cells, experimental studies and control experiments were always carried out on the same day. Significant differences were established at p < 0.05. Data were analyzed by using the mean values of all the cells during a given experiment. Data reported are mean values ± standard error of mean.
Results
Prolonged hypoxia increases cytosolic oxidation in pulmonary artery smooth muscle and endothelial cells
To assess the effects of prolonged hypoxia (1.5% O2 for 24 h) on cellular redox status, PASMCs and PAECs were transduced with a recombinant adenovirus expressing RoGFP in the cytosol. Incubation under hypoxia significantly increased RoGFP oxidation in PASMCs compared with normoxic controls (from 29.8 ± 1.3% to 39.8 ± 1.4%), indicating a shift of the redox status of the cytosol to a more oxidized state (Fig. 1). When the same hypoxic PASMCs were then superfused with normoxic solution (21% O2), a rapid and significant reversal of the hypoxia-induced RoGFP oxidation was observed, with the cells returning to near-normoxic levels (30.5 ± 1.2% oxidized) within 20 min. The increased RoGFP oxidation seen during prolonged hypoxia in PASMCs was significantly attenuated by the antioxidant EUK-134 (7 μM; 23.4 ± 1.2%, Fig. 2), indicating that ROS were involved in the hypoxia-induced oxidation.
FIG. 1.

RoGFP oxidation in PASMCs exposed to 24-h hypoxia (n = 92) compared with normoxic controls (n = 92). On restoration of normoxia (n = 92), the hypoxia-induced oxidation of RoGFP was reversed within 20 min. Values are expressed as mean ± SEM. *p < 0.05 relative to normoxia. +p < 0.05 relative to hypoxia.
FIG. 2.

Effects of the antioxidant EUK-134 on RoGFP oxidation in response to hypoxia (24 h) (n = 10) and after return to normoxia (20 min) (n = 32) compared with normoxia (n = 10) in PASMCs. Values are expressed as mean ± SEM. *p < 0.05 relative to hypoxia.
A similar pattern of redox response to prolonged hypoxia was observed in PAECs. After 24 h of hypoxic incubation, a significant increase in RoGFP oxidation was detected compared with normoxic controls (from 25.9 ± 2.1% to 43.7 ± 3.5% oxidized), indicating a shift of the redox status of the PAECs cytosol to a more-oxidized state (Fig. 3). When the same PAECs exposed to prolonged hypoxia were then superfused with normoxic solution, a significant reversal of the hypoxia-induced increase in RoGFP oxidation to near-normoxic levels was observed (27.8 ± 3.3% oxidized) over a 20-min period. The antioxidant EUK-134 (10 μM) tended to attenuate the hypoxia-induced increase RoGFP oxidation in PAECs (35.9 ± 3.0%; Fig. 4), but this did not reach statistical significance. Thus, despite the higher dose, EUK-134 was less effective in attenuating the hypoxia-induced oxidant stress in PAECs compared with PASMCs.
FIG. 3.

RoGFP oxidation in PAECs exposed to 24 h of hypoxia (n = 35) compared with normoxic controls (n = 43). On restoration of normoxia (n = 35), the hypoxia-induced oxidation of RoGFP was reversed within 20 min. Values are expressed as mean ± SEM. *p < 0.05 relative to normoxia.
FIG. 4.

Effects of the antioxidant EUK-134 on RoGFP oxidation in response to hypoxia (24 h) (n = 32) and after return to normoxia (20 min) (n = 32) compared with normoxia (n = 17) in PAECs. Values are expressed as mean ± SEM. *p < 0.05 relative to normoxia. +p < 0.05 relative to hypoxia.
Prolonged hypoxia and acute exposure to exogenous oxidants activate RhoA in PASMCs and PAECs
We then assessed the effects of prolonged hypoxia (1.5% O2 for 24 h) and exogenous oxidants on RhoA activity in PASMCs. After 24 h of hypoxia, a significant increase in RhoA activity was detected compared with normoxic controls (6.4 ± 1.2-fold increase over normoxia; Fig. 5). Exogenous oxidant treatment (25 μM H2O2 every 15 min for 1 h) also elicited a significant increase in RhoA activity (4.1 ± 0.9-fold over normoxia), indicating that ROS are capable of inducing RhoA activation in PASMCs.
FIG. 5.

RhoA activity in normoxic PASMCs (n = 14) compared with PASMCs exposed to 24 h of hypoxia (n = 19), or normoxic with acute exogenous ROS treatment (25 μM H2O2 every 15 min for 1 h) (n = 17). Treatment with the antioxidant EUK-134 (n = 16) or with complete anoxia (n = 7) did not significantly attenuate RhoA activity. Values are expressed as mean ± SEM. *p < 0.05 relative to normoxia.
In PAECs, after 24 h of hypoxia, a significant increase in RhoA activity was detected compared with normoxic controls (5.8 ± 1.6-fold over normoxia; Fig. 6). Short-term treatment with exogenous oxidants (50 μM H2O2 every 15 min for 1 h), also tended to increase RhoA activity (2.3-fold increase over normoxia), indicating that ROS are capable of activating RhoA in PAECs. Finally, thrombin treatment induced a significant increase in RhoA activation in PAECs.
FIG. 6.

RhoA activity in PAECs exposed to 24 h of normoxia (n = 7), 24 h of hypoxia (n = 5), or with acute exogenous ROS treatment (H2O2, 50 μM, every 15 min for 1 h) (n = 5) or exogenous thrombin (n = 6). Values are expressed as mean ± SEM. *p < 0.05 relative to normoxia.
Abrogation of ROS signaling does not attenuate RhoA activity in PASMCs or PAECs
To assess whether an ROS signal is required for RhoA activation, PASMCs were incubated under prolonged hypoxia (1.5% O2 for 24 h) while treated with antioxidant EUK-134 (7 μM every 3 or 6 h for 24 h). The hypoxia-induced increase in RhoA activity was not significantly attenuated with antioxidant treatment compared with hypoxic controls (4 ± 0.9-fold increase over normoxia; Fig. 5). Although EUK-134 effectively decreased the hypoxia-induced RoGFP oxidation response in PASMCs (Fig. 2), it was conceivable to us that ROS could still be involved in the hypoxia-induced activation of RhoA. To prevent ROS generation more completely, studies were repeated under conditions of complete anoxia, in which the absence of O2 would abolish all superoxide generation. However, preventing the ROS signal by incubating PASMCs in an anoxic chamber did not prevent the activation of RhoA (6.9 ± 2.6-fold increase over normoxia). These findings indicate that the ROS signal is sufficient but not required for RhoA activation in hypoxic PASMCs.
To assess the requirement of ROS for RhoA activation in PAECs, cells were incubated overnight under anoxic conditions, and cell lysates were collected for analysis of RhoA activity. As shown in Fig. 7, anoxia triggered a significant increase in RhoA activity, yet ROS production under those conditions could not have occurred. These findings indicate that ROS signals are not required for the activation of RhoA under low-oxygen conditions, although ROS are sufficient to trigger activation under normoxia (Fig. 6).
FIG. 7.

RhoA activity in PAECs exposed to 24 h of anoxia, in comparison to normoxic controls (n = 4 each). Western blot data of activated and total RhoA for each experiment also are shown. Values are expressed as mean ± SEM. *p < 0.05 relative to normoxia.
Discussion
This study extends prior work demonstrating that hypoxia increases oxidant signaling in the cytosol of cultured PASMCs. By using a ratiometric redox protein sensor, RoGFP, we show a continued cytosolic oxidative shift during prolonged hypoxia. This hypoxia-induced oxidation was reversed on restoration of normoxic conditions. Additionally, we showed that the antioxidant EUK-134 attenuated the hypoxia-induced increase in cytosolic oxidation. EUK-134, a catalase and SOD mimetic (18), acts by converting H2O2 to water and O2 and superoxide to H2O2 and O2, indicating that the hypoxia-induced oxidation was mediated by superoxide or H2O2 or both. This study therefore extends our previous work (22–24) by showing that the ROS signal generated during the Phase I component of HPV is not transient, but rather is sustained over a 24-h period.
Yang et al. (27) also detected an increase in ROS during hypoxia in porcine PAECs by using dihydrodichlorofluorescein (DCF) to measure ROS. However, the validity of DCF responses has been challenged because this compound lacks specificity and it requires peroxidase activity (7). The present study used a ratiometric redox sensor that is reversible and can therefore be calibrated, thereby eliminating problems arising from nonuniform cell uptake, autooxidation, and peroxidase-dependent responses associated with DCF (17). By using RoGFP, we also detected increases in oxidation in PAECs during prolonged hypoxia. In both cell types, a dynamic reversal of the hypoxia-induced oxidation was observed on returning to normoxic conditions, indicating that prolonged hypoxia must be associated with a continuous increase in ROS generation, lasting at least 24 h.
The duration of the increased ROS signal in hypoxia has been questioned by Mehta et al. (12), who reported finding a decreased ROS signal in human cultured PASMCs exposed to hypoxia for longer than 1 h. However, we find a continued hypoxia-induced increase in ROS signaling in PASMCs and PAECs during 24 h of hypoxia. In the studies by Mehta et al. (12), DCF, dihydroethidium, and Amplex Red were used for ROS measurement. Those tools all possess problems with lack of specificity or organelle accumulation, which may explain the discrepant results.
Chronic hypoxia has been shown to activate RhoA in PASMCs (1, 13). Jerrnigan et al. (9) demonstrated that RhoA/RhoA kinase signaling in response to ET-1 is ROS mediated in small, endothelium-disrupted pulmonary arteries isolated from chronically hypoxic rats (hypobaric chamber at 380 mm Hg for 4 weeks) with pulmonary hypertension. RhoA activity in those vessels was attenuated by the superoxide-scavenger tiron, implicating superoxide as a mediator of ET-1–mediated RhoA activation. Knock et al. (10) found that Rho kinase activation (as assessed by ROCK-2 translocation) in rat pulmonary arteries could be initiated by LY83583, a superoxide anion generator. Superoxide scavengers inhibited that response, but brief exposure to H2O2 (30 μM) did not elicit a significant increase in Rho kinase activation, leading them to conclude that RhoA is activated selectively by superoxide but not by H2O2.
Our data show that hypoxia activates RhoA, but our results are not consistent with the finding by Knock et al. (10) that H2O2 is not capable of activating Rho kinase. We found that PASMCs treated with H2O2 can elicit a significant increase in RhoA activity compared with controls. One possible explanation for this discrepancy relates to the methods they used to administer the H2O2. This molecule has a short half-life and is metabolized very quickly when administered to cells in the presence of fetal bovine serum (unpublished observation), which contains significant catalase activity. We treated PASMCs in serum-free media with 25 μM H2O2 every 15 min for 1 h, and then harvested cells for RhoA activity analysis. This protocol yielded significant increases in RhoA activity in our study, thus indicating that H2O2 is sufficient to activate RhoA.
Given the previous evidence that superoxide can activate ROCK-2 translocation, and in view of our own finding that H2O2 treatment is sufficient to activate RhoA, we sought to block RhoA activation by using an antioxidant that scavenges both H2O2 and superoxide. EUK-134 was chosen because it possesses both superoxide dismutase and catalase activities and has been reported to inhibit oxidant-mediated cell signaling (18). Nevertheless, at concentrations that did not produce toxicity, treatment with this compound did not abolish hypoxia-induced oxidation of RoGFP, nor did it prevent the hypoxia-induced RhoA activation. To provide a more definitive test of the hypothesis, we therefore used complete anoxia to prevent ROS formation by removing molecular oxygen as a potential substrate for superoxide generation. This approach was previously shown to abolish ROS-mediated signaling in hypoxia (2), but this, too, failed to prevent the activation of RhoA during prolonged incubation. These findings indicate that ROS are sufficient, but not required, for RhoA activation in PASMCs and PAECs. One possibility is that AMP-activated protein kinase (AMPK), which becomes activated during hypoxia (4), may provide a redundant mechanism leading to phosphorylation of Rho kinase. However, the ability of AMPK to phosphorylate RhoA has not been established.
Although RhoA activation in PASMCs has been implicated in calcium sensitization during prolonged hypoxia, the consequences of hypoxia-induced RhoA activation in PAEC have not been described. Our studies indicate that the increase in ROS signaling seen in PASMCs during prolonged hypoxia also occurs in PAECs, and that hypoxia and ROS also activate RhoA in those cells. It therefore appears that oxidant signals and RhoA activation contribute to the hypoxic response in the pulmonary circulation through multiple pathways in both smooth muscle and endothelial cells.
The present study adds to the mounting body of evidence linking increases in ROS signaling to the activation of hypoxic pulmonary vasoconstriction (21). Our previous work implicates mitochondria as the source of the immediate increase in ROS signaling during hypoxia (25), but other evidence also points to the involvement of ROS derived from p47phox-dependent NADPH oxidase systems in mediating the increase in [Ca2+]i in pulmonary artery cells during hypoxia (26). Recent studies suggest that mitochondrial ROS signals in hypoxia may amplify ROS signaling in PASMCs through the activation of PKCɛ, leading to the engagement of NADPH oxidase in the pulmonary artery (16). Thus, early mitochondrial ROS signals may trigger the initial activation of PKCɛ, which then results in a feed-forward amplification of ROS signaling through the activation of NADPH oxidases. This ROS-mediated ROS signaling may then contribute to the sustained oxidant signaling we observed during prolonged hypoxia in the present study.
In summary, our study demonstrates that the ROS signaling induced during hypoxia in PASMCs and PAECs is sustained over a 24-h period. By using a novel redox-sensitive ratiometric sensor, our data support the hypothesis that increases in ROS signaling contribute to the HPV response in pulmonary vascular cells. Hypoxia and exogenous H2O2 trigger the activation of RhoA in PASMCs and PAECs, but the ROS signals are not required for that response. Further studies are required to elucidate alternative pathways for RhoA activation during hypoxia.
Abbreviations Used
- ANOVA
analyses of variance
- DCF
dihydrodichlorofluorescein
- DTT
dl-dithiothreitol
- FBS
fetal bovine serum
- PA
pulmonary artery
- PAECs
pulmonary artery endothelial cells
- PASMCs
pulmonary artery smooth muscle cells
- ROS
reactive oxygen species
- tBH
tert-butyl hydroperoxide
Acknowledgments
This study was supported by grants from the National Institutes of Health (HL35440 and HL079650). We thank Dr. N.S. Chandel for the use of his anoxia workstation.
Author Disclosure Statement
The authors attest that no commercial associations might create a conflict of interest in connection with this article.
References
- 1.Barman SA. Vasoconstrictor effect of endothelin-1 on hypertensive pulmonary arterial smooth muscle involves Rho-kinase and protein kinase C. Am J Physiol Lung Cell Mol Physiol. 2007;293:L472–L479. doi: 10.1152/ajplung.00101.2006. [DOI] [PubMed] [Google Scholar]
- 2.Brunelle JK. Bell EL. Quesada NM. Vercauteren K. Tiranti V. Zeviani M. Scarpulla RC. Chandel NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Dooley CT. Dore TM. Hanson GT. Jackson WC. Remington SJ. Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- 4.Evans AM. Hardie DG. Galione A. Peers C. Kumar P. Wyatt CN. AMP-activated protein kinase couples mitochondrial inhibition by hypoxia to cell-specific Ca2+ signalling mechanisms in oxygen-sensing cells. Novartis Found Symp. 2006;272:234–252. [PubMed] [Google Scholar]
- 5.Fukata Y. Amano M. Kaibuchi K. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol Sci. 2001;22:32–39. doi: 10.1016/s0165-6147(00)01596-0. [DOI] [PubMed] [Google Scholar]
- 6.Hanson GT. Aggeler R. Oglesbee D. Cannon M. Capaldi RA. Tsien RY. Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 7.Hockberger PE. Skimina TA. Centonze VE. Lavin C. Chu S. Dadras S. Reddy JK. White JG. Activation of flavin-containing oxidases underlies light-induced production of H2O2 in mammalian cells. Proc Natl Acad Sci U S A. 1999;96:6255–6260. doi: 10.1073/pnas.96.11.6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Homma N. Nagaoka T. Morio Y. Ota H. Gebb SA. Karoor V. McMurtry IF. Oka M. Endothelin-1 and serotonin are involved in activation of RhoA/Rho kinase signaling in the chronically hypoxic hypertensive rat pulmonary circulation. J Cardiovasc Pharmacol. 2007;50:697–702. doi: 10.1097/FJC.0b013e3181593774. [DOI] [PubMed] [Google Scholar]
- 9.Jernigan NL. Walker BR. Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2008;295:L515–L529. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knock GA. Snetkov VA. Shaifta Y. Connolly M. Drndarski S. Noah A. Pourmahram GE. Becker S. Aaronson PI. Ward JP. Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca(2+) sensitization. Free Radic Biol Med. 2009;46:633–642. doi: 10.1016/j.freeradbiomed.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marshall C. Mamary AJ. Verhoeven AJ. Marshall BE. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am J Resp Cell Mol Biol. 1996;15:633–644. doi: 10.1165/ajrcmb.15.5.8918370. [DOI] [PubMed] [Google Scholar]
- 12.Mehta JP. Campian JL. Guardiola J. Cabrera JA. Weir EK. Eaton JW. Generation of oxidants by hypoxic human pulmonary and coronary smooth-muscle cells. Chest. 2008;133:1410–1414. doi: 10.1378/chest.07-2984. [DOI] [PubMed] [Google Scholar]
- 13.Nagaoka T. Morio Y. Casanova N. Bauer N. Gebb S. McMurtry I. Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2004;287:L665–L672. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- 14.Oka M. Homma N. McMurtry IF. Rho kinase-mediated vasoconstriction in rat models of pulmonary hypertension. Methods Enzymol. 2008;439:191–204. doi: 10.1016/S0076-6879(07)00415-6. [DOI] [PubMed] [Google Scholar]
- 15.Pfitzer G. Invited review: regulation of myosin phosphorylation in smooth muscle. J Appl Physiol. 2001;91:497–503. doi: 10.1152/jappl.2001.91.1.497. [DOI] [PubMed] [Google Scholar]
- 16.Rathore R. Zheng YM. Niu CF. Liu QH. Korde A. Ho YS. Wang YX. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med. 2008;45:1223–1231. doi: 10.1016/j.freeradbiomed.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Remington SJ. Fluorescent proteins: maturation, photochemistry and photophysics. Curr Opin Struct Biol. 2006;16:714–721. doi: 10.1016/j.sbi.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Rong Y. Doctrow SR. Tocco G. Baudry M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc Natl Acad Sci U S A. 1999;96:9897–9902. doi: 10.1073/pnas.96.17.9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somlyo AP. Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 20.Von Euler US. Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand. 1946;12:301–320. [Google Scholar]
- 21.Ward JP. A twist in the tail: synergism between mitochondria and NADPH oxidase in the hypoxia-induced elevation of reactive oxygen species in pulmonary artery. Free Radic Biol Med. 2008;45:1220–1222. doi: 10.1016/j.freeradbiomed.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 22.Waypa GB. Chandel NS. Schumacker PT. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res. 2001;88:1259–1266. doi: 10.1161/hh1201.091960. [DOI] [PubMed] [Google Scholar]
- 23.Waypa GB. Guzy R. Mungai PT. Mack MM. Marks JD. Roe MW. Schumacker PT. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ Res. 2006;99:970–978. doi: 10.1161/01.RES.0000247068.75808.3f. [DOI] [PubMed] [Google Scholar]
- 24.Waypa GB. Marks JD. Mack MM. Boriboun C. Mungai PT. Schumacker PT. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ Res. 2002;91:719–726. doi: 10.1161/01.res.0000036751.04896.f1. [DOI] [PubMed] [Google Scholar]
- 25.Waypa GB. Schumacker PT. O(2) sensing in hypoxic pulmonary vasoconstriction: the mitochondrial door re-opens. Respir Physiol Neurobiol. 2002;132:81–91. doi: 10.1016/s1569-9048(02)00051-4. [DOI] [PubMed] [Google Scholar]
- 26.Weissmann N. Zeller S. Schafer RU. Turowski C. Ay M. Quanz K. Ghofrani HA. Schermuly RT. Fink L. Seeger W. Grimminger F. Impact of mitochondria and NADPH oxidases on acute and sustained hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol. 2006;34:505–513. doi: 10.1165/rcmb.2005-0337OC. [DOI] [PubMed] [Google Scholar]
- 27.Yang W. Block ER. Effect of hypoxia and reoxygenation on the formation and release of reactive oxygen species by porcine pulmonary artery endothelial cells. J Cell Physiol. 1995;164:414–423. doi: 10.1002/jcp.1041640222. [DOI] [PubMed] [Google Scholar]