

Abstract
Reactive oxygen species (ROS) have become recognized for their role as second messengers in a multitude of physiologic responses. Emerging evidence points to the importance of the NADPH oxidase family of ROS-producing enzymes in mediating redox-sensitive signal transduction. However, a clear paradox exists between the specificity required for signaling and the nature of ROS as both diffusible and highly reactive molecules. We seek to understand the targets and compartmentalization of the NADPH oxidase signaling to determine how NADPH oxidase–derived ROS fit into established signaling paradigms. Herein we review recent data that link cellular NADPH oxidase enzymes to ROS signaling, with a particular focus on the mechanism(s) involved in achieving signaling specificity. Antioxid. Redox Signal. 11, 2467–2480.
Introduction
The mammalian cell constantly receives signals from its surroundings, to which it must respond appropriately. For example, growth-factor signals are integrated with internal-state information to produce decisions on cell growth, differentiation, or proliferation (47, 88, 99). The mechanisms whereby cells coordinate this information have been the subject of study for more than a century.
In one paradigm of classic receptor-mediated signaling, ligand engagement is followed by the production of a diffusible second messenger that interacts with a target to effect the signal. This arrangement supports both signal conduction over space and signal amplification, because most second messengers are produced via an enzymatic process. Typically, second messengers are small, diffusible molecules that rapidly activate effector proteins (i.e., protein kinases, protein phosphatases, ion channels) within the cell through binding or chemical modification, thus promoting signal transduction. Well-studied second messengers include cyclic nucleotides (cAMP, cGMP), modified lipids (inositol phosphates, eicosanoids), and sequestered ions (Ca2+). Generally, second messengers are specific for their effector targets and are often restricted to their organelle(s) of generation.
The discovery of nitric oxide (•NO) as an intracellular signaling molecule posed a problem for the classic signaling paradigms. Unlike other signaling molecules, •NO was both freely diffusible across membranes and (comparatively) highly reactive. Whereas Ca2+ or cyclic nucleotides are relatively specific for their targets, •NO was able to react with a number of intracellular species at considerable distances from its site of synthesis. Thus, the discovery of •NO as a signaling molecule has drastically changed contemporary thinking with regard to the properties and behavior of second messengers.
The notion that an authentic second messenger may have promiscuous reactivity and broad diffusibility is in keeping with the realization that reactive oxygen species (ROS) such as superoxide (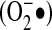 ) and hydrogen peroxide (H2O2) may also function as second messengers. Historically, ROS were simply considered a by-product of oxidative metabolism with no specific function. However, with the discovery that many signaling pathways are influenced by ROS (21, 106, 121), it became clear that ROS production must be considered within the context of how cells respond to environmental stimuli and process information. Because the reactivity of many ROS is potentially damaging to important cellular components, such as lipids, proteins, and nucleic acids, it follows that cells must have mechanisms for promoting ROS-signaling specificity and the limitation of collateral damage. The purpose of this review is to examine the literature concerning ROS-signaling specificity, with a particular focus on NADPH oxidase–derived ROS and their downstream targets.
) and hydrogen peroxide (H2O2) may also function as second messengers. Historically, ROS were simply considered a by-product of oxidative metabolism with no specific function. However, with the discovery that many signaling pathways are influenced by ROS (21, 106, 121), it became clear that ROS production must be considered within the context of how cells respond to environmental stimuli and process information. Because the reactivity of many ROS is potentially damaging to important cellular components, such as lipids, proteins, and nucleic acids, it follows that cells must have mechanisms for promoting ROS-signaling specificity and the limitation of collateral damage. The purpose of this review is to examine the literature concerning ROS-signaling specificity, with a particular focus on NADPH oxidase–derived ROS and their downstream targets.
NADPH Oxidases
The NADPH oxidases are a family of multimeric enzymes specifically designed to translocate electrons (from NADPH) across a membrane in a manner that results in the formation of ROS. Central to this function is a catalytic subunit (officially known as the Nox or Duox gene products) with a conserved general domain structure (Fig. 1) that consists of six transmembrane helices, binding sites for NADPH and FAD, and heme-coordinating histidine residues. Five isoforms (Nox 1 to 5) function strictly as oxidases, and two isoforms (Duox 1 and 2) contain peroxidase homology domains, although the functional capacity of these domains is not clear.
FIG. 1.

NADPH oxidase family members translocate electrons (from NADPH) across a membrane, which results in the formation of ROS (predominantly 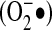 ). The seven NADPH oxidase family members (Nox 1 to 5 and Duox 1 and 2) share conserved features, including six transmembrane domains, where transmembrane domains III and V contain four heme-binding histidines. The carboxy terminus consists of an FAD-binding domain followed by an NADPH-binding domain. Nox5 includes an additional N-terminal calmodulin-like Ca2+-binding domain. Duox 1 and 2 also include peroxidase homology domains.
). The seven NADPH oxidase family members (Nox 1 to 5 and Duox 1 and 2) share conserved features, including six transmembrane domains, where transmembrane domains III and V contain four heme-binding histidines. The carboxy terminus consists of an FAD-binding domain followed by an NADPH-binding domain. Nox5 includes an additional N-terminal calmodulin-like Ca2+-binding domain. Duox 1 and 2 also include peroxidase homology domains.
For the last several years, multiple Nox genes have been described, with the gene products demonstrating wide-ranging tissue distribution, suggesting that ROS production may have broad implications for the cellular phenotype (23, 61) (Table 1). The literature regarding the Nox and Duox genes and the NADPH oxidase enzyme family is ambiguous. Many published reports refer to “NADPH oxidase” without regard to the specific gene product(s) involved. Moreover, the precise requirements for the catalytic activity of different Nox gene products (Table 1) is not yet complete. In this review, therefore, we identify specific NADPH oxidases by their Nox gene product. The term “NADPH oxidase” will be reserved for instances in which the specific gene product is not known.
Table 1.
NADPH Oxidase Family Members Exhibit a Wide Spectrum of Diversity Including Differences in Terms of Amino Acid Sequence Identity (here Compared to the Prototypical Nox2), Required Subunits for Activation, Intracellular Localization, Tissue Distribution, and Cellular Function
| Nox family member; % sequence identity to Nox2 | Necessary subunits | Localization | Tissue distribution | Known/suspected cellular functions |
|---|---|---|---|---|
| Nox 1; 60% | NOXO1, NOXA1, p22phox, Rac, p47phox*, p47phox* | cytoplasmic, nuclear, ER, caveolar | colon, smooth muscle, endothelium, uterus, placenta, prostate | maintenance of blood pressure, neurotoxic activation of microglia |
| Nox2 | p22phox, p47phox, p67phox, p40phox, Rac | cytoskeleton, synaptasomes, plasma membrane, endosomes | wide distribution; strong expression in phagocytes | host defense and inflammation |
| Nox 3; 56% | NOXO1, NOXA1, p22phox, Rac, p47phox, p67phox | ND | inner ear, fetal kidney, fetal spleen, skull bone, brain | development of inner ear vestibular system |
| Nox 4; 39% | p22phox | focal adhesions, ER, nucleus | kidney, endothelium, smooth muscle, hematopoietic stem cells, fibroblasts, neurons | angiogenesis, proliferation, differentiation, senescence, cell migration, oxygen sensor |
| Nox 5; 27% | none | ND | testis, spleen, lymph nodes, vascular smooth muscle, bone marrow, pancreas, placenta, ovary, uterus, stomach | Ca2+-dependent H+ channel, cell proliferation |
| Duox 1/2; 50% | p22phox, DUOXA1, DUOXA2 | apical membrane, ER | thyroid, airway epithelia, prostate | biosynthesis of thyroid hormones, protein cross-linking |
With regard to gene structure, the Nox/Duox family arose in concert with multicellular organization (60), suggesting that they contribute to the distinction among the functions of different cell types. Moreover, observations in primitive organisms suggest that NADPH oxidases may be important for stress responses. In the slime mold, Dictyostelium discoideum, nutrient stress induces individual amoebae to aggregate into a slug that can produce a spore-bearing fruiting body. Deletions that disable NADPH oxidase function interrupts the formation of a fruiting body (64). Thus, the NADPH oxidase family is evolutionary conserved and affects stress-induced behavior.
The first-identified and best-studied Nox isoform is known as Nox2 (originally termed gp91phox), which was identified in phagocytes for its role in antimicrobial ROS production (3). Nox 2 requires another integral membrane protein, p22phox for protein stabilization, but the activation of Nox2 is dependent on the translocation of several other cytosolic regulatory subunits, specifically p47phox, p67phox, p40phox, and Rac. As indicated in Table 1, NADPH oxidase family members have distinct regulatory patterns that do not strictly mirror those of Nox2. For example, Nox1 is found in cells that do not express p47 or p67. In such cells, Nox1 catalytic activity is supported by Nox organizer 1 (NoxO1), and Nox activator 1 (NoxA1). With regard to Nox4, constitutive activity and no requirement of cytosolic factors for activity seem to exist. Thus, it is not surprising that NADPH oxidase activity has been implicated in a host of varied cellular functions.
Understanding how NADPH oxidase–derived ROS are able to achieve target selectivity and specificity is paramount (21, 70). Conceptually, the target selectivity of these may exist at the level of the chemical characteristics of the oxidants or, perhaps, intracellular antioxidants. Alternatively, selectivity of NADPH oxidase–derived ROS could also involve their specific reactivity with putative targets in the cytosol or proteins. Finally, one could consider the intracellular localization or compartmentalization or both of NADPH oxidase catalytic activity as a means of specifying ROS signaling. In the remainder of this review, we focus our attention on mechanisms related to specificity of cell signaling.
Chemical Aspects of ROS Signaling Specificity
Reactive oxygen species
Although multiple ROS can be generated from the metabolism/excitation of oxygen, these species are not all equally reactive with respect to prospective targets. Many of these species have very short half-lives, leading to little relevance in terms of signaling. For example, the hydroxyl radical (•OH) is the most unstable ROS, with a half-life of 10−9 s (92), indicating its limited ability to transmit signals across any significant distance. Two major ROS that result from NADPH oxidase enzymes are superoxide (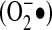 ) and hydrogen peroxide (H2O2). These two species have the most favorable chemical profiles as signaling molecules.
) and hydrogen peroxide (H2O2). These two species have the most favorable chemical profiles as signaling molecules.
Superoxide, a negatively charged molecule, has limited mobility across biologic membranes. Its diffusion is dependent on anion channels in the membrane. Thus, one aspect of its spatial specificity is related to its potential confinement within organelles such as mitochondria, endosomes, and, classically, phagosomes. This confinement is a limitation for superoxide to act as a paracrine mediator. In terms of chemical reactivity, superoxide can work both as an oxidant and as a reductant. It is the protonated form of superoxide (HO2•) that acts as an oxidant, but only a small fraction of superoxide is protonated at physiologic pH (103). Thus, the major activity of superoxide at physiologic pH is that of a reductant; it reduces iron and reacts with Fe-S centers. It is difficult to estimate the half-life of 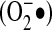 , as it is very dependent on the local concentrations of superoxide dismutase (SOD); however, it is clear that dismutation to H2O2 is its major mechanism of elimination.
, as it is very dependent on the local concentrations of superoxide dismutase (SOD); however, it is clear that dismutation to H2O2 is its major mechanism of elimination.
Peroxide is a two-electron oxidant that acts as an electrophile and can react with protein thiol moieties to produce a variety of sulfur oxidation states including disulfides, sulfenic (−SOH), sulfinic (−SO2H), or sulfonic (−SO3H) acid products (25, 30). This reactivity affords (mostly) reversible posttranslational modification of proteins that would be important for cell signaling. Moreover, the relatively longer half-life of H2O2 compared with other ROS in biologic systems affords better activity as an autocrine and paracrine messenger (21). An important question that also must be addressed is how to control the oxidizing activity of ROS and limit the potential damage to biologic molecules. One mechanism to limit excess oxidation is through cellular antioxidant defenses.
Antioxidants
The intracellular environment is generally thought to be replete with antioxidant activity. This activity is the result of both low-molecular-weight antioxidants and antioxidant proteins. Glutathione (GSH) is a tripeptide made of glutamate, cysteine (Cys), and glycine that is a ubiquitous intracellular antioxidant important for cellular protection against ROS, electrophiles, and xenobiotics. Glutathione also is important for maintaining ascorbate, itself a potent intracellular antioxidant, in a reduced state. With regard to proteins, the formation of a mixed GSH-protein disulfide (glutathionylation) has been shown to protect proteins against irreversible oxidation, as the mixed disulfide can be reduced by enzyme systems such as glutaredoxin (Grx). The intracellular space typically contains GSH in the range of 3 to 10 mM and ascorbate concentrations of 2 to 5 mM (31). Ascorbate is highly active against 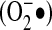 , and GSH will attenuate, but not abrogate, the reactivity of H2O2. Thus, any ROS production within the cellular cytosol is likely to encounter considerable low-molecular-weight antioxidant activity.
, and GSH will attenuate, but not abrogate, the reactivity of H2O2. Thus, any ROS production within the cellular cytosol is likely to encounter considerable low-molecular-weight antioxidant activity.
Along with direct-acting antioxidants, many antioxidant enzymes function to reduce oxidants. In mammals, three isoforms of superoxide dismutase are known: cytoplasmic SOD, which is a copper/zinc dismutase (SOD1); mitochondrial manganese SOD (SOD2); and extracellular Cu/Zn SOD (SOD3; ec-SOD). The primary function of the SODs is to catalyze the dismutation of superoxide to hydrogen peroxide, which can then either function in signaling reactions or be further reduced to water by catalase or a peroxidase. Based on the mechanism of enzyme action, it is generally believed that the principal product of the NADPH oxidase enzymes is superoxide. However, some NADPH oxidase isoforms release H2O2 (11), and a considerable amount of NADPH oxidase signaling has been linked to H2O2. The specific determinants of NADPH oxidase–derived 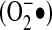 dismutation to H2O2 are not clear.
dismutation to H2O2 are not clear.
The glutathione peroxidases (GPxs) reduce peroxides by transferring electrons from GSH with the generation of oxidized glutathione (GSSG). Catalase is found primarily in the peroxisomes and efficiently converts H2O2 to water and oxygen, hence “quenching” signaling. However, it is likely that the main intracellular H2O2 scavengers are the peroxiredoxins. The peroxiredoxins (Prxs; see later) are a ubiquitous class of efficient H2O2 scavengers with much higher binding affinities than other H2O2 scavengers (116). Peroxiredoxins remove H2O2 to yield water and form an intermolecular disulfide bond, which then can be reduced by thioredoxin (Trx). Hence, Trx is not directly involved in removal of ROS, but indirectly through a supportive role to the Prxs. Oxidized Trx is then reduced by thioredoxin reductase by using NADPH as an electron donor. As discussed later, many of these antioxidant enzymes are located in close proximity to the NADPH oxidase enzymes, conceptually allowing management of the ROS response. The specific role(s) of antioxidant enzymes in NADPH oxidase signaling is(are) a relatively underdeveloped area, and many gaps exist in our knowledge.
Protein targets
Increasing evidence indicates that redox-dependent protein modification is an important mechanism in signal transduction that parallels, in part, the body of knowledge relating to classic phosphorylation. The availability and accessibility of protein targets for reaction with ROS is one mechanism for specificity in redox-sensitive signal transduction. In the following section, we discuss protein moieties typically subject to redox modification, as well as specific classes of proteins and protein complexes.
Protein Moieties Subject to Redox Modification
Iron-sulfide (Fe-S) centers
The charged molecule of 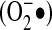 is attracted to Fe-S. These centers are involved in a variety of reactions because of their ability to maintain multiple oxidation states. One of their most highly studied reactions is their involvement in electron transfer, as in the mitochondrial electron-transport chain, which contains the biggest multi–Fe-S protein known, NADH dehydrogenase. However, Fe-S proteins are also involved in non–electron-transfer functions, such as substrate binding and catalysis, in which Fe-S clusters can lead to polarization of surrounding groups and hence serve as active sites of enzymes (12). One example of the effect
is attracted to Fe-S. These centers are involved in a variety of reactions because of their ability to maintain multiple oxidation states. One of their most highly studied reactions is their involvement in electron transfer, as in the mitochondrial electron-transport chain, which contains the biggest multi–Fe-S protein known, NADH dehydrogenase. However, Fe-S proteins are also involved in non–electron-transfer functions, such as substrate binding and catalysis, in which Fe-S clusters can lead to polarization of surrounding groups and hence serve as active sites of enzymes (12). One example of the effect 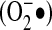 has on Fe-S centers is the protein aconitase, in which destabilization of the Fe-S cluster by superoxide inhibits enzyme activity, thereby limiting mitochondrial respiration (32, 33). The predilection of superoxide for Fe-S centers has been exploited by Escherichia coli in the form of the SoxR protein. This is a [2Fe-2S] protein that undergoes univalent oxidation of the Fe-S centers by superoxide (29). Oxidation of SoxR renders it able to bind to its target genes, known collectively as soxRS, that coordinate a complex stress response, including synthesis of proteins such as SOD (29). The SoxR protein is specific for one-electron oxidations, responding to superoxide and nitric oxide, but not to hydrogen peroxide or hydroxyl radical (34, 44). Although no orthologues have been described in higher organisms, multiple proteins contain Fe-S centers and may thus be susceptible to influence by superoxide.
has on Fe-S centers is the protein aconitase, in which destabilization of the Fe-S cluster by superoxide inhibits enzyme activity, thereby limiting mitochondrial respiration (32, 33). The predilection of superoxide for Fe-S centers has been exploited by Escherichia coli in the form of the SoxR protein. This is a [2Fe-2S] protein that undergoes univalent oxidation of the Fe-S centers by superoxide (29). Oxidation of SoxR renders it able to bind to its target genes, known collectively as soxRS, that coordinate a complex stress response, including synthesis of proteins such as SOD (29). The SoxR protein is specific for one-electron oxidations, responding to superoxide and nitric oxide, but not to hydrogen peroxide or hydroxyl radical (34, 44). Although no orthologues have been described in higher organisms, multiple proteins contain Fe-S centers and may thus be susceptible to influence by superoxide.
Cysteine residues
An attractive mechanism for the conversion of peroxide into cellular signals is through cysteine (Cys) oxidation. Almost all proteins contain Cys residues that are subject to oxidation. The inherent reactivity of these residues is due to the fact that they contain sulfur, which exists stably in multiple oxidation states, making it a versatile component in biologic systems. The most highly active and most reduced form of sulfur in biomolecules is the thiol (R-SH), present in Cys. Most protein Cys residues are protonated at physiologic pH, with a pKa of ∼8.5. The protonated thiol is not very reactive with ROS, which allows the basis of ROS specificity in signaling in which the only reactive Cys residues are those that are thiolate anions (RS−). The most reactive thiolates are characterized by pKa values in the range of 5.0 and are typically produced when the Cys residue is surrounded by basic amino acids (44, 72, 127). Cysteine thiolates are oxidized by ROS to a sulfenic acid (RSOH) intermediate, which can rapidly react with neighboring groups such as thiols [forming intra/intermolecular disulfides (RSSRs)], nitrogens [forming sulfenamides (RSNRs)], or GSH (forming S-glutathionylated mixed disulfides), all of which participate in altering protein function and signaling. However, further oxidation of sulfenic acid by ROS can lead to formation of sulfinic (RSO2H) and sulfonic (RSO3H) acid, which reactions are generally considered to be irreversible (Fig. 2) (98). The exception to this is the reactivation of some peroxiredoxins by sulfiredoxin (13, 51).
FIG. 2.

Protein cysteine (Cys) residues are fundamental in ROS signal transduction. (A) Protein Cys residues with the pKa lower than intracellular pH are readily deprotonated, leading to formation of the more-reactive thiolate anion (RS−). (B) ROS reaction with thiolate anions leads to formation of sulfenic acid (RSOH), which is highly reactive. (C) Sulfenic acid can form reversible modifications with surrounding thiols, nitrogens, or GSH to form disulfides, sulfenamides, or protein S-glutathionylation, leading to changes in protein function. (D) Reversible Cys modifications are reduced by antioxidant systems such as glutaredoxin (Grx) and thioredoxin (Trx) (E) Sulfenamides can also be further oxidized to sulfinic (RSO2H) or sulfonic (RSO3H) acid, which is generally considered irreversible.
Interconversions of methionine (Met) residues may serve a similar function, in which the sulfur in Met is present as a thioether (‱CH2‱S‱CH3) and can be oxidized to a sulfoxide. However, Met is inherently less reactive than the thiol moiety, and therefore, this review primarily focuses on the oxidation of Cys residues, which encompasses the majority of what we know about ROS regulation of signaling targets.
Direct Protein Targets
Protein tyrosine phosphatases
The best-documented targets for ROS are protein tyrosine phosphatases (PTPs), whose enzymatic activity is abolished by oxidation of a Cys residue in their active site. Protein tyrosine phosphatases are a superfamily of genes consisting of ∼100 members, which function to remove the phosphate group from proteins phosphorylated on tyrosine residues by protein tyrosine kinases (PTKs). Thus, one can promote tyrosine phosphorylation through either stimulation of the kinase or inhibition of the phosphatase. It is this latter part of the equation that is germane for redox regulation of tyrosine phosphorylation. All PTPs are characterized by an active-site motif that consists of Cys and arginine (Arg) separated by five residues (I/V-C-XX-G-X-X-R-S/T), where X is any amino acid (10). The proximity of the basic Arg residue creates a microenvironment amenable to thiolate formation (pKa =4.7‱5.4) that facilitates the Cys to function as a nucleophile and abstract a proton. In the presence of an electrophile such as H2O2, the active-site Cys residue is oxidized to a sulfenic acid intermediate followed by rapid intraprotein conversion to a cysteine sulfenyl-amide (Fig. 3). This chemistry produces an active-site conformational change that inhibits substrate binding (18, 30, 67, 98).
FIG. 3.

Protein tyrosine phosphatases (PTPs) are subject to oxidant-induced signaling. (A) In the presence of an electrophile such as H2O2, the active-site cysteine (R-S−) residue is oxidized to a sulfenic acid (R-SOH) intermediate. (B) This is followed by rapid intraprotein conversion to a cysteine sulfenyl-amide, which produces an active-site conformational change that inhibits substrate binding. (C) The activity of the PTPs is restored by antioxidant/antioxidant systems such as glutathione (GSH)/glutaredoxin (Grx).
The reversible oxidation of PTP cysteine residues lends itself to a tonic level of control, as outlined in Fig. 3. As ROS levels increase, a relative shift occurs in any local population of PTPs from the reduced to the oxidized form of the enzyme. This shift leaves the PTP unavailable for modulation of signaling and has the effect of accentuating the local activity of PTKs. In contrast, a reducing environment (such as a relative antioxidant abundance or lack of ROS) would tend to drive the population of PTPs to the reduced state, increasing enzyme activity and attenuating the activity of PTKs. Thus, the relative oxidizing tone, particularly in a localized environment, has the potential to modulate signaling tonically. This concept is in keeping with the notion that redox signaling tends to modulate pathways rather than to function as a strict binary (yes/no) proposition (52).
The proposed paradigm fits well with our understanding of NADPH oxidase enzymes and their impact on tyrosine phosphorylation and PTPs. The first indication of NADPH oxidase regulation of PTPs came from studies of the Nox2 in phagocytes, in which it was speculated that this Nox enzyme inactivated PTPs in the respiratory burst (128). Consistent with this notion, treatment of macrophages with the NADPH oxidase inhibitor DPI was associated with reduced ROS formation, increased PTP activity, and decreased PTK activation (128). The potential link between Nox2 and neutrophil PTP activity was refined by the studies demonstrating that TNF-α–mediated neutrophil activation resulted in a rapid distribution of NADPH oxidase components (Nox2, p22phox, p47phox, and p67phox) to the cytoskeleton, along with Src-family kinases (122). Similar experiments in adherent neutrophils from Nox2-incompentent patients demonstrated impaired activation and tyrosine phosphorylation of Src-family kinases (fgr and lyn) in the cytoskeletal fraction (121). Of interest, the lack of Nox2 did not completely abrogate fgr/lyn tyrosine phosphorylation, further supporting the notion that ROS tend to play a modulatory role in signaling.
Evidence indicating that NADPH oxidase regulates PTP activity also extends beyond phagocytes. For example, Nox4 is highly expressed in insulin-sensitive adipocytes (73), and insulin signaling is sensitive to PTP activity (41). This knowledge prompted Mahedev and colleagues (73) to investigate the relation between Nox4 and insulin signaling. They found that Nox4 was important in regulation of insulin-receptor signal transduction, as the Nox4-derived ROS produced by receptor-ligand engagement led to inactivation of PTP1B, enhancing downstream signal transduction. This paradigm was recently broadened to include IL-4 receptor activation that promoted downstream signaling through activation of PI3K and subsequent activation of Nox1 and Nox5L, which in turn inhibited PTP1B (101).
Other PTPs also have been implicated in NADPH oxidase–mediated regulation. In vascular smooth muscle cells, angiotensin II activation of Nox1 was shown to inhibit the Src homology protein tyrosine phosphatase-2 (PTP SHP-2), thus enhancing Akt activation. Nox4 also has been found to enhance growth factor–induced antiapoptotic action through inactivation of low-molecular-weight protein tyrosine phosphatase (LMW-PTP), thus promoting JAK2 activity and promoting survival in pancreatic cancer cells (66). In endothelial migration, the cytosolic component of the Nox2 complex, p47phox, was shown to be tethered to Hic5 (a scaffolding protein) by the small adaptor protein TRAF4, thus creating localized signaling that inactivates PTP-PEST, which also is found associated with Hic-5 (117).
Although PTP oxidation has been well documented, many gaps exist in our understanding of the mechanisms whereby this occurs and moreover how this oxidation allows specificity. As one can appreciate from the data outlined earlier, a number of ligands have been linked to activation of one or several NADPH oxidase isoforms. In addition, multiple PTPs have been identified as targets of NADPH oxidase activation. However, we do not know much about the molecular requirements that dictate signaling specificity. For example, PDGF-receptor ligand engagement is known to be specific for SHP-2 (79) inactivation, whereas blanket treatment of cultured cells with H2O2 leads to inactivation of multiple PTPs (79). Thus, the requirements for signaling specificity are not yet clear.
Recent observations from our group have begun to shed light on this issue in which endothelial Nox4 was shown to be localized to the endoplasmic reticulum (ER), and this finding was a key feature of Nox4 regulation of PTP1B, another ER-resident protein. Nox4-dependent oxidative modification of PTP1B required both proteins to be in the ER, as a mutant form of PTP1B located in the cytosol was no longer oxidized by Nox4. This was fundamentally important in the regulation of EGF signaling in which Nox4-mediated PTP1B oxidation was associated with reduced dephosphorylation of EGF receptor in proximity to the ER, resulting in accentuated EGFR-dependent signaling and cell proliferation (Fig. 4) (20).
FIG. 4.

Nox 4 mediates EGFR activity. (A)Vesiculation of internalized EGFR trafficks phosphorylated EGFR near the endoplasmic reticulum (ER). (B) Nox 4 is localized to the ER in close proximity to PTP1b, thus influencing PTP1b activity. (C) ROS emanating from ER Nox4 are then able to mediate EGFR signaling through deactivation of PTP1b, leading to increased EGFR phosphorylation and recycling of phosphorylated (active) receptor to the plasma membrane.
Protein tyrosine kinases
In addition to the aforementioned effects of NADPH oxidase–derived ROS on PTPs, evidence suggests that kinases themselves are downstream targets for ROS. The MAP kinases have been implicated in many redox-dependent signaling events; however, it is unclear whether they are direct targets of ROS or are solely activated through inactivation of upstream phosphatases or as downstream targets of protein tyrosine kinases (PTKs) (76). The PTKs themselves are potential direct targets of ROS in which ∼80% of known PTKs contain a conserved C-terminal CXXXXXXXMXXCW motif (where X is any residue), and ∼96% contain the MXXCW portion of the motif (83). These data suggest that PTKs should demonstrate direct redox regulation. UV light induces ROS production that facilitated the dimerization and activation of the receptor tyrosine kinase Ret (55, 56). This effect was due to the oxidation of C-terminal Cys residues that facilitated Ret dimerization and activation by autophosphorylation (55, 56). Given that similar Cys residues are conserved in other protein tyrosine kinases, such as Src, Abl, and Lck, one might speculate that these kinases also are subject to oxidation-induced activation. In this regard, the work of Giannoni and colleagues (37) is of particular interest. Those investigators found that cell adhesion and integrin ligand engagement was associated with cytoskeletal rearrangement and the oxidation of Src on cysteine residues at positions 245 and 487. Mutating these residues inhibited ROS-mediated Src activation resulting from integrin ligand engagement. This paradigm has also been specifically linked to Nox4 activation. Block and colleagues (14) found that angiotensin II treatment of mesangial cells resulted in Nox4 upregulation and ROS production, which resulted in the oxidation-mediated activation of Src, a critical step in angiotensin II–induced fibronectin expression (14).
These data linking Nox4 to Src oxidation are consistent with some of the information on intracellular Nox4 localization. In smooth muscle cells, Nox4 is found closely associated with focal adhesion complexes (45), where Src becomes localized on cell-surface integrin ligand engagement (95). Thus, one could certainly envision the targeted oxidation of Src in focal adhesions that result from the integrin-induced ROS signal. Thus, available data support the concept that ROS have a dual role in modulating tyrosine phoshorylation via promotion of tyrosine phosphorylation and inhibition of tyrosine dephosphorylation.
Small G proteins
Small GTPases are proteins (20 to 25 kDa) that bind to guanosine triphosphate (GTP) in their active state and are inactive when bound to GDP. Small GTPases are known to regulate a wide variety of processes, such as cell proliferation, differentiation, movement, and lipid vesicle transport (9). The Ras family of proteins are among the small GTPases, and one important mechanism of Ras protein modification involves Cys residues that are conserved in almost all Ras family members. The carboxy-terminal Cys 186 is part of the CAAX motif site for isoprenylation, but at least four other surface-exposed Cys residues (Cys 118, 181, 184, and 186) have been identified that are likely subjects to oxidation/modification. Cys 118 has been shown to interact with •NO, leading to increased nucleotide exchange and Ras activation (62), and all four sites can be modified by oxidants (74). With regard to ROS, the Cys 118 site is known to be targeted by  where it facilitates GDP dissociation from Ras and replacement with GTP (42), thus leading to increased GTPase activity (63). Angiotensin II–induced activation of NADPH oxidase and subsequent ROS production facilitates Ras activity through glutathionylation (S-SG) of Cys118, where this modification is necessary for angiotensin II–induced smooth muscle cell hypertrophy (1) (Fig. 5). Glutathionylation of Ras also occurs in cardiac myocytes, where mechanical strain induces NADPH oxidase–mediated glutathionylation of Cys118, leading to Erk activation and increased protein synthesis (91). Taken together, these data implicate Ras as a downstream target for NADPH oxidase–derived ROS. The precise mechanisms whereby NADPH oxidase catalytic activity is targeted to Ras, however, are not known.
where it facilitates GDP dissociation from Ras and replacement with GTP (42), thus leading to increased GTPase activity (63). Angiotensin II–induced activation of NADPH oxidase and subsequent ROS production facilitates Ras activity through glutathionylation (S-SG) of Cys118, where this modification is necessary for angiotensin II–induced smooth muscle cell hypertrophy (1) (Fig. 5). Glutathionylation of Ras also occurs in cardiac myocytes, where mechanical strain induces NADPH oxidase–mediated glutathionylation of Cys118, leading to Erk activation and increased protein synthesis (91). Taken together, these data implicate Ras as a downstream target for NADPH oxidase–derived ROS. The precise mechanisms whereby NADPH oxidase catalytic activity is targeted to Ras, however, are not known.
FIG. 5.

Ras proteins undergo modification of conserved Cys residues. In this paradigm, Cys118 is glutathionylated (S-SG) under oxidizing conditions. Glutathionylation of Cys118 stimulates nucleotide exchange (GDP to GTP), leading to enhanced Ras activation.
Protein disulfide isomerases
The protein disulfide isomerases (PDIs) are found predominantly in the ER and are involved in protein processing, in which they catalyze the formation of Cys disulfide bonds through thiol/disulfide exchange, allowing correct protein folding. These proteins are active in the oxidized state; thus, the maintenance of an oxidized subcellular ER compartment is necessary for proper protein folding. Studies with PDI members have demonstrated that these proteins are involved in NADPH oxidase activation (50) and as downstream targets of NADPH oxidase signaling (22). Forced overexpression of active Nox1 led to the oxidation of ERp72, an ER protein and PDI family member, on Cys residues in a thioredoxin domain (22). These Cys modifications were associated with attenuation of Erp72 reductase activity, an effect that also was seen with cell incubation with EGF.
Peroxiredoxins
The peroxiredoxin (Prx) family of peroxidases catalyze the reduction of peroxides with the aid of reducing equivalents from thiol-containing proteins. They are very conserved from an evolutionary standpoint, and all Prxs exist as homodimers (94). A conserved cysteine residue is present in the NH2-terminal region that is the primary site of peroxide-induced oxidation. The six mammalian Prx isoforms are divided into three subgroups, designated 2-Cys, atypical 2-Cys, and 1-Cys [for review, see (24)]. The best-characterized Prx isoforms are the 2-Cys types, which have been identified in the cytosol (I and II), the mitochondria (III), and the endoplasmic reticulum (IV).
The catalytic cycle of the 2-Cys Prxs is well understood and involves H2O2-mediated oxidation of the conserved N-terminal to a sulfenic acid (RSOH), prompting its subsequent reaction with the C-terminal Cys of the other homodimer to form an intermolecular disulfide. The fully reduced enzyme is reconstituted through thioredoxin-mediated reduction. Under conditions of high H2O2 stress, the Cys residues can become further oxidized to the sulfinic form (RSO2H), leading to the formation of Prx aggregates. The ATP-dependent reduction of “hyperoxidized” Prx is accomplished by sulfiredoxin, and leads to the clearing of the Prx aggregates. Recent evidence indicates that hyperoxidation of Prx and oligomer formation is associated with cell-cycle arrest, which can be reversed by clearing these aggregates via Prx reduction (89). These data suggest that the oxidation state of Prxs themselves may represent a signaling loop.
Evidence indicates that Prxs are involved in the control of NADPH oxidase signaling, principally as a means of attenuating the ROS signal. Endotoxic shock by LPS is known to be mediated, in part, by NADPH oxidase, and mice deficient in Prx-II exhibit excess mortality from LPS (124). Cell stimulation with peptide growth factors, such as insulin, PDGF, and EGF, is associated with transient NADPH oxidase–mediated inactivation of PTEN that is modulated by the cell content of PrxII. Recently, Choi and colleagues (24) demonstrated that Prx II is recruited to PDGF receptors on PDGF stimulation and attenuates the ROS-mediated PTP inactivation. These findings proved to be physiologically relevant, as Prx II overexpression attenuated the arterial neointimal thickening after injury—a process known to involve PDGF signaling (77, 77). Thus, Prxs are a downstream target of NADPH oxidase, primarily as a modulating influence on the extent of PTP inhibition and redox signaling.
Protein Complexes
Among the more intricate means of regulating redox signaling is through the formation of signaling complexes to colocalize either ROS production or ROS targets with appropriate effector proteins. This concept is in keeping with other known methods of signaling (e.g., phosphorylation, ubiquitination, acetylation), in which protein complex formation is important for the specificity of signaling.
Keap1/Nrf2
Transcription factor Nrf2 regulates the inducible expression of cytoprotective genes via cis-acting antioxidant-responsive elements or electrophile response elements (57). The expression and activity of Nrf2 is under tight control by its negative regulator, Keap1. Redox-sensitive cysteine residues in Keap1 are responsible for determining whether the local “oxidant tone” warrants the Nrf2-driven genetic program. In the normal highly reducing environment of the cell, Keap1 forms a complex with Nrf2 that facilitates its targeting by cullin family ubiquitin E3 ligases for ubiquitination and proteasomal degradation (58, 126). Oxidation of the Cys residue in Keap1 triggers its dissociation from Nrf2, preventing Nrf2 degradation. As a result, free Nrf2 is thus translocated into the nucleus for its transcriptional activity (Fig. 6). A recent study revealed the physiological significance of each reactive cysteine residue of Keap1 by using the transgene complementation analysis in vivo. Cys273 and Cys288 seemed to be keys to the constitutive repression of Nrf2 activity, whereas Cys151 was obligatory for the full activation of Nrf2 (120). By using the NADPH oxidase inhibitor, diphenyleneiodonium (DPI), NADPH oxidase was identified as acting upstream of Keap1/Nrf2, whereas Keap1 was a downstream effector for oxidase activity (100). Similarly, results were seen by using hyperoxia-induced Nrf2-dependent transcription that was blocked by diphenylene iodonium, implicating NADPH oxidase enzymes as mediators of Nrf2 activation (86).
FIG. 6.

Nrf2 is under the control of its negative regulator, Keap1. Redox-sensitive cysteine residues in Keap1 are important in Nrf2 association with Keap1. Keap1 forms a complex with Nrf2 that facilitates its targeting by cullin family ubiquitin E3 ligases for ubiquitination and proteasomal degradation. However, oxidation of Cys residues in Keap1 triggers its dissociation from Nrf2, allowing Nrf2 to translocate into the nucleus and activate stress-response genes.
Trx/ASK-1
The regulation of apoptosis-signaling kinase-1 (ASK-1) is somewhat analogous to that of Nrf2. Reduced thioredoxin 1 (Trx1) associates with ASK-1, and this complex results in inhibition of ASK-1 activity (97, 109). The oxidation of Trx1 leads to its dissociation from ASK-1, thereby freeing its enzymatic activity for activation of its downstream targets, such as JNK (48, 97). NADPH oxidase–mediated generation of H2O2 plays a critical role in this process, as the ADP-stimulated respiratory burst in alveolar macrophages leads to transient and localized oxidation of Trx, affording ASK1 activation that leads to an inflammatory response through MKK4-JNK1/c-Jun signaling (71).
NF-κB/IKK
The redox control of NF-κB signaling is complex, wherein, cytoplasmic oxidation of NF-κB subunits can activate gene transcription, but nuclear NF-κB oxidation impairs DNA binding (54). However, one component of ROS regulation of NF-κB is reminiscent of the ROS-sensitive protein sequestration seen with Nrf2/Keap1. Normally, NF-κB is sequestered in the cytosol by IκB, which, when phosphorylated, is targeted for degradation and releases NF-κB to translocate to the nucleus. It has recently been demonstrated that cytosolic ROS can promote NF-κB nuclear translocation through control of the NF-κB/IκB interaction. The dynein light chain LC8 associates with IκB and inhibits its phosphorylation by IκB kinase (IKK). However, on TNF-α stimulation, LC8 becomes oxidized by NADPH oxidase and dissociates from IκB, allowing its IKK-dependent phosphorylation and release of NF-κB to the nucleus (53). The reduced state of LC8 is maintained by thioredoxin-related protein 14 (TRP14), and thus, TRP14 is a counterregulatory component of redox-sensitive NF-κB activation.
The preceding paragraphs demonstrated the importance of locating redox-sensitive NADPH oxidase targets in proximity with their effector molecules. However, this type of protein–protein interaction does not explain how NADPH oxidase–derived ROS might affect one particular target (e.g., LC8 or Trx) in preference to another. In the following paragraphs, we focus on examples of locating ROS production in proximity to the specific ROS-sensitive targets.
Protein Localization in the Control of NOX Signaling
The localization of signaling molecules near their sites of action is a central tenet of cellular signaling. The colocalization of multicomponent pathway-specific complexes is accomplished, in part, through protein scaffolds that afford greater specificity than simple diffusion of the same limited number of components. The ability of ROS-generating enzymes to fit within this paradigm depends on the specific localization of ROS production and the limitation of ROS dissemination. The former may be accomplished through subcellular localization of Nox isoforms and their adaptor proteins. The latter is important to minimize extraneous and unintended ROS signaling. We address these two concepts in turn, realizing that considerably more data exist for localization of ROS production than for focal ROS scavenging. In this regard, we now turn our attention to mechanisms whereby ROS generation is localized to its site of signaling.
p47phox
Activation of Nox2 requires its association with adaptor proteins such as p47phox and p67phox, commensurate with their incorporation into the cytoskeletal fraction. These data, combined with observations that leading-edge membrane ruffles are enriched with Nox2 components, indicate that oxidant production is localized (43). Consistent with these observations, cytoskeletal association of p47phox has been observed in endothelial cells (40, 68), and NADPH oxidase activity appears important for endothelial cell migration (81) and angiogenesis (113). The link between NADPH oxidase activity and cell migration is critically dependent on p47phox (81). The phosphorylation of this NADPH oxidase subunit is among the earliest features of Nox2 activation, and p47phox binds moesin (115) and WAVE1(117), two proteins that are prevalent within leading-edge lamellipodia. Thus, it appears that p47phox is responsible for targeting NADPH oxidase to leading-edge lamellipodia, with a resultant localized burst of ROS activity in the leading edges of cells. This contention is supported by observations that disrupting p47 association with WAVE1, or inhibiting NADPH oxidase activity, impairs membrane ruffle formation in endothelial cells (117). The notion that NADPH oxidase activity is linked to actin dynamics is supported by observations in smooth muscle cells that cortactin, another actin-binding protein, coordinates p47phox localization to F-actin in response to angiotensin II, thereby facilitating NADPH oxidase–mediated activation of p38 MAP kinase and Akt (110). If one considers that cytoskeletal rearrangement is also highly dependent on tyrosine phosphorylation, it is not surprising that NADPH oxidase–mediated ROS production plays a critical role in this process.
IQGAP1
Focal-adhesion behavior is coordinated by the Rho family of small GTPases (85, 96) that includes Rac1 (Ras-related C3 botulinum toxin substrate 1). Nox2 has been shown to mediate endothelial cell migration in a Rac1-dependent manner (81, 113). The coordination of Nox2 with Rac1 involves a scaffolding protein known as IQGAP1 (49). IQGAP1 is involved in microtubule stabilization via binding of CLIP-170 at microtubule caps present in cell cortical regions (16, 17, 75). This scaffold is critical for directional migration and serves to coordinate Nox2 and Rac1 targeting to the leading edges of migrating cells.
TRAF4
TNF receptor–associated factor 4 is an orphan adaptor protein that binds to the focal contact scaffold Hic-5. This adaptor protein is critical for cell migration, as TRAF4-deficient mice and flies display migration defects, such as impaired neural tube closure in mice and incomplete dorsal closure in flies (19, 93). One role of TRAF-4 in migration relates to its binding to p47phox and association with Hic-5 that leads to p47phox localization in focal complexes. This localization targets NADPH oxidase activity to lamellipodia and supports cell migration as an interruption of this process using siRNA (TRAF4 or Hic-5) or oxidant-scavenging impaired migration (118). The final target appears to be PTP-PEST, as it was oxidatively modified by TRAF4 activation and independent PTP-PEST inhibition enhanced membrane ruffling (118). The interaction of TRAF4 with p47phox is also thought to be involved in the specific oxidative activation of MAPK8/JNK (119). Thus, available data indicate that TRAF4 is an important scaffold for the targeting of NADPH oxidase–derived ROS that contribute to endothelial cell migration and membrane-ruffle formation.
Lipid rafts
Lipid rafts, first proposed by Simons and Ikonen (102), are cholesterol- and sphingolipid-rich plasma-membrane domains that also contain caveolae microdomains, the latter consisting of flask-shaped membrane invaginations that use caveolin as a scaffolding protein (87). Caveolae function as signaling complexes that contain molecules such as G protein–coupled receptors, small G proteins, receptor tyrosine kinases, and protein tyrosine kinases. These lipid rafts are thought to provide for spatial organization of specific signaling pathways and, as one might expect, have been implicated in ROS signaling. Angiotensin II stimulation of vascular smooth muscle cells is associated with caveolae incorporation of the angiotensin II, type I receptor (AT1R) (129) that is accompanied by Rac1 recruitment and NADPH oxidase–mediated EGF receptor transactivation, which is Src-dependent (78). The involvement of lipid rafts has also been extended to endothelial cells. Endothelial cell treatment with TNF-α results in p47phox recruitment into raft domains that helps co-localize NADPH oxidase with eNOS to support the generation of peroxynitrite (OONO−), resulting in protein tyrosine nitration (123). Thus, it appears that existing paradigms for lipid raft–mediated signaling compartmentalization also apply to ROS-mediated signal transduction.
Endosomes
Another paradigm of cell signaling involves the formation of endosomes to facilitate the transport of signaling complexes among cellular compartments. One classic example of this pathway is the cycling of EGF receptors from the cell surface to the ER for PTP1b-mediated dephosphorylation, thereby providing for regulation of growth-factor signaling. As shown in Fig. 4, this particular process is subject to regulation by Nox4 (20). As an intriguing twist on this paradigm, Li et al. (69) demonstrated that formation of an active interleukin-1 (IL-1) receptor complex in the endosomal compartment required Nox2-derived ROS (69), perhaps via endosomal anionic transporters that facilitate superoxide release (82). Consistent with these observations, Miller and colleagues (80) recently found that TNF-α– and IL-1β–induced Nox1 activation occurs in early endosomes, where Nox1 is colocalized with CIC-3, an anion channel that affords charge neutralization from the electron flow of superoxide generation (80). The activation of NF-κB in this system was dependent on endosomal activation of Nox1.
Conclusions and Knowledge Gaps
Available data indicate that ROS signaling is a local process, consistent with existing paradigms for phosphorylation-based signaling. The specificity of ROS signaling appears to involve both the localization of ROS production and the limitation of extraneous ROS dissemination. The preceding paragraphs reviewed the evidence that ROS production can be localized via the characteristics of specific ROS, the protein targets for modification, and the adaptor proteins for NADPH oxidase localization. The mechanisms for the prevention of ROS “leaking” away from these redox microdomains, however, remain unclear. The cytosol may constrain ROS “leaking” from their intended targets, although this phenomenon would be very difficult to test because of the presence of multiple antioxidant species. Thus, it seems clear that more study will be needed to clarify the means of restricting ROS production to the site of action. Recent studies indicating that peroxiredoxin localization to the PDGF receptor is important for the limitation of PDGF signaling (24) may provide clues for future investigation. It seems likely that we will find “antioxidant scaffolds” that could restrict the ROS flux in a manner that would maximize its signal-to-noise ratio. Ultimately, a clearer understanding of ROS localization will help us unravel the cellular mechanism(s) for processing of information.
Abbreviations Used
- Abl
Abelson tyrosine kinase
- Arg
arginine
- ASK-1
apoptosis signaling kinase-1
- AT1R
angiotensin II receptor type I
- cAMP
cyclic adenosine monophosphate
- cGMP
cyclic guanosine monophosphate
- CLIP-170
CAP-Gly domain-containing linker protein 170
- DPI
diphenyliodonium
- EGF
epidermal growth factor
- ER
endoplasmic reticulum
- GPxs
glutathione peroxidases
- Grx
glutaredoxin
- GSH
glutathione
- GSSG
oxidized glutathione
- Hic5
hydrogen peroxide-induced clone 5
- IKK
IκB kinase
- IL-4
interleukin-4
- IQGAP1
IQ motif containing GTPase-activating protein 1
- JAK2
Janus kinase 2
- Keap1
Kelch-like ECH-associated protein 1
- Lck
lymphocyte-specific protein tyrosine kinase
- LMW-PTP
low-molecular-weight protein tyrosine phosphatase
- NF-κB
nuclear factor κB
- NoxA1
Nox activator 1
- NoxO1
Nox organizer 1
- Nrf2
NF-E2–related factor-2
- PDGF
platelet-derived growth factor
- PDIs
protein disulfide isomerases
- PI3K
phosphoinositide 3-kinase
- Prx
peroxiredoxin
- PTKs
protein tyrosine kinases
- PTPs
protein tyrosine phosphatases
- PTP SHP-2
Src homology protein tyrosine phosphatase-2
- Rac1
Ras-related C3 botulinum toxin substrate 1
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TNF
tumor necrosis factor
- TRAF4
TNF receptor–associated factor
- TRP14
thioredoxin-related protein 14
- Trx
thioredoxin
Acknowledgments
K. Chen is the recipient of a Scientist Development Grant form the American Heart Association, and this work was partially supported by NIH grants AG027081; HL67266; HL081587 and HL68758 to J. F. Keaney Jr.
References
- 1.Adachi T. Pimentel DR. Heibeck T. Hou X. Lee YJ. Jiang B. Ido Y. Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 2.Ago T. Kitazono T. Kuroda J. Kumai Y. Kamouchi M. Ooboshi H. Wakisaka M. Kawahara T. Rokutan K. Ibayashi S. Iida M. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke. 2005;36:1040–1046. doi: 10.1161/01.STR.0000163111.05825.0b. [DOI] [PubMed] [Google Scholar]
- 3.Babior BM. Kipnes RS. Curnutte JT. Biological defense mechanisms: the production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest. 1973;52:741–744. doi: 10.1172/JCI107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banfi B. Clark RA. Steger K. Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. 2003;278:3510–3513. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 5.Banfi B. Malgrange B. Knisz J. Steger K. Dubois-Dauphin M. Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- 6.Banfi B. Maturana A. Jaconi S. Arnaudeau S. Laforge T. Sinha B. Ligeti E. Demaurex N. Krause KH. A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science. 2000;287:138–142. doi: 10.1126/science.287.5450.138. [DOI] [PubMed] [Google Scholar]
- 7.Banfi B. Molnar G. Maturana A. Steger K. Hegedus B. Demaurex N. Krause KH. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J Biol Chem. 2001;276:37594–37601. doi: 10.1074/jbc.M103034200. [DOI] [PubMed] [Google Scholar]
- 8.Banfi B. Tirone F. Durussel I. Knisz J. Moskwa P. Molnar GZ. Krause KH. Cox JA. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5) J Biol Chem. 2004;279:18583–18591. doi: 10.1074/jbc.M310268200. [DOI] [PubMed] [Google Scholar]
- 9.Bar-Sagi D. Hall A. Ras and Rho GTPases: a family reunion. Cell. 2000;103:227–238. doi: 10.1016/s0092-8674(00)00115-x. [DOI] [PubMed] [Google Scholar]
- 10.Barford D. Jia Z. Tonks NK. Protein tyrosine phosphatases take off. Nat Struct Biol. 1995;2:1043–1053. doi: 10.1038/nsb1295-1043. [DOI] [PubMed] [Google Scholar]
- 11.Bedard K. Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 12.Beinert H. Iron-sulfur proteins: ancient structures, still full of surprises. J Biol Inorg Chem. 2000;5:2–15. doi: 10.1007/s007750050002. [DOI] [PubMed] [Google Scholar]
- 13.Biteau B. Labarre J. Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 14.Block K. Eid A. Griendling KK. Lee DY. Wittrant Y. Gorin Y. Nox4 NAD(P)H oxidase mediates Src-dependent tyrosine phosphorylation of PDK-1 in response to angiotensin II: role in mesangial cell hypertrophy and fibronectin expression. J Biol Chem. 2008;283:24061–24076. doi: 10.1074/jbc.M803964200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borregaard N. Heiple JM. Simons ER. Clark RA. Subcellular localization of the b-cytochrome component of the human neutrophil microbicidal oxidase: translocation during activation. J Cell Biol. 1983;97:52–61. doi: 10.1083/jcb.97.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brandt DT. Grosse R. Get to grips: steering local actin dynamics with IQGAPs. EMBO Rep. 2007;8:1019–1023. doi: 10.1038/sj.embor.7401089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Briggs MW. Sacks DB. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep. 2003;4:571–574. doi: 10.1038/sj.embor.embor867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caselli A. Marzocchini R. Camici G. Manao G. Moneti G. Pieraccini G. Ramponi G. The inactivation mechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2. J Biol Chem. 1998;273:32554–32560. doi: 10.1074/jbc.273.49.32554. [DOI] [PubMed] [Google Scholar]
- 19.Cha GH. Cho KS. Lee JH. Kim M. Kim E. Park J. Lee SB. Chung J. Discrete functions of TRAF1 and TRAF2 in Drosophila melanogaster mediated by c-Jun N-terminal kinase and NF-kappaB-dependent signaling pathways. Mol Cell Biol. 2003;23:7982–7991. doi: 10.1128/MCB.23.22.7982-7991.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen K. Kirber MT. Xiao H. Yang Y. Keaney JF., Jr Regulation of ROS signal transduction by NADPH oxidase 4 localization. J Cell Biol. 2008;181:1129–1139. doi: 10.1083/jcb.200709049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen K. Thomas SR. Keaney JF., Jr Beyond LDL oxidation: ROS in vascular signal transduction. Free Radic Biol Med. 2003;35:117–132. doi: 10.1016/s0891-5849(03)00239-9. [DOI] [PubMed] [Google Scholar]
- 22.Chen W. Shang WH. Adachi Y. Hirose K. Ferrari DM. Kamata T. A possible biochemical link between NADPH oxidase (Nox) 1 redox-signalling and ERp72. Biochem J. 2008;416:55–63. doi: 10.1042/BJ20071259. [DOI] [PubMed] [Google Scholar]
- 23.Cheng G. Cao Z. Xu X. van Meir EG. Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269:131–140. doi: 10.1016/s0378-1119(01)00449-8. [DOI] [PubMed] [Google Scholar]
- 24.Choi MH. Lee IK. Kim GW. Kim BU. Han YH. Yu DY. Park HS. Kim KY. Lee JS. Choi C. Bae YS. Lee BI. Rhee SG. Kang SW. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. 2005;435:347–353. doi: 10.1038/nature03587. [DOI] [PubMed] [Google Scholar]
- 25.Claiborne A. Miller H. Parsonage D. Ross RP. Protein-sulfenic acid stabilization and function in enzyme catalysis and gene regulation. FASEB J. 1993;7:1483–1490. doi: 10.1096/fasebj.7.15.8262333. [DOI] [PubMed] [Google Scholar]
- 26.Cucoranu I. Clempus R. Dikalova A. Phelan PJ. Ariyan S. Dikalov S. Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 27.Cui XL. Brockman D. Campos B. Myatt L. Expression of NADPH oxidase isoform 1 (Nox1) in human placenta: involvement in preeclampsia. Placenta. 2006;27:422–431. doi: 10.1016/j.placenta.2005.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De DX. Wang D. Many MC. Costagliola S. Libert F. Vassart G. Dumont JE. Miot F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J Biol Chem. 2000;275:23227–23233. doi: 10.1074/jbc.M000916200. [DOI] [PubMed] [Google Scholar]
- 29.Demple B. Hidalgo E. Ding H. Transcriptional regulation via redox-sensitive iron-sulphur centres in an oxidative stress response. Biochem Soc Symp. 1999;64:119–128. [PubMed] [Google Scholar]
- 30.Denu JM. Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 31.Frei B. Reactiv oxygen species and antioxidant vitamins: mechanisms of action. Am J Med. 1994;97(suppl 3A):3A. doi: 10.1016/0002-9343(94)90292-5. [DOI] [PubMed] [Google Scholar]
- 32.Gardner PR. Nguyen DD. White CW. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc Natl Acad Sci U S A. 1994;91:12248–12252. doi: 10.1073/pnas.91.25.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gardner PR. Raineri I. Epstein LB. White CW. Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem. 1995;270:13399–13405. doi: 10.1074/jbc.270.22.13399. [DOI] [PubMed] [Google Scholar]
- 34.Gaudu P. Weiss B. SoxR, a [2Fe-2S] transcription factor, is active only in its oxidized form. Proc Natl Acad Sci U S A. 1996;93:10094–10098. doi: 10.1073/pnas.93.19.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geiszt M. Kopp JB. Varnai P. Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci U S A. 2000;97:8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geiszt M. Witta J. Baffi J. Lekstrom K. Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003;17:1502–1504. doi: 10.1096/fj.02-1104fje. [DOI] [PubMed] [Google Scholar]
- 37.Giannoni E. Buricchi F. Raugei G. Ramponi G. Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol Cell Biol. 2005;25:6391–6403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grasberger H. Refetoff S. Identification of the maturation factor for dual oxidase: evolution of an eukaryotic operon equivalent. J Biol Chem. 2006;281:18269–18272. doi: 10.1074/jbc.C600095200. [DOI] [PubMed] [Google Scholar]
- 39.Groemping Y. Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J. 2005;386:401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu Y. Xu YC. Wu RF. Souza RF. Nwariaku FE. Terada LS. TNFalpha activates c-Jun amino terminal kinase through p47(phox) Exp Cell Res. 2002;272:62–74. doi: 10.1006/excr.2001.5404. [DOI] [PubMed] [Google Scholar]
- 41.Haj FG. Zabolotny JM. Kim YB. Kahn BB. Neel BG. Liver-specific protein-tyrosine phosphatase 1B (PTP1B) re-expression alters glucose homeostasis of PTP1B-/- mice. J Biol Chem. 2005;280:15038–15046. doi: 10.1074/jbc.M413240200. [DOI] [PubMed] [Google Scholar]
- 42.Heo J. Campbell SL. Superoxide anion radical modulates the activity of Ras and Ras-related GTPases by a radical-based mechanism similar to that of nitric oxide. J Biol Chem. 2005;280:12438–12445. doi: 10.1074/jbc.M414282200. [DOI] [PubMed] [Google Scholar]
- 43.Heyworth PG. Robinson JM. Ding J. Ellis BA. Badwey JA. Cofilin undergoes rapid dephosphorylation in stimulated neutrophils and translocates to ruffled membranes enriched in products of the NADPH oxidase complex: evidence for a novel cycle of phosphorylation and dephosphorylation. Histochem Cell Biol. 1997;108:221–233. doi: 10.1007/s004180050162. [DOI] [PubMed] [Google Scholar]
- 44.Hidalgo E. Ding H. Demple B. Redox signal transduction via iron-sulfur clusters in the SoxR transcription activator. Trends Biochem Sci. 1997;22:207–210. doi: 10.1016/s0968-0004(97)01068-2. [DOI] [PubMed] [Google Scholar]
- 45.Hilenski LL. Clempus RE. Quinn MT. Lambeth JD. Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 46.Huang J. Hitt ND. Kleinberg ME. Stoichiometry of p22-phox and gp91-phox in phagocyte cytochrome b558. Biochemistry. 1995;34:16753–16757. doi: 10.1021/bi00051a024. [DOI] [PubMed] [Google Scholar]
- 47.Hunter T. Signaling: 2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 48.Ichijo H. Nishida E. Irie K. ten Dijke P. Saitoh M. Moriguchi T. Takagi M. Matsumoto K. Miyazono K. Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 49.Ikeda S. Yamaoka-Tojo M. Hilenski L. Patrushev NA. Anwar GM. Quinn MT. Ushio-Fukai M. IQGAP1 regulates reactive oxygen species-dependent endothelial cell migration through interacting with Nox2. Arterioscler Thromb Vasc Biol. 2005;25:2295–2300. doi: 10.1161/01.ATV.0000187472.55437.af. [DOI] [PubMed] [Google Scholar]
- 50.Janiszewski M. Lopes LR. Carmo AO. Pedro MA. Brandes RP. Santos CX. Laurindo FR. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J Biol Chem. 2005;280:40813–40819. doi: 10.1074/jbc.M509255200. [DOI] [PubMed] [Google Scholar]
- 51.Jeong W. Park SJ. Chang TS. Lee DY. Rhee SG. Molecular mechanism of the reduction of cysteine sulfinic acid of peroxiredoxin to cysteine by mammalian sulfiredoxin. J Biol Chem. 2006;281:14400–14407. doi: 10.1074/jbc.M511082200. [DOI] [PubMed] [Google Scholar]
- 52.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jung Y. Kim H. Min SH. Rhee SG. Jeong W. Dynein light chain LC8 negatively regulates NF-kappaB through the redox-dependent interaction with IkappaBalpha. J Biol Chem. 2008;283:23863–23871. doi: 10.1074/jbc.M803072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kabe Y. Ando K. Hirao S. Yoshida M. Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7:395–403. doi: 10.1089/ars.2005.7.395. [DOI] [PubMed] [Google Scholar]
- 55.Kato M. Iwashita T. Akhand AA. Liu W. Takeda K. Takeuchi K. Yoshihara M. Hossain K. Wu J. Du J. Oh C. Kawamoto Y. Suzuki H. Takahashi M. Nakashima I. Molecular mechanism of activation and superactivation of Ret tyrosine kinases by ultraviolet light irradiation. Antioxid Redox Signal. 2000;2:841–849. doi: 10.1089/ars.2000.2.4-841. [DOI] [PubMed] [Google Scholar]
- 56.Kato M. Iwashita T. Takeda K. Akhand AA. Liu W. Yoshihara M. Asai N. Suzuki H. Takahashi M. Nakashima I. Ultraviolet light induces redox reaction-mediated dimerization and superactivation of oncogenic Ret tyrosine kinases. Mol Biol Cell. 2000;11:93–101. doi: 10.1091/mbc.11.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Katsuoka F. Motohashi H. Ishii T. Aburatani H. Engel JD. Yamamoto M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol. 2005;25:8044–8051. doi: 10.1128/MCB.25.18.8044-8051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kobayashi A. Kang MI. Okawa H. Ohtsuji M. Zenke Y. Chiba T. Igarashi K. Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kobayashi S. Nojima Y. Shibuya M. Maru Y. Nox1 regulates apoptosis and potentially stimulates branching morphogenesis in sinusoidal endothelial cells. Exp Cell Res. 2004;300:455–462. doi: 10.1016/j.yexcr.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 60.Lalucque H. Silar P. NADPH oxidase: an enzyme for multicellularity? Trends Microbiol. 2003;11:9–12. doi: 10.1016/s0966-842x(02)00007-0. [DOI] [PubMed] [Google Scholar]
- 61.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 62.Lander HM. Hajjar DP. Hempstead BL. Mirza UA. Chait BT. Campbell S. Quilliam LA. A molecular redox switch on p21(ras): structural basis for the nitric oxide-p21(ras) interaction. J Biol Chem. 1997;272:4323–4326. doi: 10.1074/jbc.272.7.4323. [DOI] [PubMed] [Google Scholar]
- 63.Lander HM. Ogiste JS. Pearce SF. Levi R. Novogrodsky A. Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J Biol Chem. 1995;270:7017–7020. doi: 10.1074/jbc.270.13.7017. [DOI] [PubMed] [Google Scholar]
- 64.Lardy B. Bof M. Aubry L. Paclet MH. Morel F. Satre M. Klein G. NADPH oxidase homologs are required for normal cell differentiation and morphogenesis in Dictyostelium discoideum. Biochim Biophys Acta. 2005;1744:199–212. doi: 10.1016/j.bbamcr.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 65.Lassegue B. Sorescu D. Szocs K. Yin Q. Akers M. Zhang Y. Grant SL. Lambeth JD. Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 66.Lee JK. Edderkaoui M. Truong P. Ohno I. Jang KT. Berti A. Pandol SJ. Gukovskaya AS. NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterology. 2007;133:1637–1648. doi: 10.1053/j.gastro.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 67.Lee SR. Kwon KS. Kim SR. Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 68.Li JM. Shah AM. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem. 2002;277:19952–19960. doi: 10.1074/jbc.M110073200. [DOI] [PubMed] [Google Scholar]
- 69.Li Q. Harraz MM. Zhou W. Zhang LN. Ding W. Zhang Y. Eggleston T. Yeaman C. Banfi B. Engelhardt JF. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol Cell Biol. 2006;26:140–154. doi: 10.1128/MCB.26.1.140-154.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Linnane AW. Kios M. Vitetta L. Healthy aging: regulation of the metabolome by cellular redox modulation and prooxidant signaling systems: the essential roles of superoxide anion and hydrogen peroxide. Biogerontology. 2007;8:445–467. doi: 10.1007/s10522-007-9096-4. [DOI] [PubMed] [Google Scholar]
- 71.Liu H. Zhang H. Iles KE. Rinna A. Merrill G. Yodoi J. Torres M. Forman HJ. The ADP-stimulated NADPH oxidase activates the ASK-1/MKK4/JNK pathway in alveolar macrophages. Free Radic Res. 2006;40:865–874. doi: 10.1080/10715760600758514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lohse DL. Denu JM. Santoro N. Dixon JE. Roles of aspartic acid-181 and serine-222 in intermediate formation and hydrolysis of the mammalian protein-tyrosine-phosphatase PTP1. Biochemistry. 1997;36:4568–4575. doi: 10.1021/bi963094r. [DOI] [PubMed] [Google Scholar]
- 73.Mahadev K. Motoshima H. Wu X. Ruddy JM. Arnold RS. Cheng G. Lambeth JD. Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol. 2004;24:1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mallis RJ. Buss JE. Thomas JA. Oxidative modification of H-ras: S-thiolation and S-nitrosylation of reactive cysteines. Biochem J. 2001;355:145–153. doi: 10.1042/0264-6021:3550145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mateer SC. Wang N. Bloom GS. IQGAPs: integrators of the cytoskeleton, cell adhesion machinery, and signaling networks. Cell Motil Cytoskeleton. 2003;55:147–155. doi: 10.1002/cm.10118. [DOI] [PubMed] [Google Scholar]
- 76.Matsuzawa A. Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta. 2008;1780:1325–1336. doi: 10.1016/j.bbagen.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 77.Mehta D. George SJ. Jeremy JY. Izzat MB. Southgate KM. Bryan AJ. Newby AC. Angelini GD. External stenting reduces long-term medial and neointimal thickening and platelet derived growth factor expression in a pig model of arteriovenous bypass grafting. Nat Med. 1998;4:235–239. doi: 10.1038/nm0298-235. [DOI] [PubMed] [Google Scholar]
- 78.Mehta PK. Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 79.Meng TC. Fukada T. Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 80.Miller FJ., Jr Filali M. Huss GJ. Stanic B. Chamseddine A. Barna TJ. Lamb FS. Cytokine activation of nuclear factor kappa B in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ Res. 2007;101:663–671. doi: 10.1161/CIRCRESAHA.107.151076. [DOI] [PubMed] [Google Scholar]
- 81.Moldovan L. Moldovan NI. Sohn RH. Parikh SA. Goldschmidt-Clermont PJ. Redox changes of cultured endothelial cells and actin dynamics. Circ Res. 2000;86:549–557. doi: 10.1161/01.res.86.5.549. [DOI] [PubMed] [Google Scholar]
- 82.Mumbengegwi DR. Li Q. Li C. Bear CE. Engelhardt JF. Evidence for a superoxide permeability pathway in endosomal membranes. Mol Cell Biol. 2008;28:3700–3712. doi: 10.1128/MCB.02038-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakashima I. Takeda K. Kawamoto Y. Okuno Y. Kato M. Suzuki H. Redox control of catalytic activities of membrane-associated protein tyrosine kinases. Arch Biochem Biophys. 2005;434:3–10. doi: 10.1016/j.abb.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 84.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122:277–291. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 85.Nobes CD. Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 86.Papaiahgari S. Kleeberger SR. Cho HY. Kalvakolanu DV. Reddy SP. NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells. J Biol Chem. 2004;279:42302–42312. doi: 10.1074/jbc.M408275200. [DOI] [PubMed] [Google Scholar]
- 87.Parton RG. Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–194. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 88.Pawson T. Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 89.Phalen TJ. Weirather K. Deming PB. Anathy V. Howe AK. van der Vliet A. Jonsson TJ. Poole LB. Heintz NH. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J Cell Biol. 2006;175:779–789. doi: 10.1083/jcb.200606005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Piccoli C. Ria R. Scrima R. Cela O. D'Aprile A. Boffoli D. Falzetti F. Tabilio A. Capitanio N. Characterization of mitochondrial and extra-mitochondrial oxygen consuming reactions in human hematopoietic stem cells: novel evidence of the occurrence of NAD(P)H oxidase activity. J Biol Chem. 2005;280:26467–26476. doi: 10.1074/jbc.M500047200. [DOI] [PubMed] [Google Scholar]
- 91.Pimentel DR. Adachi T. Ido Y. Heibeck T. Jiang B. Lee Y. Melendez JA. Cohen RA. Colucci WS. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J Mol Cell Cardiol. 2006;41:613–622. doi: 10.1016/j.yjmcc.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 92.Pryor WA. Oxy-radicals and related species: their formation, lifetimes, and reactions. Annu Rev Physiol. 1986;48:657–667. doi: 10.1146/annurev.ph.48.030186.003301. [DOI] [PubMed] [Google Scholar]
- 93.Regnier CH. Masson R. Kedinger V. Textoris J. Stoll I. Chenard MP. Dierich A. Tomasetto C. Rio MC. Impaired neural tube closure, axial skeleton malformations, and tracheal ring disruption in TRAF4-deficient mice. Proc Natl Acad Sci U S A. 2002;99:5585–5590. doi: 10.1073/pnas.052124799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rhee SG Kang SW. Jeong W. Chang TS. Yang KS. Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 95.Roach T. Slater S. Koval M. White L. Cahir McFarland ED. Okumura M. Thomas M. Brown E. CD45 regulates Src family member kinase activity associated with macrophage integrin-mediated adhesion. Curr Biol. 1997;7:408–417. doi: 10.1016/s0960-9822(06)00188-6. [DOI] [PubMed] [Google Scholar]
- 96.Rottner K. Hall A. Small JV. Interplay between Rac and Rho in the control of substrate contact dynamics. Curr Biol. 1999;9:640–648. doi: 10.1016/s0960-9822(99)80286-3. [DOI] [PubMed] [Google Scholar]
- 97.Saitoh M. Nishitoh H. Fujii M. Takeda K. Tobiume K. Sawada Y. Kawabata M. Miyazono K. Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Salmeen A. Andersen JN. Myers MP. Meng TC. Hinks JA. Tonks NK. Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 99.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 100.Sekhar KR. Crooks PA. Sonar VN. Friedman DB. Chan JY. Meredith MJ. Starnes JH. Kelton KR. Summar SR. Sasi S. Freeman ML. NADPH oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 2003;63:5636–5645. [PubMed] [Google Scholar]
- 101.Sharma P. Chakraborty R. Wang L. Min B. Tremblay ML. Kawahara T. Lambeth JD. Haque SJ. Redox regulation of interleukin-4 signaling. Immunity. 2008;29:551–564. doi: 10.1016/j.immuni.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Simons K. Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 103.Stocker R. Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 104.Suh YA. Arnold RS. Lassegue B. Shi J. Xu X. Sorescu D. Chung AB. Griendling KK. Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 105.Sumimoto H. Miyano K. Takeya R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun. 2005;338:677–686. doi: 10.1016/j.bbrc.2005.08.210. [DOI] [PubMed] [Google Scholar]
- 106.Sundaresan M. Yu ZX. Ferrans VJ. Irani K. Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 107.Szanto I. Rubbia-Brandt L. Kiss P. Steger K. Banfi B. Kovari E. Herrmann F. Hadengue A. Krause KH. Expression of NOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J Pathol. 2005;207:164–176. doi: 10.1002/path.1824. [DOI] [PubMed] [Google Scholar]
- 108.Tejada-Simon MV. Serrano F. Villasana LE. Kanterewicz BI. Wu GY. Quinn MT. Klann E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol Cell Neurosci. 2005;29:97–106. doi: 10.1016/j.mcn.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tobiume K. Saitoh M. Ichijo H. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J Cell Physiol. 2002;191:95–104. doi: 10.1002/jcp.10080. [DOI] [PubMed] [Google Scholar]
- 110.Touyz RM. Yao G. Quinn MT. Pagano PJ. Schiffrin EL. p47phox associates with the cytoskeleton through cortactin in human vascular smooth muscle cells: role in NAD(P)H oxidase regulation by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25:512–518. doi: 10.1161/01.ATV.0000154141.66879.98. [DOI] [PubMed] [Google Scholar]
- 111.Ueno N. Takeya R. Miyano K. Kikuchi H. Sumimoto H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. J Biol Chem. 2005;280:23328–23339. doi: 10.1074/jbc.M414548200. [DOI] [PubMed] [Google Scholar]
- 112.Ueyama T. Geiszt M. Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol Cell Biol. 2006;26:2160–2174. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ushio-Fukai M. Tang Y. Fukai T. Dikalov SI. Ma Y. Fujimoto M. Quinn MT. Pagano PJ. Johnson C. Alexander RW. Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res. 2002;91:1160–1167. doi: 10.1161/01.res.0000046227.65158.f8. [DOI] [PubMed] [Google Scholar]
- 114.Vallet P. Charnay Y. Steger K. Ogier-Denis E. Kovari E. Herrmann F. Michel JP. Szanto I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience. 2005;132:233–238. doi: 10.1016/j.neuroscience.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 115.Wientjes FB. Reeves EP. Soskic V. Furthmayr H. Segal AW. The NADPH oxidase components p47(phox) and p40(phox) bind to moesin through their PX domain. Biochem Biophys Res Commun. 2001;289:382–388. doi: 10.1006/bbrc.2001.5982. [DOI] [PubMed] [Google Scholar]
- 116.Wood ZA. Poole LB. Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 117.Wu RF. Gu Y. Xu YC. Nwariaku FE. Terada LS. Vascular endothelial growth factor causes translocation of p47phox to membrane ruffles through WAVE1. J Biol Chem. 2003;278:36830–36840. doi: 10.1074/jbc.M302251200. [DOI] [PubMed] [Google Scholar]
- 118.Wu RF. Xu YC. Ma Z. Nwariaku FE. Sarosi GA., Jr Terada LS. Subcellular targeting of oxidants during endothelial cell migration. J Cell Biol. 2005;171:893–904. doi: 10.1083/jcb.200507004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xu YC. Wu RF. Gu Y. Yang YS. Yang MC. Nwariaku FE. Terada LS. Involvement of TRAF4 in oxidative activation of c-Jun N-terminal kinase. J Biol Chem. 2002;277:28051–28057. doi: 10.1074/jbc.M202665200. [DOI] [PubMed] [Google Scholar]
- 120.Yamamoto T. Suzuki T. Kobayashi A. Wakabayashi J. Maher J. Motohashi H. Yamamoto M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yan SR. Berton G. Regulation of Src family tyrosine kinase activities in adherent human neutrophils: evidence that reactive oxygen intermediates produced by adherent neutrophils increase the activity of the p58c-fgr and p53/56lyn tyrosine kinases. J Biol Chem. 1996;271:23464–23471. doi: 10.1074/jbc.271.38.23464. [DOI] [PubMed] [Google Scholar]
- 122.Yan SR. Fumagalli L. Dusi S. Berton G. Tumor necrosis factor triggers redistribution to a Triton X-100-insoluble, cytoskeletal fraction of beta 2 integrins, NADPH oxidase components, tyrosine phosphorylated proteins, and the protein tyrosine kinase p58fgr in human neutrophils adherent to fibrinogen. J Leukoc Biol. 1995;58:595–606. doi: 10.1002/jlb.58.5.595. [DOI] [PubMed] [Google Scholar]
- 123.Yang B. Rizzo V. TNF-alpha potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H954–H962. doi: 10.1152/ajpheart.00758.2006. [DOI] [PubMed] [Google Scholar]
- 124.Yang CS. Lee DS. Song CH. An SJ. Li S. Kim JM. Kim CS. Yoo DG. Jeon BH. Yang HY. Lee TH. Lee ZW. El-Benna J. Yu DY. Jo EK. Roles of peroxiredoxin II in the regulation of proinflammatory responses to LPS and protection against endotoxin-induced lethal shock. J Exp Med. 2007;204:583–594. doi: 10.1084/jem.20061849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang S. Madyastha P. Bingel S. Ries W. Key L. A new superoxide-generating oxidase in murine osteoclasts. J Biol Chem. 2001;276:5452–5458. doi: 10.1074/jbc.M001004200. [DOI] [PubMed] [Google Scholar]
- 126.Zhang DD. Lo SC. Cross JV. Templeton DJ. Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang ZY. Dixon JE. Active site labeling of the Yersinia protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry. 1993;32:9340–9345. doi: 10.1021/bi00087a012. [DOI] [PubMed] [Google Scholar]
- 128.Zor U. Ferber E. Gergely P. Szucs K. Dombradi V. Goldman R. Reactive oxygen species mediate phorbol ester-regulated tyrosine phosphorylation and phospholipase A2 activation: potentiation by vanadate. Biochem J. 1993;295:879–888. doi: 10.1042/bj2950879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zuo L. Ushio-Fukai M. Ikeda S. Hilenski L. Patrushev N. Alexander RW. Caveolin-1 is essential for activation of Rac1 and NAD(P)H oxidase after angiotensin II type 1 receptor stimulation in vascular smooth muscle cells: role in redox signaling and vascular hypertrophy. Arterioscler Thromb Vasc Biol. 2005;25:1824–1830. doi: 10.1161/01.ATV.0000175295.09607.18. [DOI] [PubMed] [Google Scholar]