

Abstract
Myeloperoxidase (MPO) binds H2O2 in the absence and presence of chloride (Cl−) and catalyzes the formation of potent oxidants through 1e− and 2e− oxidation pathways. These potent oxidants have been implicated in the pathogenesis of various diseases including atherosclerosis, asthma, arthritis, and cancer. Thus, inhibition of MPO and its byproducts may have a much wide application in biological systems. Using direct rapid kinetic measurements and H2O2-selective electrodes, we showed that tryptophan (Trp), an essential amino acid, was linked kinetically to the inhibition of MPO catalysis under physiological-like conditions. Trp inactivated MPO in the absence and presence of plasma levels of chloride (Cl−), in various degrees, through binding to MPO forming the inactive complexes, Trp-MPO and Trp-MPO-Cl, respectively; and accelerating MPO Compound II formation, an inactive form of MPO. Inactivation of MPO was mirrored by the direct conversion of MPO-Fe(III) to MPO Compound II without any sign of Compound I accumulation. This behavior indicates that Trp binding modulates the formation of MPO intermediates and their decay rates. Importantly, Trp is a poor substrate for MPO Compound II and has no role in destabilizing complex formation. Thus, the overall MPO catalytic activity will be limited by: 1) the dissociation of Trp from Trp-MPO and Trp-MPO-Cl complexes; 2) the affinity of MPO Compound I towards Cl− versus Trp; and 3) the slow conversion of MPO Compound II to MPO-Fe(III). Importantly, Trp dependent-inhibition of MPO occurred with a wide range of concentrations that span various physiological and supplemental ranges.
Keywords: Hydrogen peroxide, hypochlorous acid, hypohalous acid, mammalian peroxidase, sleep rhythms, tryptophan
INTRODUCTION
Inflammation and immune activation are remarkably involved in the pathogenesis of various diseases including atherosclerosis, cardiovascular, and lung disease [1,2]. Accordingly, potential markers of inflammation, such as myeloperoxidase (MPO) and its oxidative byproducts, as well as enhancement of tryptophan (Trp) degradation, are found in patients with coronary heart disease, HIV infection, autoimmune syndromes, malignant diseases, and neurodegeneration [1–9]. Several studies have shown that modulated MPO levels and/or the acceleration of the rate of Trp degradation correlate with the extent of disease and predicting disease progression [3–9]. For example, cancer patients have enhanced degradation of Trp are not only found to predict shorter lifespan but are also associated with impaired quality of life [10–12]. In obesity, plasma tryptophan concentrations have been shown to be decreased and to be independent of weight reduction or dietary intake [13–14]. The Trp metabolic changes may subsequently reduce serotonin production and cause mood disturbances, depression, and impaired satiety ultimately leading to increased caloric uptake and obesity [15–17].
MPO is a hemoprotein with a molecular mass of 150–165 kDa synthesized during myeloid differentiation and constitutes the major component of neutrophil azurophilic granules [18]. MPO and other related hemoproteins such as eosinophil peroxidase and lactoperoxidase share the ability to catalyze H2O2 - dependent peroxidation of halides and pseudo halides to produce anti-microbial agents, hypohalous acids, through the formation of a ferryl π cation radical (E-Fe(IV)=O+π) intermediate Compound I [19–23]. Hypohalous acids are potent cytotoxic oxidants, which directly oxidize reactive groups, including sulphhydryls, iron-sulfur centers and hemes, or react with amines forming chloramines [24,25]. The three enzymes also oxidize multiple organic and inorganic molecules by two successive sequential 1e− transition generating oxidant and diffusible radical species through the activated intermediates Compound II (E-Fe(IV)=O) and Fe(III), respectively [26–31]. Compound II is a long living intermediate whose decay to ground state is considered to be the rate-limiting step during steady-state catalysis [32,33]. Structural studies and a variety of spectroscopic techniques have shown that heme pockets of mammalian peroxidases are envisioned to form the catalytic site where the stepwise reduction of H2O2 takes place [34–38]. Therefore, by blocking the entrance of heme, ligand binding to MPO heme iron or perturbation in the heme pocket geometry may promote the inhibition of the enzyme [32–40].
Since modulated MPO levels and higher degradation of Trp are major contributors to inflammation, we therefore, hypothesized that there is a potential link between levels of Trp and the catalytic activity of MPO. In the current report, we explore this hypothesis by characterizing the effect of increasing concentration of Trp on the catalytic activity of human MPO utilizing a combination of H2O2- selective electrode, optical absorbance, and direct rapid kinetics measurements. Our results clearly demonstrated that increasing concentration of Trp promotes MPO inhibition in a concentration that spans both physiological and supplemental ranges.
MATERIALS AND METHODS
Materials
L-Tryptophan used was of the highest purity grades obtained from Sigma Chemical Co. (St. Louis, MO).
Enzyme purification
MPO was purified from detergent extracts of human leukocytes as described [41–43]. Trace levels of contaminating eosinophil peroxidase were then removed by passage over a sulphopropyl Sephadex column [44]. Purity of isolated MPO was established by demonstrating a Reinheitzhal (RZ) value of >0.85 (A430/A280), SDS PAGE analysis was achieved with Coomassie blue staining, and in-gel tetramethylbenzidine peroxidase staining to confirm absence of observable contaminating eosinophil peroxidase activity [45]. Enzyme concentration was determined spectrophotometrically utilizing extinction coefficients of 89,000 M−1cm−1/heme of MPO [46].
H2O2 selective electrode measurements
H2O2 measurements were carried out using an H2O2-selective electrode (Apollo 4000 Free Radical Analyzer; World Precision Instruments, Sarasota, FL). Experiments were performed at 25 °C by immersing the electrode in 3 ml of 0.2 M sodium phosphate buffer, pH 7.0, under air. 20 μM H2O2 was added to a continuously stirred buffer solution containing various levels of Trp and/or Cl− (100 mM) during which the rise and fall of H2O2 concentration was continuously monitored. Where indicated, 20 μl MPO (20 nM final) was added to the reaction mixture.
Absorbance measurements
Spectra were recorded with a Cary 100 Bio UV-visible spectrophotometer, at 25 °C, in phosphate buffer pH 7.0. Experiments were performed with a 1 ml cuvette containing MPO (0.7–1.0 μM) pre-incubated with increasing concentration of Trp (6 – 400 μM, final concentrations), in the absence and presence of 100 mM Cl−. Concentrated volumes of H2O2 solution were added to the sample cuvette (20 μM final), and absorbance changes were recorded from 300 to 700 nm.
Rapid kinetic measurements
The kinetic measurements of MPO Compound I and/or compound II formation and decay in the absence and presence of different Trp and/or Cl− concentrations were performed using a dual syringe stopped-flow instrument obtained from Hi-Tech, Ltd. (Model SF-61). Experiments were initially performed under conditions identical to those recently reported for MPO and other related hemoproteins to facilitate comparison [46–48]. Measurements were carried out under an aerobic atmosphere at 10 °C following rapid mixing of equal volumes of an H2O2-containing buffer solution and a peroxidase solution that contained 100 mM Cl− and/or different Trp concentrations. Reactions were monitored at both 432 and 452 nm. The time course of the absorbance change was fit to a single-exponential, (Y = 1 − e−kt), or a double exponential (Y = Ae−k1t + Be−k2t) functions as indicated. Signal-to-noise ratios for all kinetic analyses were improved by averaging at least six to eight individual traces. In some experiments, the stopped-flow instrument was attached to a rapid scanning diode array device (Hi-Tech) designed to collect multiple numbers of complete spectra (200–800 nm) at specific time ranges. The detector was automatically calibrated relative to a holmium oxide filter, as it has spectral peaks at 360.8, 418.5, 446.0, 453.4, 460.4, 536.4, and 637.5 nm, which were used by the software to correctly align pixel positions with wavelength. Rapid scanning experiments involve mixing solutions of peroxidase (1–2 μM) pre-incubated with 100 mM Cl− in the absence or in the presence of increasing (25–400 μM, final) Trp concentrations with buffer solutions containing 40 μM H2O2, at 10 °C.
RESULTS
Ability of Trp to inhibit MPO catalytic activity
We first utilized an H2O2-selective electrode to determine whether Trp serves as a potent inhibitor for MPO. Following addition of 20 μM H2O2 (final) to the continuously stirred reaction mixture, the H2O2 signal rose rapidly, achieved a maximum after ~30 s, and fell gradually as H2O2 was depleted by autoreduction (2 H2O2 = 2 H2O+ O2) (Fig. 1). In a competitive assay, addition of different Trp levels (5, 7, 14, 28, 60, and 100 μM) and/or plasma level of Cl− (100 mM) to a stirred H2O2 buffer solution had little or no effect on H2O2 autoreduction rates, indicating that Trp alone did not significantly consume H2O2. In the absence of Cl−, addition of MPO to the reaction mixture caused an immediate rapid decay in the level of free H2O2, followed by a slower decay indicating that H2O2 was consumed as a substrate by MPO during steady-state catalysis, as previously reported (Fig. 1) [47]. The first step occurred immediately after the enzyme addition and was attributed to the formation of MPO compound I. This initial step occurs through multiple cycles before complete conversion of Compound I to Compound II. The second step was much slower and was attributed to the reaction of MPO with H2O2 after the conversion of Compound II to MPO-Fe(III). Adding MPO 20 μl MPO (200 nM final) to continuously stirred H2O2 buffer (20 μM) solutions supplemented with 0.15, 30, and 100 μM Trp caused rapid conversion of Compound I to Compound II, as judged by the attenuation of the first phase of H2O2 consumption. Increasing Trp concentration progressively decreased the rate of H2O2 consumption (Fig. 1) consistent with the fact that Trp is a poor 1e- substrate of MPO Compound II [49].
Fig. 1.
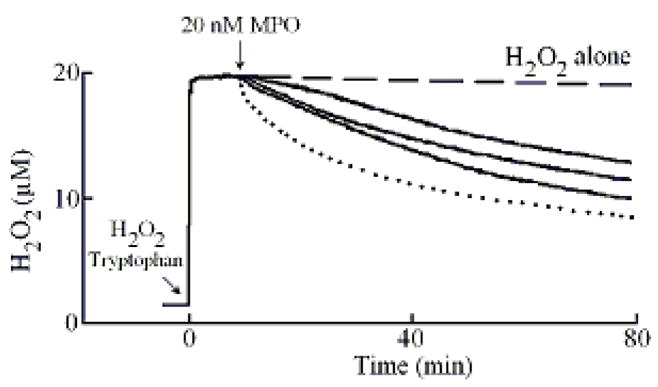
Effect of Trp on H2O2 consumption by MPO. A typical recording by an H2O2-selective electrode demonstrating the autoreduction of H2O2 (20 μM) following addition to a stirred 0.2 M phosphate buffer (pH 7.0), at 25 °C (dashed line). Addition of MPO (20 nM) to H2O2 solution in the absence (dotted line) and the presence of 15, 30 and 100 μM of Trp (solid lines bottom to the top) caused decrease in the rate of H2O2 consumption. Tracings shown are from a typical experiment performed at least three times.
We next characterized Trp binding to MPO. Addition of MPO (20 μl, 20 nm final) solution to a continuously stirred H2O2 (20 μM) solution supplemented with 100 mM Cl− caused a rapid consumption of H2O2 (Fig. 2A, dotted line). Under these experimental conditions, MPO underwent steady-state catalysis metabolizing Cl− through a 2e− oxidation pathway generating HOCl. In contrast, two kinetically distinct species could be distinguished when MPO solution was added to a solution containing 5 μM Trp and 20 μM H2O2. As this trace shows that ~70% of H2O2 was consumed by original fast phase, while the rest by slow phase probably when both Trp and Cl− bound to the enzyme. Increasing Trp concentration (7, 14, 28, 60, and 100 μM) caused an increase in the amplitude of the slow phase, and with higher Trp concentration, the slower phase represents a majority (approximately 80–90%). Together, this makes MPO to operate at a fraction of its maximum activity in the steady state and significantly alters its activity response within the physiologic concentration range of Trp (0–400 μM). As shown in Fig. 2B, increasing the added enzyme concentration to 200 nM caused a progressive increase in the rate of H2O2 consumption, but the reaction behavior stayed the same.
Fig. 2.

Trp/Cl− inhibit H2O2 consumption by MPO. (Panel A) A typical recording by an H2O2-selective electrode demonstrating the autoreduction of H2O2 (20 μM) following addition to a stirred 0.2 M phosphate buffer (pH 7.0) supplemented with 100 mM Cl−, at 25 °C (dashed line). Addition of 20 μl (20 nm in 3ml solution, final) of MPO solution to the H2O2/Cl− stirred buffer caused rapid H2O2 consumption (dotted line). Addition of 20 μl (30 nM final) MPO solution to the H2O2/Cl− solution supplemented with increasing Trp concentrations 5, 7, 14, 28, 60 and 100 μM (solid lines from left to right, respectively) causing slower H2O2 removal. (Panel B) Similar reactions were repeated by adding 20 μl (200 nM) MPO to the H2O2/Cl− solution supplemented with increasing Trp concentrations (60, 100, 400, 800, 1600 μM, sold lines, left to right, respectively). Tracings shown are from a typical experiment performed at least three times.
Rapid kinetic measurements
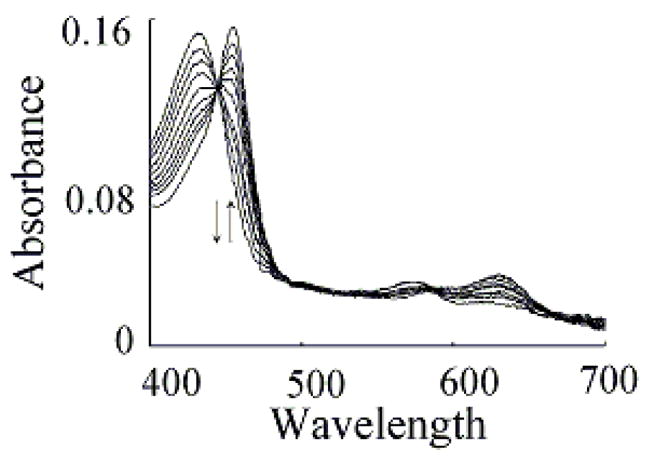
We characterized Trp binding to MPO-Fe(III) on MPO catalytic activity during steady-state catalysis at 10 °C. The effect of Trp on MPO Compound I formation and its conversion to Compound II were studied by rapid mixing MPO-free or Trp-bound MPO with a solution containing H2O2. As shown in Fig. 3A, the starting spectrum was characteristic of MPO-Fe(III), which displays a Soret absorbance peak at 432 nm and visible peaks at 573, 630, and 694 nm. Fig. 3B also shows that the formation of MPO Compound I in the absence of Trp was very fast and required less than 0.05 s to reach completion, whereas, its conversion to Compound II was slow and reached completion in the next 3 s (Fig. 3B). In both cases, the spectral change was monophasic and best fit to a single exponential function with rate constant of 350 and 3 s−1, respectively. Replica experiments were run in the presence of 25, 50, 400 μM Trp to monitor the effect of Trp on MPO intermediates’ distributions. The rate of the spectral change at 432 nm was monophasic, essentially identical to that observed at 452 nm, a wavelength that detects MPO Compound II accumulation, and consistent with the direct conversion of MPO-Fe(III) to Compound II without the build-up of Compound I (Fig. 3C). We next examined the MPO Compound II complex formation as a function of Trp concentration using single wavelength stopped-flow methods. At all Trp concentration tested, there was a monophasic increase in absorbance at 452 nm and attributed to buildup of MPO Compound II. As shown in Fig. 4, plot of kobs versus Trp concentration was linear and showed a sign of saturation above 200 μM Trp. To help interpret these results, we measured the rate of the initial decrease in absorbance at 432 nm in the absence and presence of saturated amounts of Trp (400 μM). As shown in Fig. 5, the rate of decrease in absorbance at 432 nm in the absence of Trp had a rate constant of 400 s−1 at 10 °C, and was attributed to the formation of MPO Compound I. This rate was decreased nearly 2-fold in the presence of saturated amount of Trp (400 μM) which was identical to that observed at 452 nm, and consistent with direct conversion of MPO-Fe(III) to Compound II occurring in this circumstance. Collectively, this behavior indicates that Trp binds MPO to form Trp-MPO complex and limits the overall catalytic cycle by the dissociation of Trp from the complex.
Fig. 3.

Trp accelerates the conversion rates of MPO Compound I to Compound II. Absorbance spectra of MPO recorded by diode array stopped-flow in the absence (Panels A and B) and the presence of 400 μM Trp (Panel C). Experiments were performed under aerobic condition when a phosphate buffer solution (200 mM, pH 7.0) containing 2.4 μM MPO in the absence and presence of either 200 μM were rapidly mixed with a buffer solution supplemented with 40 μM H2O2 at 10 °C as described under the Experimental Conditions section. Panel A contains spectra collected at 0.00, 0.001, 0.004, 0.07 s after mixing showing the formation of MPO Compound I. Panel B contains spectra collected at 0.015, 0.025, 0.105, 0.205, 0.305, 0.495, 0.950, 02.95 and 4.00 s after initiating the reaction showing the conversion of MPO Compound I to Compound II. Panel C contains spectra collected at 0.01, 0.07, 0.016, 0.022, 0.031, 0.049 and 2.85 s after mixing showing the direct conversion of MPO-Fe(III) to Compound II without the accumulation of Compound I. Arrows indicate the direction of spectral change over time.
Fig. 4.
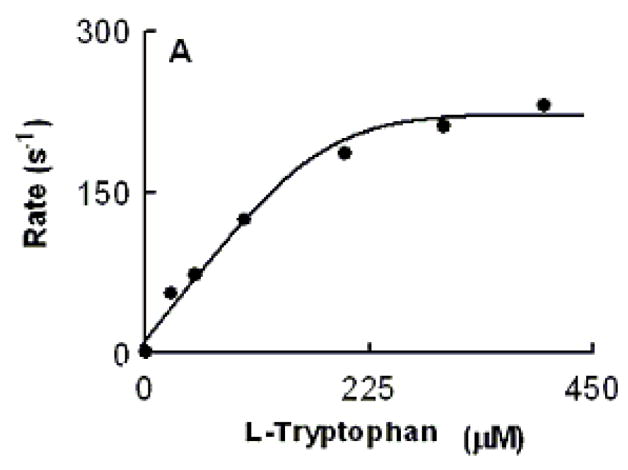
Relationship between Trp concentration and the observed rate constant of MPO Compound II formation monitored at 10 °C. An anaerobic solution containing phosphate buffer (200 mM, pH 7.0) supplemented with H2O2 (40 μM) was rapidly mixed with an equal volume of buffer supplemented with 1.4 μM of MPO-Fe(III) in the presence of increasing Trp concentrations using single wavelength stopped-flow methods. Spectral changes were monitored at 432 nm, a characteristic wavelength for Compound II, as a function of time. Data were best fitted to a single exponential function.
Fig. 5.

Kinetics of absorbance changes at 432 nm in the absence and presence of saturated amounts of Trp as measured by stopped-flow spectroscopy at 10 °C. One syringe contained MPO-Fe(III) (μM) (left trace) or MPO pre-incubated with saturated amounts of Trp (400 μM) (right trace) and was rapidly mixed with a syringe containing 40 μM H2O2. Note that in the absence of Trp, the change in absorbance was attributed to the formation of MPO Compound I. In the presence of Trp, the change in absorbance was attributed to the direct conversion of MPO-Fe(III) to MPO Compound II. The smooth line running through each experimental trace is the line of best fit according to a one exponential equation.
We next investigated how the plasma level of Cl− in the reaction mixture changes the MPO intermediates distribution during steady-state catalysis. In the absence of Trp, addition of H2O2 to MPO solution in the presence of 100 mM Cl− (final) caused rapid small decreases in absorbance (not shown), indicating that the decay of MPO Compound I is faster than its formation. When the same reactions were repeated in the presence of saturated amounts of Trp (200 μM), MPO-Fe(III) was converted directly to Compound II without any sign of Compound I accumulation (Fig. 6). Under these circumstances, the formation rate of MPO Compound I became faster than its decay, therefore, Compound I did not accumulate. We next investigated the effect of various Trp and H2O2 concentrations on the conversion rate of MPO-Fe(III) to Compound II by following the increase in absorbance at 452 nm, using single wavelength stopped-flow methods. It was evident that as Trp concentration increased, MPO Compound II formation rate constant decreased and was saturated above 200 μM Trp (Fig. 7). When the same reactions were monitored at 432 nm, the direction of absorbance change reversed, but otherwise, proceeded with identical kinetics (Fig. 7).
Fig. 6.
Trp competes with plasma level of Cl- and switches the MPO reaction from a 2e− oxidation to a 1e− oxidation pathway. Absorbance spectra of MPO recorded by diode array stopped-flow when a phosphate buffer (200 mM, pH 7.0) containing 1.9 μM MPO, 200 mM Cl−, and 400 μM Trp was rapidly mixed with a buffer solution supplemented with 40 μM H2O2 at 10 °C. This figure contains spectra collected at 0.001, 0.016 0.031, 0.073, 0.103, 0.180, 0.240 and 1.425 s after initiating the reaction. Arrows indicate the direction of spectral change over time.
Fig. 7.
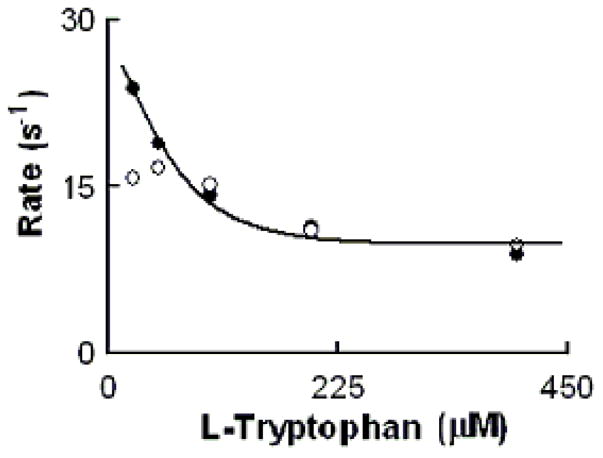
Rate of MPO-Fe(III) conversions to Compound II as a function of Trp concentration. Experiments were carried out by rapid mixing of EPO-Fe(III) solution supplemented with fixed levels of Cl− (200 mM) and increasing concentration of Trp with the same volume of a buffer solution supplemented with 40 μM H2O2. The observed rate of MPO Compound II formation monitored at both 452 (close circles) and 432 nm (open circles) were plotted as a function of Trp. Data represent the mean of triplicate determinations from an experiment performed three times.
To see if increased H2O2 concentration alters the rate of MPO-Fe(III) conversion to Compound II, we measured the conversion rate in the presence of increasing H2O2 concentration and a fixed amount of Trp (400 μM). Again the observed rate constants measured at both 432 and 452 nm were identical, and increased in a concentration-dependent and saturable manner (Fig. 8). The first-order rate constant of the saturation largely matched the rate constant of the saturation levels when MPO Compound II formation rates were plotted against Trp concentrations, suggesting that the overall catalytic activity is limited by the dissociation rate of Trp from Trp-MPO-Cl complex.
Fig. 8.
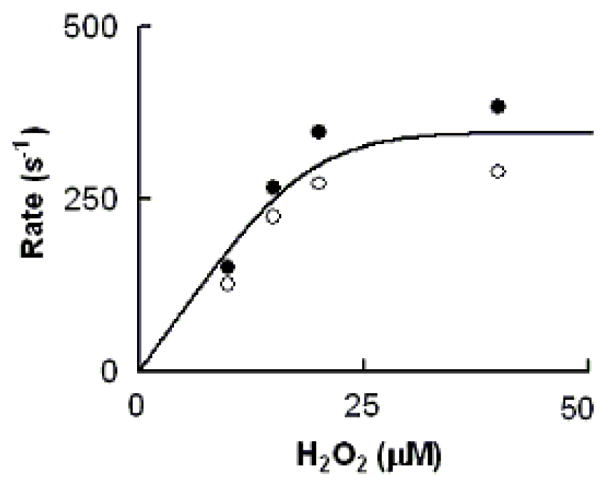
Rate of MPO-Fe(III) conversions to Compound II as a function of H2O2 concentration. The observed rate of MPO Compound II formation monitored at both 452 (close circles) and 432 nm (open circles) when a buffer solution containing 1.2 μM MPO, 400μM Trp, and 100 mM Cl− was rapidly mixed with a buffer solution supplemented with increasing H2O2 concentrations. Data represent the mean of triplicate determinations from an experiment performed three times.
DISCUSSION
Tryptophan is a natural constituent of the diet, antioxidant, displays low toxicity, and produces few side effects [50]. Based on these advantages, high doses of dietary Trp supplementation have been used in the management of neuropsychiatric disorders with variable success [51]. For example, 50 mg to 6 g tryptophan has been used as an adjunct therapy for smoking cessation, sleep disorders, mood swings, tension, and irritability in women with premenstrual dysphoria [49,51–54]. Trp plasma or serum concentration normally ranges from 30–70 μM in human and 60–180 μM in rats [55]. In this report, our central finding is that physiological Trp concentration inhibited the MPO catalytic activity, which may have a beneficially role in modulating the pathogenesis of several disorders. This was independently demonstrated by monitoring rates of H2O2 reduction by MPO and subsequent buildup of MPO Compound II, the inactive form of MPO, or the duration of the steady-state as a function of the Trp concentration in the presence and absence of Cl−. Direct conversion of MPO-Fe(III) to Compound II and the acceleration of MPO Compound II formation were shown to be Trp- and chloride-dependent. Our data may indicate that this is an allosteric coupling between Trp, MPO heme pocket entrance binding (regulatory) site, and the catalytic center that induces detectable inhibition of MPO. Consequently, Trp release is required before the enzyme restores its catalytic activity. Importantly, Trp is a poor substrate of MPO Compound II [56] and has no role in destabilizing complex formation. Thus, the overall MPO catalytic activity is limited by: 1) dissociation of Trp from Trp- MPO and Trp-MPO-Cl complexes; 2) the affinity of MPO Compound I towards Cl− versus Trp; and 3) the slow conversion of MPO Compound II to the ground state. Because MPO-mediated reactive oxidants promote oxidative tissue damage in a variety of inflammatory conditions [3,4,57–61], inhibition of MPO and its downstream inflammatory byproducts can be attractive targets in the development of therapeutic interventions of various conditions ranging from lung to heart diseases.
Our results fit nicely within a hypothetical model of MPO catalysis (Fig. 9). MPO can partition between two major cycles during steady-state catalysis: cycle A, the binding of Trp and/or Cl− to MPO-Fe(III) and cycle B, the classic peroxidase cycle that involves the formation of MPO Compounds I and II. In cycle A, Trp may bind to MPO-Fe(III) in the presence and absence of Cl− leading to inhibition of MPO activity and formation of Trp-MPO and Trp-MPO-Cl complexes. Examination of the Trp concentration dependence for the rate of Compound II formation revealed saturable kinetic levels of Trp >200 μM in both in the absence (Fig. 4) and presence of (Fig. 7) of the competitor Cl−. The first order rate constant observed in the absence of Cl− and high levels of Trp (230 s−1) is 23-fold higher than that in the presence of Cl−. Thus, in the presence of Cl− as well as high levels of Trp, the rate-limiting step in catalysis becomes the relatively slow dissociation Trp from Trp-MPO-Cl complex, resulting in the peroxidase inhibition. In contrast, in the absence of physiological levels of alternative Cl−, the dissociation rate of Trp from the Trp-MPO complex is relatively high (230 s−1) which is significantly less than the formation rate of MPO Compound I. Thus, Cl− presence significantly enhances the affinity of MPO towards Trp. Since Trp is a bulky molecule and binds on the entrance of MPO heme pocket, there is a direct communication between the active site of the enzyme, where HOCl is generated, and the heme pocket entrance binding (regulatory) site. Our current results are consistent with the hypothesis that Cl− promotes a significant alteration in the heme pocket of ground-state MPO [41,62], as reflected in the observed rates of Compound II formation.
Fig. 9.
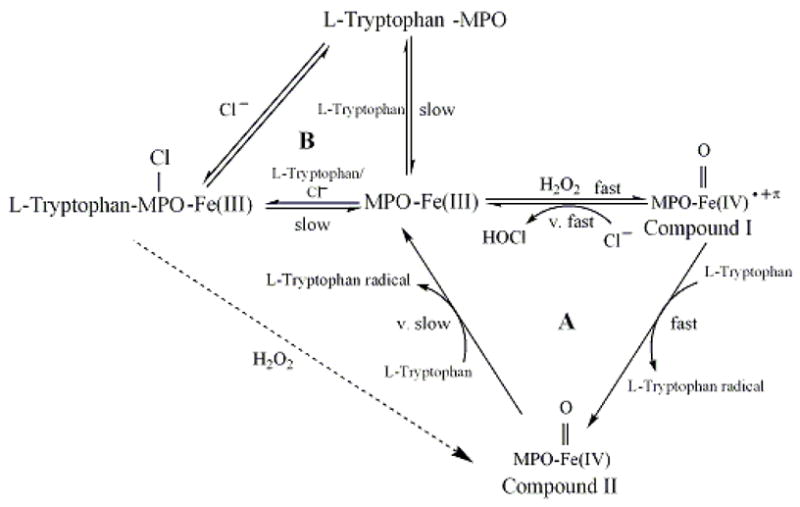
Working kinetic model for Trp interactions with MPO.
In the presence of sub-saturated amounts of Trp, most of MPO-Fe(III) is free and rapidly reacts with H2O2 to form Compound I, a 2− electron oxidized intermediate possessing a Fe(IV)=O group and a resonance-stabilized porphyrin cation radical [19–23]. Compound I has a reduction potential (approximately 1.1 V) enabling it to oxidize halides via a single 2e− oxidation step to their respective hypohalous acids [24,25]. Physiological levels of Trp can compete with Cl− and accelerate the formation of MPO Compound II through a 1e− oxidation pathway. Under these conditions, the decay of Compound I becomes faster than its formation rate, therefore Compound I can not be accumulated during catalysis, instead Compound II. Our results did not exclude the possibility that Cl-MPO-Trp complex might have reacted directly with H2O2 generating Compound II and Trp. Since Trp is a poor substrate for MPO Compound II [56], the catalytic activity of MPO is limited by the slow conversion of MPO Compound II to the ground state. The ability of Cl− to enhance Trp performance in inhibiting MPO suggests there is a cross-communication between the MPO active site, where HOCl is generated, and the MPO heme pocket entrance binding (regulatory) site. This suggestion was emphasized by the slower formation rate of Compound II in the presence of Cl− in contrast to its absence. Collectively, Trp significantly inhibited H2O2 consumption in levels that govern both (patho)physiological, and supplemental ranges [49,51–55].
MPO X-ray structure is presently available [34,35]. These structural analyses confirm that the heme prosthetic group lies on a deep narrow heme pocket were the H2O2 reduction takes place [34,35]. Related studies by Hallingback et al. utilizing computational docking have shown that melatonin could be docked to MPO-Fe(III), Compound I, and Compound II forms [63]. To achieve stacking, they have shown that the indole ring is docked parallel to the heme plane and close enough to the D pyrrole ring. The distance of the closest indole atom to the center of the D ring is averaged between 3.3–3.5 Å. Comparison between MPO-Fe(III), Compound I, and Compound II modeling indicated that melatonin was pushed approximately 1Å away from the ferryl oxygen in both compounds I and II. In all three cases, the side chain of the indole compound was directed toward the outside of the distal cavity [63]. Early studies by Hori et al. have shown that salicylhydroxamic acid and benzohydroxamic acid [64] is a relatively bulky aromatic molecule that may influence MPO steady-state catalysis by binding to the entrance of the hydrophobic pocket of the distal heme cavity, preventing the access of H2O2 to the catalytic site of the enzyme. However, using rapid kinetic measurements and NO binding to MPO heme iron, we have recently shown that Cl− may bind at two different binding sites on MPO [62]. Both sites have distinct effects on the MPO heme iron microenvironment. Our current results are consistent with this hypothesis, since much effective inhibition of MPO requires the occupation of both catalytic sites of MPO. Our findings provide further evidence for an allosteric coupling between Trp, MPO heme pocket entrance binding site, and the catalytic center that induces detectable inhibition of MPO. They also provide new role of Cl− in facilitating Trp binding that mediates MPO inhibition. Collectively, Trp and peroxidase activities are apparently coupled through complex and interdependent pathways.
Recently, we have demonstrated that melatonin can serve as a potent inhibitor of MPO [65]. Although Trp-MPO and melatonin-MPO complex formations inhibit MPO catalysis in a similar way, there are important differences in the fundamental aspects. For example, melatonin and Trp can serve as a 1e− substrate for Compound I largely in the same efficiency, but for MPO Compound II, only melatonin can serve as a 1e− substrate [65]. This melatonin behavior causes a shift in the MPO activity from peroxidation to catalase- like activity [65]. In contrast, Trp limits MPO catalytic activity by the slow conversion rate of MPO Compound II, an inactive form of MPO, to the ground state. The ability of melatonin to serve as 1e- substrate for Compound II in the absence of Cl− drives MPO to accelerate H2O2 consumption upon increasing melatonin concentration [65]. As opposed to this, increasing Trp concentration drive MPO to consume H2O2 in a slower rate. In addition, the dissociation rate of both melatonin-MPO and Cl-MPO-melatonin complexes to generate free MPO and melatonin seem likely to be much lower than that of Trp complexes with MPO. Therefore, in the absence of Cl−, higher concentrations of Trp are required to promote MPO inhibition.
Trp-dependent inhibition of MPO is physiologically relevant since only trace levels of Trp are required, relative to the concentrations of Cl− and the co-substrate H2O2 used. Thus, inactivation of MPO and its duration can be controlled effectively by Trp supplementation. In addition of its potential capacity to inhibit MPO, its supplements have been thought to keep good health, act as an antidepressants, relieving poor mood and irritability, reduce the cravings for carbohydrates, provide a relief of chronic pain and the symptoms of manic behavior, and normalize sleep [53,66,67]. Tryptophan serves as a biosynthetic precursor for a wide variety of indole-containing metabolites, which are involved in a large and essential number of physiological functions [68]. For example, it increases levels of serotonin on the brain as well as melatonin [69]. It is tempting to speculate that under normal condition, there is a balance between levels of MPO and Trp. Disturbing this balance by either elevated MPO levels or accelerated Trp degradation poses an increased risk of inflammatory disorders. Direct reaction between MPO and Trp would be anticipated to generate MPO Compound II, an inactive form of the enzyme, which can be driven to MPO-Fe(III) by good alternative 1e− substrates such as NO and NO2− [37,57,70–72].
Acknowledgments
This work was supported by the National Institutes of Health grant RO1 HL066367 and a grant from Children’s Hospital of Michigan (to H. M. A.-S.); and by an award from the American Heart Association (S. G.).
ABBREVIATIONS
- Cl−
Chloride
- H2O2
hydrogen peroxide
- MPO
myeloperoxidase
- Trp
tryptophan
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hansson GH. Inflammation, atherosclerosis, and coronary artery disease. NEJM. 2005;35:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 2.Gao H, Neff T, Ward PA. Regulation of lung inflammation in the model of IgG immune-complex injury. Annu Rev Pathol Mech Dis. 2006;1:215–242. doi: 10.1146/annurev.pathol.1.110304.100155. [DOI] [PubMed] [Google Scholar]
- 3.Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 4.Shao B, Oda MN, Oram JF, Heinecke JW. Myeloperoxidase: an inflammatory enzyme for generating dysfunctional high density lipoprotein. Curr Opin Cardiol. 2006;21:322–328. doi: 10.1097/01.hco.0000231402.87232.aa. [DOI] [PubMed] [Google Scholar]
- 5.Nauseef WM. Lessons from MPO deficiency about functionally important structural features. Jpn J Infect Dis. 2004;57:S4–5. [PubMed] [Google Scholar]
- 6.Everse J, Coates PW. Neurodegeneration and peroxidases. Neurobiol Aging. 2007 November 27; doi: 10.1016/j.neurobiolaging.2007.10.007. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 7.Boasso A, Shearer GM. How does indoleamine 2,3-dioxygenase contribute to HIV-mediated immune dysregulation. Curr Drug Metab. 2007;8:217–223. doi: 10.2174/138920007780362527. [DOI] [PubMed] [Google Scholar]
- 8.Brandacher G, Winkler C, Schroecksnadel K, Margreiter R, Fuchs D. Antitumoral activity of interferon-gamma involved in impaired immune function in cancer patients. Curr Drug Metab. 2006;7:599–612. doi: 10.2174/138920006778017768. [DOI] [PubMed] [Google Scholar]
- 9.Leonard BE. Inflammation, depression and dementia: are they connected? Neurochem Res. 2007;32:1749–1756. doi: 10.1007/s11064-007-9385-y. [DOI] [PubMed] [Google Scholar]
- 10.Huang A, Fuchs D, Widner B, Glover C, Henderson DC, Allen-Mersh TG. Tryptophan decrease in advanced colorectal cancer correlates with immune activation and impaired quality of life. Br J Cancer. 2002;86:1691–1696. doi: 10.1038/sj.bjc.6600336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroecksnadel K, Fiegl M, Prassl K, Winker C. Diminished quality of life in patients with cancer correlates with tryptophan degradation. Cancer Res Clin Oncol. 2007;133:477–485. doi: 10.1007/s00432-007-0191-3. [DOI] [PubMed] [Google Scholar]
- 12.Brandacher G, Margreiter R, Fuchs D. Interferons, immunity and cancer immunoediting leading to impaired immune function in cancer patients. Nature Reviews of Immunology. 2008 In press. [Google Scholar]
- 13.Brandacher G, Hoeller E, Fuchs D, Weiss HG. Chronic immune activation underlies morbid obesity: is IDO a key player? Curr Drug Metab. 2007;8:289–295. doi: 10.2174/138920007780362590. [DOI] [PubMed] [Google Scholar]
- 14.Guijarro A, Laviano A, Meguid MM. Hypothalamic integration of immune function and metabolism. Prog Brain Res. 2006;153:367–340. doi: 10.1016/S0079-6123(06)53022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatry. 2002;7:468–473. doi: 10.1038/sj.mp.4000995. [DOI] [PubMed] [Google Scholar]
- 16.Owens MJ, Nemeroff CB. Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin Chem. 1994;40:288–295. [PubMed] [Google Scholar]
- 17.Widner B, Laich A, Sperner-Unterweger B, Ledochowski M, Fuchs D. Neopterin production tryptophan degradation and mental depression: what is the link? Brain Behav Immun. 2002;16:590–595. doi: 10.1016/s0889-1591(02)00006-5. [DOI] [PubMed] [Google Scholar]
- 18.Nauseef WM, Malech HL. Analysis of the peptide subunits of human neutrophil myeloperoxidase. Blood. 1986;67:1504–1507. [PubMed] [Google Scholar]
- 19.Jong EC, Henderson WR, Klebanoff SJ. Bactericidal activity of eosinophil peroxidase. J Immunol. 1980;124:1378–1382. [PubMed] [Google Scholar]
- 20.Jong EC, Klebanoff SJ. Eosinophil-mediated mammalian tumor cell cytotoxicity: role of the peroxidase system. J Immuno. 1980;124:1949–1953. [PubMed] [Google Scholar]
- 21.Weiss SJ, Test ST, Eckmann CM, Roos D, Regiani S. Brominating oxidants generated by human eosinophils. Science. 1986;234:200–203. doi: 10.1126/science.3018933. [DOI] [PubMed] [Google Scholar]
- 22.Mayeno AN, Curran AJ, Roberts RL, Foote CS. Eosinophils preferentially use bromide to generate halogenating agents. J Biol Chem. 1989;264:5660–5668. [PubMed] [Google Scholar]
- 23.Klebanoff SJ, Waltersdorph AM, Rosen H. Antimicrobial activity of myeloperoxidase. Methods Enzymol. 1984;105:399–403. doi: 10.1016/s0076-6879(84)05055-2. [DOI] [PubMed] [Google Scholar]
- 24.Albrich JM, McCarthy CA, Hurst JK. Biological reactivity of hypochlorous acid: implications for microbicidal mechanisms of leukocyte myeloperoxidase. Proc Natl Acad Sci USA. 1981;78:210–214. doi: 10.1073/pnas.78.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosen H, Klebanoff SJ. Oxidation of Escherichia coli iron centers by the myeloperoxidase-mediated microbicidal system. J Biol Chem. 1982;257:13731–13735. [PubMed] [Google Scholar]
- 26.van der Vliet A, Eiserich JP, Halliwell B, Cross CE. Formation of reactive nitrogen species during peroxidase-catalyzed oxidation of nitrite. A potential additional mechanism of nitric oxide-dependent toxicity. J Biol Chem. 1997;272:7617–7625. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 27.Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, van der Vliet A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 28.Weiss SL. Tissue destruction by neutrophils. NEJM. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 29.Heinecke JW, Li W, Daehnke HL, III, Goldstein JA. Dityrosine, a specific marker of oxidation, is synthesized by the myeloperoxidase-hydrogen peroxide system of human neutrophils and macrophages. J Biol Chem. 1993;268:4069–4077. [PubMed] [Google Scholar]
- 30.Hazen SL, Hsu FF, Mueller DM, Crowley J, Heinecke JW. Human neutrophils employ chlorine gas as an oxidant during phagocytosis. J Clin Invest. 1996;98:1283–1289. doi: 10.1172/JCI118914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hazen SL, d’Avignon A, Anderson MM, Hsu FF, Heinecke JW. Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to oxidize alpha-amino acids to a family of reactive aldehydes. Mechanistic studies identifying labile intermediates along the reaction pathway. J Biol Chem. 1998;273:4997–5005. doi: 10.1074/jbc.273.9.4997. [DOI] [PubMed] [Google Scholar]
- 32.Kettle AJ, Winterbourn CC. Myeloperoxidase: a key regulator of neutrophil oxidant production. Redox Rep. 1997;3:3–15. doi: 10.1080/13510002.1997.11747085. [DOI] [PubMed] [Google Scholar]
- 33.Furtmuller PG, Burner U, Regelsberger G, Obinger C. Spectral and kinetic studies on the formation of eosinophil peroxidase compound I and its reaction with halides and thiocyanate. Biochemistry. 2000;39:15578–15584. doi: 10.1021/bi0020271. [DOI] [PubMed] [Google Scholar]
- 34.Zeng J, Fenna RE. X-ray crystal structure of canine myeloperoxidase at 3 A resolution. J Mol Biol. 1992;226:185–207. doi: 10.1016/0022-2836(92)90133-5. [DOI] [PubMed] [Google Scholar]
- 35.Davey CA, Fenna RE. 2.3 A resolution X-ray crystal structure of the bisubstrate analogue inhibitor salicylhydroxamic acid bound to human myeloperoxidase: a model for a prereaction complex with hydrogen peroxide. Biochemistry. 1996;35:10967–10974. doi: 10.1021/bi960577m. [DOI] [PubMed] [Google Scholar]
- 36.Abu-Soud HM, Hazen SL. Interrogation of heme pocket environment of mammalian peroxidases with diatomic ligan. Biochemistry. 2001;40:10747–10755. doi: 10.1021/bi010478v. [DOI] [PubMed] [Google Scholar]
- 37.Abu-Soud HM, Hazen SL. Nitric oxide modulates the catalytic activity of myeloperoxidase. J Biol Chem. 2000;275:5425–5430. doi: 10.1074/jbc.275.8.5425. [DOI] [PubMed] [Google Scholar]
- 38.Bolscher BG, Wever RA. A kinetic study of the reaction between human myeloperoxidase, hydroperoxides and cyanide. Inhibition by chloride and thiocyanate. Biochim Biophys Acta. 1984;788:1–10. doi: 10.1016/0167-4838(84)90290-5. [DOI] [PubMed] [Google Scholar]
- 39.Hori H, Fenna RE, Kimura S, Ikeda-Saito M. Aromatic substrate molecules bind at the distal heme pocket of myeloperoxidase. J Biol Chem. 1994;269:8388–8392. [PubMed] [Google Scholar]
- 40.Abu-Soud HM, Raushel FM, Hazen SL. A novel multistep mechanism for oxygen binding to ferrous hemoproteins: rapid kinetic analysis of ferrous-dioxy myeloperoxidase (compound III) formation. Biochemistry. 2004;43:11589–11595. doi: 10.1021/bi049541h. [DOI] [PubMed] [Google Scholar]
- 41.Tahboub YR, Galijasevic S, Diamond MP, Abu-Soud HM. Thiocyanate modulates the catalytic activity of mammalian peroxidases. J Biol Chem. 2005;280:26129–26136. doi: 10.1074/jbc.M503027200. [DOI] [PubMed] [Google Scholar]
- 42.Rakita RM, Michel BR, Rosen H. Differential inactivation of Escherichia coli membrane dehydrogenases by a myeloperoxidase-mediated antimicrobial system. Biochemistry. 1990;29:1075–1080. doi: 10.1021/bi00456a033. [DOI] [PubMed] [Google Scholar]
- 43.Galijasevic S, Saed GM, Diamond MP, Abu-Soud HM. Myeloperoxidase up-regulates the catalytic activity of inducible nitric oxide synthase by preventing nitric oxide feedback inhibition. Proc Natl Acad Sci USA. 2003;100:14766–14771. doi: 10.1073/pnas.2435008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Dalen CJ, Whitehouse MW, Winterbourn CC, Kettle AJ. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem J. 1997;327:487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agner KA. kinetic analysis the interaction of human myeloperoxidase with hydrogen peroxide, chloride ion and protons. Acta Chem Scand. 1963;17:S332–338. [Google Scholar]
- 46.Galijasevic S, Saed GM, Diamond MP, Abu-Soud HM. High dissociation rate constant of ferrous-dioxy complex linked to the catalase-like activity in lactoperoxidase. J Biol Chem. 200;279:39465–39470. doi: 10.1074/jbc.M406003200. [DOI] [PubMed] [Google Scholar]
- 47.Galijasevic S, Proteasa G, Abdulhamid I, Abu-Soud HM. The potential role of nitric oxide in substrate switching in eosinophil peroxidase. Biochemistry. 2007;46:406–415. doi: 10.1021/bi061177u. [DOI] [PubMed] [Google Scholar]
- 48.Galijasevic S, Saed GM, Hazen SL, Abu-Soud HM. Myeloperoxidase metabolizes thiocyanate in a reaction driven by nitric oxide. Biochemistry. 2006;45:1255–1262. doi: 10.1021/bi051438k. [DOI] [PubMed] [Google Scholar]
- 49.Bowen DJ, Spring B, Fox E. Tryptophan and high-carbohydrate diets as adjuncts to smoking cessation therapy. J Behav Med. 1991;14:97–110. doi: 10.1007/BF00846173. [DOI] [PubMed] [Google Scholar]
- 50.Wurtman RJ. Nutrients affecting brain composition and behavior. Integr Psychiatry. 1987;5:226–238. [PubMed] [Google Scholar]
- 51.Sandyk R. L-tryptophan in neuropsychiatric disorders: a review. Int J Neurosci. 1992;67:127–144. doi: 10.3109/00207459208994781. [DOI] [PubMed] [Google Scholar]
- 52.Steinberg S, Annable L, Young SN, Liyanage N. A placebo-controlled study of the effects of L-tryptophan in patients with premenstrual dysphoria. Adv Exp Med Biol. 1999;467:85–88. doi: 10.1007/978-1-4615-4709-9_11. [DOI] [PubMed] [Google Scholar]
- 53.Schneider-Helmert D, Spinweber CL. Evaluation of L-tryptophan for treatment of insomnia: a review. Psychopharmacology (Berl) 1986;89:1–7. doi: 10.1007/BF00175180. [DOI] [PubMed] [Google Scholar]
- 54.Korner E, Bertha G, Flooh E, Reinhart B, Wolf R, Lechner H. Sleep-inducing effect of L-tryptophane. Eur Neurol. 1986;25:75–81. doi: 10.1159/000116087. [DOI] [PubMed] [Google Scholar]
- 55.Richard J, Wurtman RJ. In: Trptophan encyclopedia of neuroscience. 3. Adelman G, Smith BH, editors. Burlington, MA: Elsevier Science; 2004. [Google Scholar]
- 56.Kettle AJ, Candaeis LP. Oxidation of tryptophan by redox intermediates of myeloperoxidase and inhibition of hypochlorous acid production. Redox Rep. 2000;5:179–184. doi: 10.1179/135100000101535726. [DOI] [PubMed] [Google Scholar]
- 57.Podrez EA, Abu-Soud HM, Hazen SL. Myeloperoxidase-generated oxidants and atherosclerosis. Free Radic Biol Med. 2000;28:1717–1725. doi: 10.1016/s0891-5849(00)00229-x. [DOI] [PubMed] [Google Scholar]
- 58.Shao B, Oda MN, Vaisar T, Oram JF, Heinecke JW. Pathways for oxidation of high-density lipoprotein in human cardiovascular disease. Curr Opin Mol Ther. 2006;8:198–205. [PubMed] [Google Scholar]
- 59.Xu W, Zheng S, Dweik RA, Erzurum SC. Role of epithelial nitric oxide in airway viral infection. Free Radic Biol Med. 2006;41:19–28. doi: 10.1016/j.freeradbiomed.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Westman E, Lundberg K, Erlandsson Harris H. Arthritogenicity of collagen type II is increased by chlorination. Clin Exp Immunol. 2006;145:339–345. doi: 10.1111/j.1365-2249.2006.03129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arinci A, Ademoglu E, Aslan A, Mutlu-Turkoglu U, Karabulut AB, Karan A. Molecular correlates of temporomandibular joint disease. Oral Sur Oral Med Oral Pathol Oral Radiol Endo. 2005;99:666–670. doi: 10.1016/j.tripleo.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 62.Proteasa G, Tahboub YR, Galijasevic S, Raushel FM, Abu-Soud HM. Kinetic evidence supports the existence of two halide binding sites that have a distinct impact on the heme iron microenvironment in myeloperoxidase. Biochemistry. 2007;46:398–405. doi: 10.1021/bi0609725. [DOI] [PubMed] [Google Scholar]
- 63.Hallingbäck HR, Gabdoulline RR, Wade RC. Comparison of the binding and reactivity of plant and mammalian peroxidases to indole derivatives by computational docking. Biochemistry. 2006;45:2940–2950. doi: 10.1021/bi051510e. [DOI] [PubMed] [Google Scholar]
- 64.Hori H, Fenna RE, Kimura S, Ikeda-Saito M. Aromatic substrate molecules bind at the distal heme pocket of myeloperoxidase. J Biol Chem. 1994;269:8388–8392. [PubMed] [Google Scholar]
- 65.Galijasevic S, Abdulhamid I, Abu-Soud HM. Melatonin is a potent inhibitor of myeloperoxidase. Biochemistry. 2008 doi: 10.1021/bi702016q. In press. [DOI] [PubMed] [Google Scholar]
- 66.Wurtman JJ. The involvement of brain serotonin in excessive carbohydrate snacking by obese carbohydrate craves. J Am Diet Assoc. 1984;84:1004–1007. [PubMed] [Google Scholar]
- 67.Lieberman HR, Corkin S, Spring BJ, Growdon JH, Wurtman RJ. Mood, performance, and pain sensitivity: changes induced by food constituents. J Psychiatr Res. 1982;17:135–145. doi: 10.1016/0022-3956(82)90015-2. [DOI] [PubMed] [Google Scholar]
- 68.Lehnert H, Wurtman RJ. Amino acid control of neurotransmitter synthesis and release: physiological and clinical implications. Psychother Psychosom. 1993;60:18–32. doi: 10.1159/000288676. [DOI] [PubMed] [Google Scholar]
- 69.Minneman KP, Wurtman RJ. The pharmacology of the pineal gland. Annu Rev Pharmacol Toxicol. 1976;16:33–51. doi: 10.1146/annurev.pa.16.040176.000341. [DOI] [PubMed] [Google Scholar]
- 70.Abu-Soud HM, Hazen SL. Nitric oxide is a physiological substrate for mammalian peroxidases. J Biol Chem. 2000;275:37524–3732. doi: 10.1074/jbc.275.48.37524. [DOI] [PubMed] [Google Scholar]
- 71.Abu-Soud HM, Khassawneh MY, Sohn JT, Murray P, Haxhiu MA, Hazen SL. Peroxidases inhibit nitric oxide (NO) dependent bronchodilation: development of a model describing NO-peroxidase interactions. Biochemistry. 2001;40:11866–11875. doi: 10.1021/bi011206v. [DOI] [PubMed] [Google Scholar]
- 72.Burner U, Furtmuller PG, Kettle AJ, Koppenol WH, Obinger C. Mechanism of reaction of myeloperoxidase with nitrite. J Biol Chem. 2000;275:20597–20601. doi: 10.1074/jbc.M000181200. [DOI] [PubMed] [Google Scholar]