

Summary
As a major intracellular degradation pathway, autophagy is tightly regulated to prevent cellular dysfunction in all eukaryotic cells. The rapamycin-sensitive Tor kinase complex 1 is a major regulator of autophagy. Several other nutrient-sensory kinases also play critical roles to precisely modulate autophagy; however, the network of regulatory mechanisms remains largely elusive. We used genetic analyses to elucidate the mechanism by which the stress-responsive, cyclin-dependent kinase, Pho85 and its corresponding cyclin complexes antagonistically modulate autophagy in Saccharomyces cerevisiae. When complexed with cyclins Pho80 and Pcl5, Pho85 negatively regulates autophagy through downregulating the protein kinase Rim15, and the transcription factors Pho4 and Gcn4. The cyclins Clg1, Pcl1 and Pho80, in concert with Pho85, positively regulate autophagy through promoting the degradation of Sic1, a negative regulator of autophagy that targets Rim15. Our results suggest a model in which Pho85 and its cyclin complexes have opposing roles in autophagy regulation.
Keywords: autophagy, Pho85, Sic1, Rim15, TORC1, yeast
Introduction
Eukaryotic cells confronted by various stresses in their intracellular and extracellular environment can initiate an adaptive response, autophagy, for survival (Levine and Klionsky, 2004). Autophagy involves a dynamic rearrangement of subcellular membranes to sequester portions of cytoplasm into a double-membrane vesicle, an autophagosome, which is delivered to a degradative organelle, the vacuole/lysosome, resulting in the breakdown of the contents; the resulting macromolecules are released back into the cytosol for recycling (Huang and Klionsky, 2007; Yang et al., 2006). Autophagy is an evolutionarily conserved pathway that occurs in all eukaryotic cells. In yeast, autophagy helps cells survive nutrient limitation; in mammalian cells, autophagy is also implicated in development and differentiation, cancer, and innate and adaptive immunity (Levine and Deretic, 2007; Levine and Klionsky, 2004).
Autophagy is tightly regulated in order to prevent its occurring at insufficient or excess levels, both of which are harmful for cells. Studies in the budding yeast Saccharomyces cerevisiae have provided important insight into the mechanism of autophagy regulation (Fig. 1A). The target of rapamycin (Tor) proteins form two functionally distinct complexes, Tor complex 1 and 2 (TORC1 and TORC2) (Loewith et al., 2002). TORC1 has primary functions in regulating autophagy induction, and is particularly sensitive to the drug rapamycin (Wullschleger et al., 2006). Under nutrient starvation conditions, TORC1 is inhibited and autophagy is induced. In addition to TORC1, the Ras/cAMP-dependent protein kinase A (PKA) signaling pathway, and Sch9, a homologue of mammalian protein kinase B (PKB)/Akt, also negatively regulate autophagy (Budovskaya et al., 2004; Schmelzle et al., 2004; Yorimitsu et al., 2007). In contrast, the Gcn2 kinase pathway is involved in the positive regulation of autophagy (Talloczy et al., 2002). In response to amino acid starvation, Gcn2 derepresses GNC4 mRNA translation. Gcn4 is a master transcriptional activator and initiates transcriptional induction of nearly all amino acid biosynthetic genes. Upon loss of Gcn2 or Gcn4, autophagy is impaired.
Figure 1.

Pho80 and Pcl5 are the cyclins of Pho85 that participate in the negative regulation of autophagy.
(A) Schematic overview of the key components in autophagy regulation. Arrows represent positive regulation; bars represent negative regulation. Both Clg1 and Pcl1 (not shown) are cyclins that form positive regulatory complexes with Pho85 through the inhibition of Sic1.
(B), (C), (D) and (E) Cells expressing Pho8Δ60 were grown in YPD to midlog phase and then treated with rapamycin for 4 h. The Pho8Δ60 activity was measured as described in Supplemental Experimental Procedures, and was normalized to the activity of wild-type cells with rapamycin treatment, which was set to 100%. Error bars indicate the standard deviation (SD) of three independent experiments. Strains used were wild type (TN124), and atg1Δ (HAY572), and in (B) pho85Δ (ZFY089), pho80Δ (ZFY105), pcl5Δ (ZFY099) and pcl5Δ pho80Δ (ZFY128); in (C) rim15Δ (ZFY100), pho80Δ rim15Δ (ZFY102) and pcl5Δ rim15Δ (ZFY103); in (D) pho4Δ (ZFY135), pho4Δ pho80Δ (ZFY137) and pcl5Δ pho4Δ (ZFY143); in (E) gcn4Δ (ZFY111), gcn4Δ pcl5Δ (ZFY112). See also Figure S1.
Pho85, a yeast cyclin-dependent kinase (CDK), is structurally and functionally related to the mammalian kinase CDK5. It has ten different cyclin regulatory subunits, each of which potentially direct Pho85 to different target substrates that regulate numerous biological functions, including phosphate metabolism, cell cycle control and autophagy regulation (Huang et al., 2007). Pho85 negatively regulates starvation-induced autophagy, antagonistically with a positive regulator of autophagy, Snf1, the closest yeast homologue of the mammalian AMP-activated protein kinase (AMPK) (Wang et al., 2001a). It is unknown, however, which potential cyclin(s) associates with Pho85 to carry out this function, and which downstream target(s) of Pho85 is involved.
Based on sequence alignment within a region called the “cyclin box”, ten Pho85 cyclin (Pcl) partners are grouped into two subfamilies: the Pcl1, 2 subfamily (Pcl1, Pcl2, Pcl5, Pcl9 and Clg1) and the Pho80 family (Pho80, Pcl6, Pcl7, Pcl8, Pcl10; (Carroll and O'Shea, 2002). The Pho80-Pho85 complex signals a response to the stress of phosphate starvation through controlling the activity of the transcription factor Pho4. The Pho80-Pho85 complex also negatively regulates cell entry into a quiescent state (G0) in response to nutrient availability, through direct phosphorylation and retention of Rim15 in the cytosol (Wanke et al., 2005). Rim15 is a protein kinase, which functions as a key controller of many aspects of the G0 program through its ability to integrate signaling from TORC1, PKA, Sch9 and Pho80-Pho85 (Swinnen et al., 2006). Moreover, Rim15 is also required for the induction of autophagy that occurs upon inhibition of PKA and Sch9 (Yorimitsu et al., 2007). Another well-studied ancillary partner of Pho85 is the cyclin Pcl5. Pcl5 targets Pho85 specifically to Gcn4, eventually causing the degradation of this transcription factor (Shemer et al., 2002).
A role of the CDK inhibitor in the regulation of autophagy is suggested by several studies in mammalian cells. p27, a mammalian CDK2-cyclin E inhibitor, has similar functions with the yeast CDK inhibitor Sic1, such as a role in orchestrating the G1/S transition, although they do not share significant sequence similarity (Bloom and Cross, 2007). In cancer cells, overexpression of p27, or expression of a stabilized, active p27, induces autophagy (Komata et al., 2003; Liang et al., 2007). A recently identified small molecule (CpdA) stabilizes p27, in association with the induction of autophagy (Chen et al., 2008). Nonetheless, the mechanism by which p27 positively regulates autophagy remains largely elusive. Using budding yeast as a model system may help to elucidate the mechanisms by which CDK inhibitors regulate autophagy. In yeast, the major kinases involved in controlling Sic1 stability are the Cln-Cdc28 Cdks (Verma et al., 1997); Pho85 is also involved in this process because it phosphorylates Sic1 in vitro and prompts Sic1 degradation in vivo (Nishizawa et al., 1998). However, at this point, it is still unclear which cyclin(s) of Pho85 is required in vivo for the phosphorylation and destabilization of Sic1 (Carroll and O'Shea, 2002). In addition, if Sic1 is involved in the control of autophagy, it is unknown whether Pho85 regulates this process.
Here, we examined the mechanism by which the Pho85 signaling pathway is involved in the control of autophagy. Our work implicates that Pho85 and its corresponding cyclin complexes function as either negative or positive regulators of autophagy. Our results indicate that the yeast CDK inhibitor, Sic1, functions as a negative regulator of autophagy, which is opposite from the mammalian ortholog, p27. Based on our observations, we propose a model for the regulation of autophagy by Pho85 (Fig. 1A).
Results
Pho80 and Pcl5 are the cyclins of Pho85 that participate in the negative regulation of autophagy
Although Pho85 serves as a negative regulator of autophagy (Wang et al., 2001a), it is unclear which Pho85 partner cyclin(s) may be involved, and the mechanism through which Pho85 exerts its effect. Pho80 directs Pho85 to inhibit Rim15 function, and Rim15 is needed for autophagy that is induced by PKA and Sch9 inactivation (Wanke et al., 2005; Yorimitsu et al., 2007). Pcl5 directs Pho85 to initiate the degradation of Gcn4, a protein required for both starvation- and rapamycin-induced autophagy (Shemer et al., 2002; Talloczy et al., 2002). Thus, Pho80 and Pcl5 are potential Pho85 cyclins involved in negative autophagy regulation. In order to measure the level of autophagy induction in the presence of PHO85 or cyclin knockouts, we used the Pho8Δ60 assay (Noda et al., 1995), which measures autophagy-dependent activation of an altered alkaline phosphatase marker.
Wild-type cells grown in rich medium displayed a basal level of Pho8Δ60-dependent alkaline phosphatase activity, and rapamycin treatment increased the level of activity substantially (Fig. 1B). In contrast, atg1Δ cells showed no increase after rapamycin treatment. In agreement with previous studies (Wang et al., 2001a), deletion of PHO85 caused a markedly elevated activity of Pho8Δ60 after rapamycin treatment, being ∼40-50% higher than that of wild-type cells. As expected, upon rapamycin treatment, pho80Δ cells and pcl5Δ cells displayed a significant increase of Pho8Δ60 activity, with values similar to that of pho85Δ cells (Fig. 1B). Thus, Pho80 and Pcl5 are cyclins of Pho85 that negatively regulate autophagy. Double deletion of both PHO80 and PCL5 resulted in ∼80% higher activity of Pho8Δ60 than that of wild-type cells, after rapamycin treatment, suggesting that Pho80-Pho85 and Pcl5-Pho85 kinase complexes may have an additive effect on autophagy and that they presumably function in parallel pathways (Fig. 1A).
As part of our effort to elucidate how Pho80-Pho85 and Pcl5-Pho85 negatively regulate autophagy, we wanted to determine which downstream targets might be involved. We first examined the role of downstream targets of Pho80-Pho85, Rim15 and Pho4. The corresponding higher increase of Pho8Δ60 activity beyond the wild type level in pho80Δ cells depended to a large extent on the presence of Rim15 and Pho4 (Fig. 1C, 1D). However, deletion of RIM15 or PHO4 did not affect the increase of Pho8Δ60 activity in pcl5Δ cells, in agreement with the fact that Rim15 and Pho4 are bona fide targets of Pho80-Pho85, but not Pcl5-Pho85 (Fig. 1A, 1C, 1D). Next, we examined the effect on autophagy of loss of Gcn4, a downstream target of Pcl5-Pho85. Knockout of GCN4 significantly reduced the level of Pho8Δ60 activity in pcl5Δ cells (Fig. 1E). Together, these data suggested that the Pho80-Pho85 and Pcl5-Pho85 kinase complexes contribute appreciably to the negative regulation of autophagy, through their inhibitory roles on Rim15 and Pho4, and on Gcn4, respectively. Further analyses showed that compared to single rim15Δ, pho4Δ, or gcn4Δ mutants or the double gcn4Δ pho4Δ mutant, the triple gcn4Δ pho4Δ rim15Δ mutant displayed the lowest activity of Pho8Δ60, being ∼50% relative to that of wild-type cells, after rapamycin treatment (Fig. S1), suggesting that Rim15, Pho4 and Gcn4 have partially additive effects on autophagy, and presumably regulate autophagy in parallel.
Sic1 functions as a negative regulator of autophagy
In cancer cells, overexpression of a mammalian CDK inhibitor, p27, induces autophagy (Liang et al., 2007). A recently identified small molecule (CpdA) stabilizes p27, in association with the induction of autophagy (Chen et al., 2008). The yeast CDK inhibitor Sic1 is negatively regulated by Pho85 through phosphorylation (Nishizawa et al., 1998). To gain more insight into the mechanism by which this CDK inhibitor regulates autophagy, we decided to investigate the role of Sic1 in autophagy.
We first asked whether overexpression of Sic1 would induce autophagy in yeast, using a GFP-Atg8 processing assay; Atg8 is associated with autophagosomes and, when delivered into the vacuole, GFP-Atg8 is hydrolyzed to yield free GFP, which thus reflects the level of autophagy (Shintani and Klionsky, 2004). The wild-type strain was transformed with a plasmid expressing GFP-Atg8 and a plasmid expressing Sic1 driven by the GAL1 promoter, or an empty vector. After 12 h overexpression of Sic1 in galactose-containing medium, we found that no free GFP was detected in cells, suggesting that autophagy was not induced under this condition (Fig. 2A, lanes 4 and 10). After rapamycin treatment or starvation for nitrogen, cells harboring the empty vector displayed free GFP, whereas the level of free GFP was substantially reduced in the cells overexpressing Sic1 (Fig. 2A). Furthermore, overexpression of Sic1 resulted in a markedly reduced activity of Pho8Δ60 after rapamycin treatment or following nitrogen starvation (Fig. 2B). A similar result was observed when we examined the effect of Sic1 overexpression on the delivery of GFP-Atg8 to the vacuole by fluorescence microscopy. Wild-type cells grown in SMGal displayed a prominent GFP-Atg8 punctum with a perivacuolar localization that corresponded to the phagophore assembly site (PAS; the organizing site for the autophagosome), as well as diffuse cytosolic staining (Fig. 2C). When shifted to SG-N, GFP-Atg8 could also be detected within the vacuole lumen. The atg1Δ mutant prevented the movement of GFP-Atg8 into the lumen. The overexpression of Sic1 also blocked movement of GFP-Atg8 into the vacuole lumen, indicating a defect in autophagy. Thus, these results suggested that the CDK inhibitors Sic1 and p27 have opposite roles (inhibitory and stimulatory, respectively) in the control of autophagy in yeast and mammalian cells. Moreover, deletion of SIC1 caused a significantly elevated activity of Pho8Δ60 after rapamycin treatment and a modest increase following starvation, compared to wild-type cells (Fig. 2D). Taken together, our data indicated that Sic1 functions as a negative regulator of autophagy in parallel with TORC1, although we cannot rule out other models.
Figure 2.
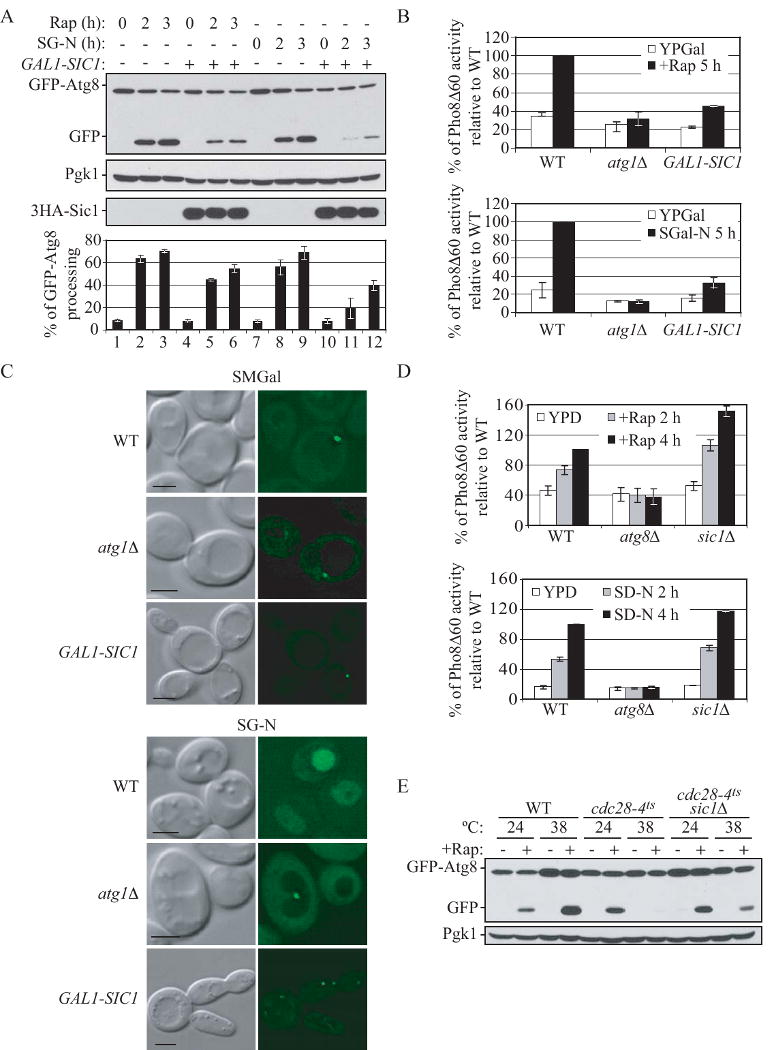
Sic1 functions as a negative regulator of autophagy.
(A) and (B) Overexpression of Sic1 inhibits rapamycin- and nitrogen starvation-induced autophagy. (A) Wild-type (W303-1B) cells expressing GFP-Atg8 (pCU-GFP-AUT7(414)) and expressing either 3HA-Sic1 (pZY011), or an empty vector (pTY006), were grown in SMD and shifted to SMGal for 12 h, and then subjected to either rapamycin treatment or starvation treatment (SG-N). At the indicated times, proteins were TCA-precipitated and subjected to immunoblotting with anti-YFP, anti-HA and anti-Pgk1 (loading control) antisera. Percentage of GFP-Atg8 processing was calculated as described in Supplemental Experimental Procedures. Error bars indicate the SD of three independent experiments.
(B) Wild-type (ZFY202), atg1Δ (TYY181) and GAL1-SIC1 (ZFY203) cells expressing Pho8Δ60 were grown in YPD and shifted to YPGal for 12 h, and were then treated with rapamycin or shifted to nitrogen starvation (SG-N) medium for 5 h. The Pho8Δ60 activity was normalized to the activity of wild-type cells treated with rapamycin or nitrogen starved, which was set to 100%. Error bars indicate the SD of three independent experiments.
(C) Overexpression of Sic1 inhibits delivery of GFP-Atg8 to the vacuole. Wild type (W303-1B), atg1Δ (TYY164), and GAL1-SIC1 (ZFY184) cells expressing GFP-Atg8 (pCU-GFP-AUT7(416)), were analyzed by fluorescence microscopy as described in Supplemental Experimental Procedures. Bar, 5 μm.
(D) Nonspecific autophagy is elevated upon deletion of SIC1. Wild-type (TN124), atg8Δ (YZX200) and sic1Δ (ZFY098) cells expressing Pho8Δ60 were grown to midlog phase and treated with rapamycin, or shifted to nitrogen starvation conditions (SD-N). At the indicated times, the Pho8Δ60 activity was measured, and it was normalized to the activity of wild-type cells with nitrogen starvation or rapamycin treatment for 4 h, which was set to 100%. Error bars indicate the SD of three independent experiments.
(E) The autophagic defect in the cdc28-4ts mutant is partially suppressed by deletion of SIC1. Wild-type cells (BY4742) and temperature-sensitive mutants cdc28-4ts (D4), and cdc28-4ts sic1Δ (ZFY258) expressing GFP-Atg8 (pCU-GFP-AUT7(416)), were grown at 24°C or 38°C for 3 h, and treated with rapamycin for 2 h. TCA-precipitated proteins were subjected to immunoblotting with anti-YFP and anti-Pgk1 (loading control) antisera. See also Figure S2.
Sic1 is controlled by ubiquitin-dependent protein degradation. Cln-Cdc28 can phosphorylate Sic1, which allows Sic1 to be specifically recognized by the F-box protein Cdc4. Cdc4, Cdc53 and Skp1 constitute a ubiquitin ligase complex (SCFCdc4) that cooperates with the ubiquitin-conjugating enzyme Cdc34 to promote the ubiquitination of Phospho-Sic1, leading to its degradation, and S phase entry (Feldman et al., 1997; Schwob et al., 1994). Sic1 accumulates in temperature-sensitive mutants such as cdc28 and cdc34, leading to cell cycle arrest at the G1 phase; whereas deletion of SIC1 allows cdc34 mutant to enter into S phase (Schneider et al., 1998; Schwob et al., 1994). To further examine the negative regulation of autophagy by Sic1, we decided to monitor this process in cdc28 and cdc34 mutants that stabilize Sic1. Cells expressing GFP-Atg8 were grown at permissive temperature and then shifted to non-permissive temperature, followed by rapamycin treatment or nitrogen starvation. Wild-type cells displayed processing of GFP-Atg8 when autophagy was induced by either condition (Fig. S2). In contrast, cdc28-4ts and cdc28-13ts mutants displayed a significant defect in GFP-Atg8 processing at the non-permissive temperature (Fig. S2). cdc34-2ts mutant also displayed an autophagic defect at the non-permissive temperature when autophagy was induced by nitrogen starvation, but not rapamycin treatment (Fig. S2). We next addressed whether accumulation of Sic1 in these cdc mutants is responsible for the autophagic defect. We found that deletion of SIC1 in the cdc28-4ts mutant partially suppressed the defect in GFP-Atg8 processing at non-permissive temperature after rapamycin treatment (Fig. 2E). Thus, these data suggested that autophagy is inhibited in these cdc mutants that are defective in Sic1 degradation and accumulate Sic1.
Pho85 positively regulates autophagy
Pho85 negatively regulates Sic1 (Nishizawa et al., 1998), and our data suggested that Sic1 is a negative regulator of autophagy. Thus, it was tempting to speculate that Pho85 might promote autophagy through downregulation of Sic1, even though Pho85 is also a negative regulator of autophagy. Consistent with this idea, the level of autophagy induced by rapamycin treatment was higher in the double pho80Δ pcl5Δ mutant than in the single pho85Δ mutant (Fig. 1B). This result indicated that loss of PHO85 might have an inhibitory effect on autophagy when the Pho80 and Pcl5 cyclins that exert a negative effect are deleted (Fig. 1A). To test this hypothesis, we used the Pho8Δ60 assay to monitor the induction of autophagy in pho80Δ pcl5Δ cells in the presence and absence of PHO85. When treated with rapamycin, or after nitrogen starvation, pcl5Δ pho80Δ pho85Δ cells displayed ∼1.5-fold lower activity of Pho8Δ60 than that of pcl5Δ pho80Δ cells (Fig. 3A). This result suggested that Pho85 has a positive role in the induction of autophagy, at least in the pho80Δ pcl5Δ background. To further substantiate the premise that Pho85 promotes autophagy, we chose a different genetic background, the gcn4Δ pho4Δ rim15Δ strain, lacking all three downstream targets of Pho80-Pho85 and Pcl5-Pho85 to negatively regulate autophagy (Fig. 1A). Again, we found that deletion of PHO85 decreased the level of autophagy, in this case in the gcn4Δ pho4Δ rim15Δ cells, particularly during nitrogen starvation (Fig. S3).
Figure 3.

Pho85 positively regulates autophagy.
(A) Deletion of PHO85 suppresses autophagy in pcl5Δ pho80Δ cells. Wild-type (TN124), atg1Δ (HAY572), pho85Δ (ZFY089), pcl5Δ pho80Δ (ZFY128) and pcl5Δ pho80Δ pho85Δ (ZFY172) cells were analyzed by the Pho8Δ60 assay, as described in Figure 2D. See also Figure S3.
(B) Addition of the inhibitor 1-Na-PP1 to block Pho85F82G kinase activity partially suppressed autophagy in the background of pcl5Δ pho80Δ, and further deletion of SIC1 relieved the autophagic defect that is seen upon loss of Pho85 activity. Wild-type (ZFY155), pho85-F82G (ZFY145), pho85-F82G atg1Δ (ZFY213), pho85-F82G pcl5Δ pho80Δ (ZFY215) and pho85-F82G pcl5Δ pho80Δ sic1Δ (ZFY214) cells expressing Pho8Δ60, were grown in YPD to midlog phase, and treated (+) or untreated (−) for 1 h with 1-Na-PP1, followed by additional treatment with rapamycin for 5 h to induce autophagy as indicated (total treatment with 1-Na-PP1 was 6 h). Samples were collected and analyzed by the Pho8Δ60 assay. The Pho8Δ60 activity in wild-type cells was normalized to the activity of wild-type cells treated with rapamycin alone, which was set to 100%. The Pho8Δ60 activity in the other four strains was normalized to the activity of pho85-F82G cells treated with rapamycin alone, which was set to 100%. Error bars indicate the SD of three independent experiments. * indicates a statistically non-significant difference (p > 0.05), *** indicates a statistically significant effect of 1-Na-PP1 versus untreated cells (p < 0.01).
Next, we decided to use a chemical genetics approach to eliminate potential confounding effects of the chronic pho85Δ mutation. We took advantage of a strain carrying a mutation in the ATP binding pocket of Pho85 (Pho85F82G). This mutant retains wild-type function but is rapidly inactivated when treated with the cell-permeable ATP-analogue inhibitor 1-Na-PP1 (Carroll et al., 2001). Cells expressing Pho85F82G were treated with or without 1-Na-PP1, followed by additional treatment with rapamycin to induce autophagy. Consistent with our previous finding that Pho85 is partly a negative regulator of autophagy, PHO85-F82G cells displayed an increase of Pho80Δ60 activity when treated with both 1-Na-PP1 and rapamycin, compared to rapamycin alone (Fig. 3B). In contrast, PHO85-F82G pcl5Δ pho80Δ cells showed a decrease of Pho8Δ60 activity in the presence of 1-Na-PP1, in agreement with our result that loss of Pho85 inhibited autophagy in the pcl5Δ pho80Δ background. Notably, 1-Na-PP1 treatment had essentially no effect on rapamycin-induced autophagy in wild-type cells. Taken together, we propose a model in which Pho85 and its corresponding cyclin complexes antagonistically regulate autophagy; these different complexes exert either a negative or a positive regulatory effect (Fig. 1A).
Pho85 inhibits Sic1 to positively regulate autophagy
Having established a role of Pho85 as a positive regulator of autophagy, we decided to determine whether Sic1, which is a known Pho85 substrate, was the downstream target of Pho85 for autophagy induction. 1-Na-PP1 treatment caused a decrease in the level of autophagy that was induced by rapamycin in PHO85-F82G pho80Δ pcl5Δ cells (Fig. 3B). We further extended our analysis by examining the effect of deletion of SIC1. Without 1-Na-PP1 treatment, there was a marked increase in the level of autophagy induced by rapamycin treatment in PHO85-F82G pcl5Δ pho80Δ sic1Δ cells (Fig. 3B), with approximately 1.25-fold higher activity of Pho8Δ60 than in PHO85-F82G pcl5Δ pho80Δ cells. This result suggested that Pho80-Pho85, Pcl5-Pho85, and Sic1, at least in part, negatively regulate autophagy in parallel pathways. When treated with 1-Na-PP1 followed by rapamycin, the PHO85-F82G pcl5Δ pho80Δ sic1Δ cells did not display any decrease in the activity of Pho8Δ60, compared to rapamycin treatment alone (Fig. 3B). This result suggested that the deletion of SIC1 in the PHO85-F82G pcl5Δ pho80Δ cells suppressed the inhibition of autophagy that was caused by inactivation of Pho85 after addition of 1-Na-PP-1.
To further confirm our chemical genetics data, we utilized a complementary approach. Instead of using mutants, we decided to overexpress Pho85 and/or Sic1. To this end, we first examined the effect caused by overexpression of Pho85 alone. GAL1∷HIS3 chromosomal tagging was used to replace the endogenous PHO85 promoter. After 12 h overexpression of Pho85 in galactose-containing medium, GFP-Atg8 was processed in nutrient-rich conditions even without rapamycin treatment (Fig. 4A), although the level of autophagy induction was low. Additional deletion of PHO80 and PCL5, increased the level of free GFP by ∼30-40%, which further confirmed the view that Pho80-Pho85 and Pcl5-Pho85 negatively regulate autophagy. Deletion of ATG1 completely abolished the induction of autophagy upon overexpression of Pho85, indicating that the induction was Atg1-dependent. A similar result was observed when we examined the effect of Pho85 overexpression on the delivery of GFP-Atg8 to the vacuole by fluorescence microscopy (Fig. 4B). We then extended our analysis to examine the ability of Pho85 to induce autophagy by using the quantitative Pho8Δ60 assay. Overexpression of Pho85 caused a significant induction of autophagy in pcl5Δ pho80Δ GAL1-PHO85 cells, with approximately 2-fold higher activity of Pho8Δ60 than that of pcl5Δ pho80Δ cells (Fig. 4C, open bars). After additional treatment with rapamycin, the activity of Pho8Δ60 dramatically increased, being ∼40-50% higher than that of pcl5Δ pho80Δ cells (Fig. 4C, closed bars). Taken together, these results further supported the model that Pho85 and its cyclin complexes have opposing roles in autophagy regulation.
Figure 4.
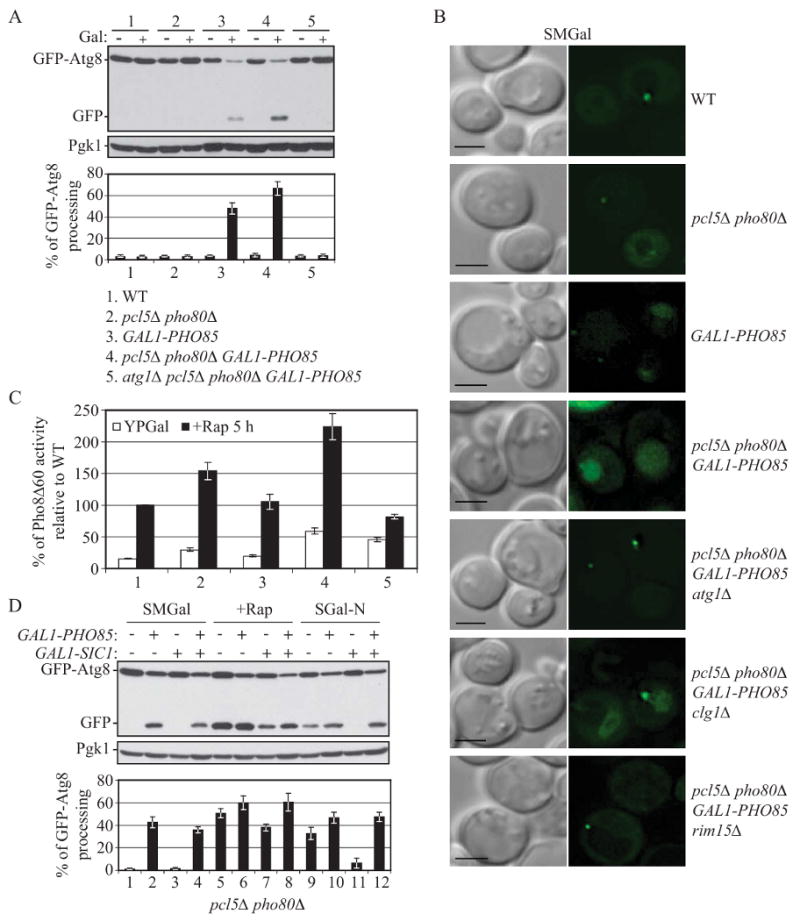
Overexpression of Pho85 induces autophagy and relieves the inhibitory effect of Sic1 overexpression on autophagy.
(A), (B) and (C) Overexpression of Pho85 induces autophagy. (A) and (B) Wild-type (W3030-1B), pcl5Δ pho80Δ (ZFY207), GAL1-PHO85 (ZFY209), pcl5Δ pho80Δ GAL1-PHO85 (ZFY208), atg1Δ pcl5Δ pho80Δ GAL1-PHO85 (ZFY217), clg1Δ pcl5Δ pho80Δ GAL1-PHO85 (ZFY246) and pcl5Δ pho80Δ rim15Δ GAL1-PHO85 (ZFY216) cells expressing GFP-Atg8 (pCU-GFP-AUT7(416)), were grown in SMD and shifted to SMGal for 12 h. In (A), cells that were grown in SMD (−) or SMGal (+) were collected and subjected to immunoblotting, as described in Figure 2A. In (B), cells that were grown in SMGal were analyzed by fluorescence microscopy as described in Supplemental Experimental Procedures. Bar, 2.5 μm.
(C) Wild type (ZFY202), pcl5Δ pho80Δ (ZFY206), GAL1-PHO85 (ZFY224), pcl5Δ pho80Δ GAL1-PHO85 (ZFY225) and atg1Δ pcl5Δ pho80Δ GAL1-PHO85 (ZFY227) cells were grown in YPD and shifted to YPGal for 12 h, then treated with rapamycin for 5 h and analyzed by the Pho8Δ60 assay. Values were normalized to the activity of wild-type cells with rapamycin treatment, which was set to 100%. Error bars indicate the SD of three independent experiments.
(D) In pcl5Δ pho80Δ cells, overexpression of Pho85 suppresses the autophagic defect resulting from Sic1 overexpression. pcl5Δ pho80Δ (ZFY207) and pcl5Δ pho80Δ GAL1-PHO85 (ZFY208) cells were analyzed by the GFP-Atg8 processing assay, as described in Figure 2A. See also Figure S4.
Next, we examined the effect of overexpressing Sic1, or both Pho85 and Sic1. We again used the pcl5Δ pho80Δ mutant to simplify the analysis by removing the Pho80-Pho85- and Pcl5-Pho85-mediated negative regulation pathways (Fig. 1A). To this end, the pcl5Δ pho80Δ cells and pcl5Δ pho80Δ GAL1-PHO85 cells were co-transformed with a plasmid expressing GFP-Atg8, and a plasmid allowing galactose-inducible expression of SIC1, or an empty vector. As expected, after 12 h in galactose-containing medium, in pcl5Δ pho80Δ GAL1-PHO85 cells containing an empty vector, autophagy was induced without any additional treatment (Fig. 4D, lane 2); whereas treatment with rapamycin or nitrogen starvation resulted in a significantly elevated level of free GFP (Fig. 4D, compare lane 5 to 6, and 9 to 10). Sic1 overexpression inhibited autophagy induction (Fig. 4D, compare lane 5 to 7, and 9 to 11), although the inhibition was only partial in the case of rapamycin treatment. In contrast, when both Sic1 and Pho85 were overexpressed, the inhibition of autophagy caused by Sic1 overexpression was abolished (Fig. 4D, compare lane 3 to 4, 7 to 8, and 11 to 12). Taken together, we infer that Pho85 targeted the CDK inhibitor Sic1 for degradation to relieve the inhibitory effect of Sic1 overexpression on autophagy, hence promoting autophagy induction.
Clg1 targets Pho85 to antagonize Sic1 inhibition of autophagy
Having established the positive regulatory role of Pho85 in autophagy, we sought to determine which particular Pho85 cyclin(s) may be involved. A pcl6Δ pcl7Δ double mutant displays normal induction of autophagy (Wang et al., 2001b). By using the Pho8Δ60 assay, we found essentially normal induction of autophagy in pcl8Δ, pcl10Δ or pcl8Δ pcl10Δ double mutant cells (Fig. S4A). Similarly, pcl1Δ, pcl2Δ, pcl9Δ or triple pcl1Δ pcl2Δ pcl9Δ cells did not display any discernable difference in Pho8Δ60 activity relative to wild-type cells (Fig. S4B, 5A). We then turned to the cyclin Clg1, which belongs to the Pcl1, 2 subfamily; the role of Clg1 has not yet been clearly characterized. When treated with rapamycin, cells deleted for CLG1 displayed a significant reduction in Pho8Δ60 activity, being ∼30% lower than that of wild-type cells (Fig. 5A). This result suggested that Clg1 is a potential cyclin of Pho85 to positively regulate autophagy. When autophagy was induced by nitrogen starvation, however, clg1Δ cells displayed no appreciable difference in Pho8Δ60 activity compared to the wild type although further deletion of PCL1, PCL2 and PCL9 significantly reduced the level of autophagy (Fig. 5A). When autophagy was induced by overexpression of Pho85, deletion of CLG1 significantly reduced the level of free GFP by ∼70-80% (Fig. 5B). Similarly, there was a reduction in the delivery of GFP-Atg8 to the vacuole (Fig. 4B). These results suggested that Clg1 is a major cyclin that targets Pho85 to positively regulate autophagy, whereas Pcl1, Pcl2 and Pcl9 might function redundantly with Clg1 in autophagy regulation. To test the possibility that the autophagic defect seen in clg1Δ and clg1Δ pcl1Δ pcl2Δ pcl9Δ cells was due to the accumulation of Sic1, we decided to examine the stability of protein A (PA)-tagged Sic1 in the presence of PHO85 or cyclin knockouts. Deletion of PHO85 largely stabilized PA-Sic1 (Fig. S5A), in agreement with previous studies (Nishizawa et al., 1998). Although single deletion of CLG1, PCL1, PCL2 or PCL9 did not appreciably stabilize PA-Sic1, triple knockout pcl1Δ pcl2Δ pcl9Δ cells and quadruple knockout clg1Δ pcl1Δ pcl2Δ pcl9Δ cells displayed an approximately 2-fold and 2.5-fold higher amount of PA-Sic1, respectively, compared to wild-type cells (Fig. S5A). These results suggested that Clg1, Pcl1, Pcl2 and Pcl9 might be functionally redundant cyclins of Pho85 to promote Sic1 degradation.
Figure 5.
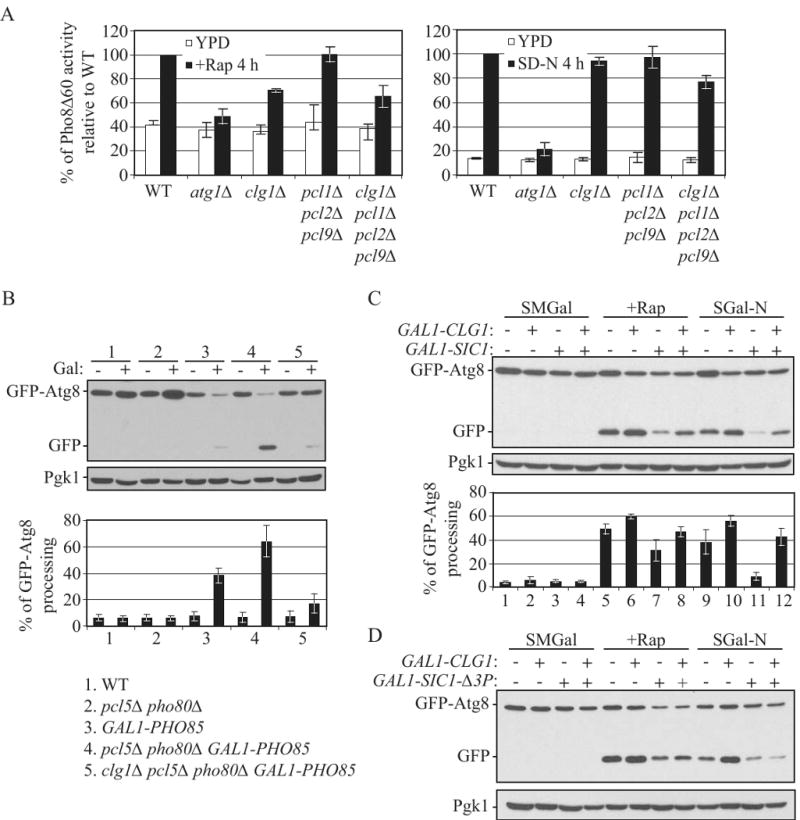
Clg1 targets Pho85 to antagonize Sic1 inhibition of autophagy.
(A) Deletion of CLG1 inhibits autophagy induced by rapamycin treatment. Wild-type (TN124), atg1Δ (HAY572), clg1Δ (ZFY182), pcl1Δ pcl2Δ pcl9Δ (ZFY129) and clg1Δ pcl1Δ pcl2Δ pcl9Δ (ZFY183) cells were analyzed by the Pho8Δ60 assay, as described in Figure 2D.
(B) Deletion of CLG1 inhibits autophagy induced by overexpression of Pho85 in pcl5Δ pho80Δ cells. Wild-type (W3030-1B), pcl5Δ pho80Δ (ZFY207), GAL1-PHO85 (ZFY209), pcl5Δ pho80Δ GAL1-PHO85 (ZFY208) and clg1Δ pcl5Δ pho80Δ GAL1-PHO85 (ZFY246) cells were analyzed by the GFP-Atg8 processing assay, as described in Figure 4A.
(C) and (D) In pcl5Δ pho80Δ cells, overexpression of Clg1 partially suppresses the autophagic defect resulting from overexpression of wild-type Sic1 but not the degradation-resistant mutant, Sic1-Δ3P. pcl5Δ pho80Δ (ZFY207) and pcl5Δ pho80Δ GAL1-CLG1 (ZFY220) cells expressing either 3HA-Sic1 (pZY011), 3HA- Sic1-Δ3P (pZY016), or an empty vector (pTY006), were analyzed by the GFP-Atg8 processing assay, as described in Figure 2A. See also Figure S5.
To further assess whether Clg1-Pho85 regulates autophagy through targeting Sic1 for degradation, we examined the phenotypes associated with co-overexpression of Sic1 and Clg1. pcl5Δ pho80Δ cells bearing a plasmid expressing Sic1 driven by the GAL1 promoter displayed a slow growth phenotype on a galactose-containing plate, compared to vector alone; whereas Clg1 overexpression partially suppressed the slow growth phenotype resulting from overexpression of Sic1 (Fig. S5B). We then asked whether overexpression of Clg1 suppressed the inhibition of autophagy caused by Sic1 overexpression. After 12 h in galactose-containing medium followed by treatment with rapamycin or nitrogen starvation, a pcl5Δ pho80Δ GAL1-CLG1 strain overexpressing both Clg1 and Sic1, displayed a substantially greater level of free GFP than the pcl5Δ pho80Δ strain overexpressing Sic1 alone (Fig. 5C, compare lane 7 to 8, and 11 to 12). A similar result was observed when we examined the effect of combined overexpression of Clg1 and Sic1on the delivery of GFP-Atg8 to the vacuole by fluorescence microscopy (Fig. S5C). Thus, Clg1-Pho85 potentially targets Sic1 for degradation to relieve inhibition of autophagy caused by Sic1 overexpression.
An in vivo phosophorylation study suggests that Pho85 phosphorylates one of the consensus sites (Thr5) of Sic1, and at least one of another two sites (Val33 and Ser76), for efficient degradation of Sic1 (Nishizawa et al., 1998). To further clarify the mechanism of Clg1-Pho85 in the destabilization of Sic1, we generated a mutant version of Sic1, Sic1-Δ3P (T5A, T33V, and S76A). This mutant has a severe ubiquitination defect and is largely stabilized; overexpression of Sic1-Δ3P driven by the GAL1 promoter strongly inhibits cell growth (Verma et al., 1997). As expected, overexpression of Sic1-Δ3P strongly inhibited cell proliferation in both wild-type cells and GAL1-CLG1 cells (Fig. S5B). Moreover, overexpression of Clg1 failed to suppress the autophagic defect when Sic1-Δ3P was overexpressed (Fig. 5D and S5C), suggesting that Sic1 is degraded in a Clg1-dependent manner, and that this degradation requires phosphorylation of Sic1. Taken together, these results let us propose a model in which the Clg1-Pho85 kinase complex (as well as Pcl1/2/9-Pho85) targets Sic1 for degradation, exerting a positive regulatory effect on autophagy induction (Fig. 1A).
Phosphorylation of Sic1 by Pho85 associated with Clg1, Pcl1 and Pho80
When complexed with Pcl1, Pho85 phosphorylates Sic1 in vitro (Nishizawa et al., 1998), although there is no evidence to show that Pcl1 is the actual cyclin required in vivo for the phosphorylation and hence destabilization of Sic1. Based on our model that the Clg1-Pho85 kinase complex exerts a positive regulatory effect on autophagy through destabilizing Sic1, we decided to examine whether this complex is able to directly phosphorylate Sic1 in an in vitro phosphorylation assay. Pho85 associates with its cyclin partners and hence should co-immunoprecipitate with them (Measday et al., 1997). Immunoprecipitated HA-tagged Clg1 obtained from PHO85 cells but not from pho85Δ cells phosphorylated Sic1, indicating specific phosphorylation by the Clg1-Pho85 complex (Fig. 6A). As our genetic data suggested that Clg1 is not the sole cyclin that targets Pho85 to positively regulate autophagy, we decided to extend our in vitro phosphorylation analysis to cyclins Pcl1, Pcl2, Pcl9, Pho80 and Pcl5. Consistent with the result from Nishizawa et al. (1998), immunoprecipitated HA-tagged Pcl1, but not Pcl2, obtained from PHO85 cells but not from pho85Δ cells, phosphorylated Sic1 in vitro (Fig. 6A and S6A). Phosphorylated Sic1 was also obtained with Pho80 (Fig. 6A); however, we could not detect phosphorylation of Sic1 by immunoprecipitated HA-tagged Pcl9 or Pcl5 in vitro (Fig. S6A). Further analysis demonstrated that the Clg1-Pho85, Pcl1-Pho85 or Pho80-Pho85-mediated phosphorylation of Sic1 was significantly reduced by the introduction of the T5A, T33V, and S76A mutations (Sic1-Δ3P) (Fig. 6A), suggesting that T5, T33 and/or S76 are the Pho85 targets in vitro (notably, the residual phosphorylation level in Sic1-Δ3P suggests the presence of an additional site(s) in Sic1 that may be target in vitro by Pho85). Taken together, our data suggested that the Pho85 cyclins Clg1, Pcl1 and Pho80 contribute to the kinase activity of Pho85 towards the T5, T33 and/or S76 sites on Sic1.
Figure 6.
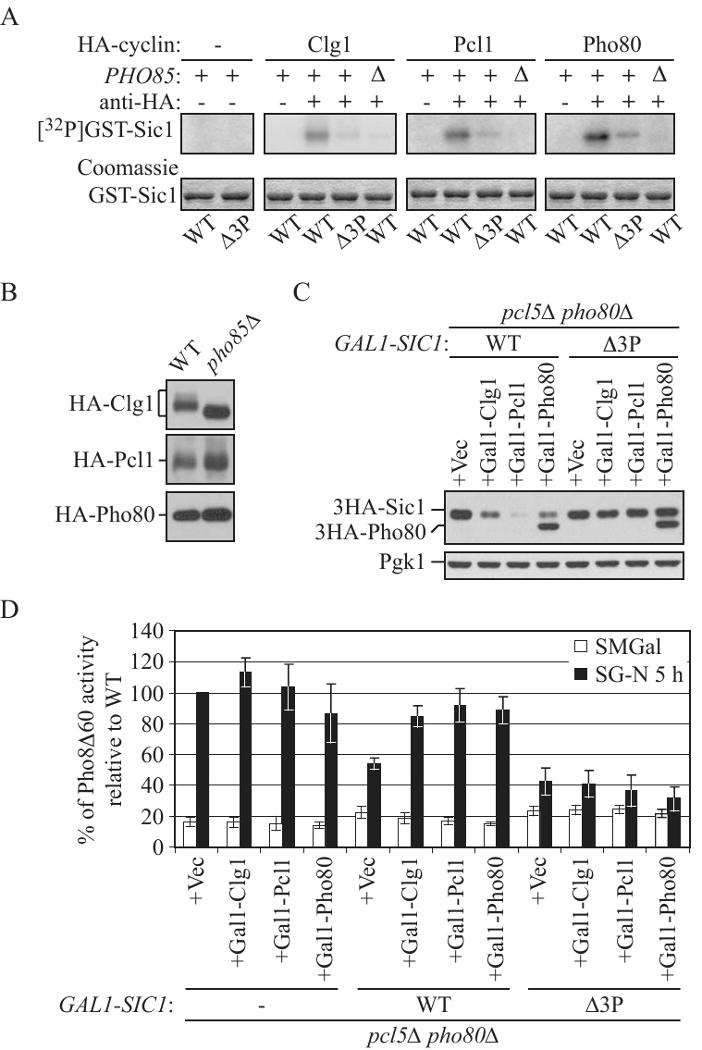
Clg1, Pcl1 and Pho80 are cyclin partners of Pho85 for Sic1 phosphorylation.
(A) In vitro phosphorylation of T5, T33 and/or S76 in Sic1 by Clg1-, Pcl1- or Pho80-associated Pho85 complexes. GST-Sic1 (pZY037) and GST-Sic1-Δ3P (pZY040), expressed and purified from E. coli, were used as substrates in in vitro kinase assay using immunoprecipitated HA-tagged Clg1 (pZY017), Pcl1 (pZY018), and Pho80 (pZY022) from wild-type (W303-1B) or pho85Δ (ZFY088) cells. As a negative control, the antibody was not added (-) to the reaction. Phosphorylated proteins were detected by autoradiography (top) and the protein input was shown by Coomassie Blue staining (bottom).
(B) The immunoprecipitates used in (A) were analyzed for the presence of HA-cyclin by immunoblotting using anti-HA antibody.
(C) and (D) Overexpression of Clg1, Pcl1 or Pho80 reduced the abundance of overexpressed Sic1 wild type, but not the Sic1-Δ3P mutant, and suppressed the inhibitory effect of overexpression of wild-type Sic1, but not Sic1-Δ3P, on autophagy. pcl5Δ pho80Δ (ZFY255), pcl5Δ pho80Δ GAL1-3HA-SIC1 (ZFY256) and pcl5Δ pho80Δ GAL1-3HA-SIC1-Δ3P (ZFY257) cells expressing either 3HA-Clg1 (pZY017), 3HA-Pcl1 (pZY018), 3HA-Pho80 (pZY022) or an empty vector (pTY006), were grown in SMD and shifted to SMGal for 12 h. In (C), samples were collected and analyzed by immunoblotting using anti-HA and anti-Pgk1 (loading control) antisera; in (D) cells were shifted to SG-N and analyzed by the Pho8Δ60 assay. Values were normalized to the activity of ZFY255 cells bearing pTY006, and subjected to nitrogen starvation, which was set to 100%. Error bars indicate the SD of three independent experiments. See also Figure S6.
To assess the relevance of Sic1 phosphorylation by Clg1-, Pcl1- and Pho80-associated Pho85 complexes in vivo, we assayed the effects of overexpression of these cyclins on the stability of Sic1 or Sic1-Δ3P, and hence the effect on autophagy. pcl5Δ pho80Δ cells bearing chromosomally integrated GAL1 promotor-driven HA-tagged Sic1 or Sic1-Δ3P, were transformed with a plasmid expressing GAL1 promotor-driven HA-tagged Clg1, Pcl1, Pcl2, Pcl9, Pho80 or Pcl5, or an empty vector. Although all of the various cyclin-CDK complexes displayed similar stability (Fig. 6B and S6B), after 12 h in galactose-containing medium, cells that overexpressed Sic1 alone accumulated Sic1, whereas cells that additionally overexpressed Clg1, Pcl1 or Pho80, but not Pcl9, Pcl2 or Pcl5, displayed a markedly reduced level of Sic1 (Fig. 6C and S6C). Furthermore, the defect of nitrogen starvation-induced autophagy caused by overexpression of Sic1 was almost completely abolished by overexpression of Clg1, Pcl1 or Pho80, but not Pcl9, Pcl2 or Pcl5 (Fig. 6D and S6D). In contrast, in the cells that overexpressed Sic1-Δ3P, the level of Sic1-Δ3P did not decrease appreciably when combined with overexpression of Clg1, Pcl1 or Pho80 (Fig. 6C). Correspondingly, these cells were unable to relieve the autophagic defect caused by accumulation of Sic1-Δ3P (Fig. 6D). Taken together, the above results suggested that Pho85 forms complexes with cyclins Clg1, Pcl1 and Pho80 to directly phosphorylate T5, T33 and/or S76 of Sic1, and hence promote autophagy by targeting Sic1 for degradation.
Rim15 is a downstream target of Sic1
To better understand how Sic1 negatively regulates autophagy, we decided to identify which downstream effectors of Sic1 might be involved in autophagy regulation. The data presented above suggested that Pho80-Pho85, Pcl5-Pho85, and Sic1, at least in part, negatively regulate autophagy in parallel pathways. If this is true, one would expect to see that loss of the three downstream targets of Pho80-Pho85 and Pcl5-Pho85 (Fig. 1A) would not suppress the upregulation of autophagy that was seen in the sic1Δ mutant. When treated with rapamycin, deletion of PHO4 had little, if any, effect on Pho8Δ60 activity in sic1Δ cells (Fig. 7A). In contrast, deletion of RIM15 or GCN4 resulted in a dramatic decrease in the Pho8Δ60 activity of sic1Δ cells (Fig. 7A). This result suggested that Rim15 and/or Gcn4, but not Pho4, might be downstream targets of Sic1.
Figure 7.
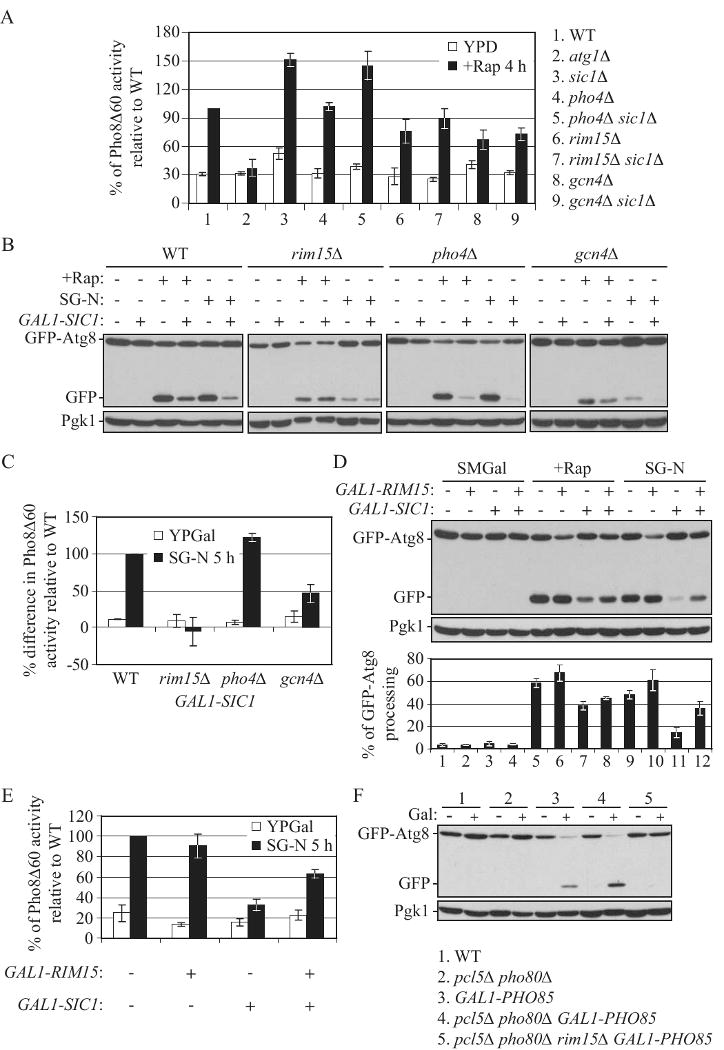
Rim15 is a downstream target of Sic1.
(A) Deletion of RIM15 or GCN4, but not PHO4, suppresses the upregulation of autophagy in sic1Δ cells. Wild-type (TN124), atg1Δ (HAY572), sic1Δ (ZFY098), pho4Δ (ZFY135), pho4Δ sic1Δ (ZFY141), rim15Δ (ZFY100), rim15Δ sic1Δ (ZFY116), gcn4Δ (ZFY111) and gcn4Δ sic1Δ (ZFY115) cells were analyzed by the Pho8Δ60 assay, as described in Figure 2D.
(B) and (C) Overexpression of Sic1 inhibits autophagy in wild-type, pho4Δ and gcn4Δ cells, but not rim15Δ cells. (B) Wild type (W303-1B), rim15Δ (ZFY132), pho4Δ (ZFY170), gcn4Δ (ZFY131), GAL1-SIC1 (ZFY184), rim15Δ GAL1-SIC1 (ZFY187), pho4Δ GAL1-SIC1 (ZFY188) and gcn4Δ GAL1-SIC1 (ZFY185) cells were analyzed by the GFP-Atg8 processing assay, as described in Figure 2A. (C) Wild-type (ZFY202), rim15Δ (ZFY204), pho4Δ (ZFY252), gcn4Δ (ZFY251), GAL1-SIC1 (ZFY203), rim15Δ GAL1-SIC1 (ZFY249), pho4Δ GAL1-SIC1 (ZFY250) and gcn4Δ GAL1-SIC1 (ZFY248) cells were analyzed by the Pho8Δ60 assay, as described in Figure 2B. The difference of the Pho8Δ60 activity between the absence and presence of Sic overexpression was normalized to the difference in wild-type cells treated with nitrogen starvation, which was set to 100%. Error bars indicate the SD of three independent experiments.
(D) and (E) Overexpression of Rim15 partially suppresses the inhibitory effect of Sic1 overexpression on autophagy. (D) Wild-type (W303-1B), GAL1-RIM15 (ZFY192), GAL1-SIC1 (ZFY184) and GAL1-SIC1 GAL1-RIM15 (ZFY193) cells were analyzed by the GFP-Atg8 processing assay, as described in Figure 2A. (E) Wild-type (ZFY202), GAL1-RIM15 (ZFY253), GAL1-SIC1 (ZFY203) and GAL1-SIC1 GAL1-RIM15 (ZFY254) cells were analyzed by the Pho8Δ60 assay, as described in Figure 2B.
(F) Deletion of RIM15 blocks the induction of autophagy upon overexpression of Pho85 in pcl5Δ pho80Δ cells. Wild-type (W303-1B), pcl5Δ pho80Δ (ZFY207), GAL1-PHO85 (ZFY209), pcl5Δ pho80Δ GAL1-PHO85 (ZFY208) and pcl5Δ pho80Δ rim15Δ GAL1-PHO85 (ZFY216) cells were analyzed by the GFP-Atg8 processing assay, as described in Figure 4A. See also Figure S7.
We reasoned that if Sic1 inhibits autophagy through downregulating Rim15 and/or Gcn4, then overexpression of Sic1 in mutants lacking Rim15 or Gcn4 would not exaggerate the defect in autophagy. To this end, the GAL1 promoter was integrated at the SIC1 locus to allow conditional overexpression of SIC1 in wild-type, rim15Δ, pho4Δ and gcn4Δ cells. With rapamycin or nitrogen starvation, the level of free GFP processed from GFP-Atg8 was significantly reduced upon overexpression of Sic1 in wild-type, pho4Δ and gcn4Δ cells (Fig. 7B). In contrast, in rim15Δ cells, there was no discernable difference in GFP-Atg8 processing between the presence and absence of Sic1 overexpression (Fig. 7B). Furthermore, overexpression of Sic1 resulted in a markedly reduced activity of Pho8Δ60 that was induced by nitrogen starvation, in wild-type, pho4Δ and gcn4Δ cells, but not rim15Δ cells (Fig. 7C). Notably, deletion of RIM15, PHO4 or GCN4 does not affect the stability of Sic1 (Fig. S7A). Thus, these results indicated that Rim15, but not Gcn4, might be a downstream target of Sic1.
To further examine this possibility, we generated a strain in which the GAL1 promoter was integrated in front of both RIM15 and SIC1. Overexpression of Sic1 driven by the GAL1 promoter displayed a slow growth phenotype on a galactose-containing plate, compared to wild-type cells; whereas Rim15 overexpression significantly suppressed the slow growth phenotype resulting from overexpression of Sic1 (Fig. S7B and S7C). When cells were shifted to nitrogen starvation, the percentage of GFP-Atg8 processing was dramatically increased in cells co-overexpressing Rim15 and Sic1, being ∼2.5-fold higher than that of cells overexpressing Sic1 alone (Fig. 7D, compare lane 11 to 12). Furthermore, the level of Pho8Δ60 activity was significantly elevated (∼2-fold increase) in cells co-overexpressing Rim15 and Sic1, compared to that of cells overexpressing Sic1 alone (Fig. 7E). Thus, these data let us propose a model in which Rim15 acts as a downstream target of Sic1, and overexpression of Rim15 acts to antagonize the inhibition of autophagy caused by Sic1 overexpression.
Based on the above data, Pho85 negatively regulated Sic1, and Sic1 negatively regulated Rim15, which indicated that Rim15 may be essential for autophagy induced by Pho85 overexpression. However, since Rim15 is also under the negative regulation of Pho80-Pho85, a negative regulator of autophagy, loss of Rim15 would presumably suppress both negative and positive regulation of Pho85 (Fig. 1A). To this end, we decided to examine the effect associated with deletion of RIM15 in the background of pcl5Δ pho80Δ GAL1-PHO85. We found that loss of Rim15 completely abolished the appearance of vacuolar GFP-Atg8 (Fig. 4B) and free GFP induced by Pho85 overexpression (Fig. 7F). Thus, these data further strengthened our model that Rim15 is a downstream target of Sic1.
Discussion
The nutritional environment is a critical determinant of cellular behavior. As an intracellular degradative process, autophagy is tightly regulated by intracellular and extracellular nutrient levels. Several key intracellular regulators, many of which are protein kinases, play roles in the regulation of autophagy, including Tor, PKA, Sch9 and Pho85. In this study, we elucidated the mechanism of Pho85 in autophagy regulation, and demonstrated: (1) Pho80 and Pcl5 target Pho85 to negatively regulate autophagy via downregulating Rim15, Pho4 and Gcn4; (2) Sic1 is a negative regulator of autophagy; (3) Clg1, Pcl1 and Pho80 target Pho85 to positively regulate autophagy via destabilizing Sic1; (4) Rim15 acts as a downstream target of Sic1; although we have not been able to show that Rim15 is directly downstream of Sic1.
Pho85 seems to be involved in at least two types of sensing, including roles in environmental signaling and cell cycle regulation (Huang et al., 2007). The main role of Pho85 in environmental signaling is to turn off activities that are needed only under specific stress conditions. For example, when inorganic phosphate becomes scarce, Pho80-Pho85 is inhibited, thereby allowing activation of Pho4 which promotes the expression of genes involved in phosphate metabolism, and activation of Rim15 which is a key controller of many aspects of the G0 program. In response to amino acid starvation, the level of the Pcl5 protein is substantially reduced, resulting in hypophosphorylation and stabilization of Gcn4, which activates amino acid biosynthesis genes. We found that both of these two nutrient sensors, Pho80-Pho85 and Pcl5-Pho85, negatively regulate autophagy. Thus, Pho85 links the regulation of autophagy to phosphate and amino acid starvation.
In addition to serving as a repressor of environmental stress responses, Pho85 has important roles in regulating cell-cycle progression (Carroll and O'Shea, 2002). We investigated one of the targets of Pho85 with well-defined functions in G1/S progression, the CDK inhibitor Sic1. While most previous work has linked only the roles of Sic1 to cell cycle regulation, our observations indicate that it also functions as a negative regulator of autophagy. Moreover, we found that Pho85 is a positive regulator of autophagy through targeting Sic1 for degradation, and apparently several of the cyclins, including Clg1, form complexes with Pho85 to fulfill this role. Thus, these data demonstrated that Pho85 and its corresponding cyclin complexes have two separate and opposite roles in autophagy regulation.
One question is to ask which cyclin partner(s) function together with Pho85 to promote autophagy. We reasoned that since Pho85 promotes the induction of autophagy, then the corresponding cyclin(s) must also be involved in autophagy regulation. We found that deletion of CLG1 alone, but not other cyclins, significantly reduced the level of autophagy that was induced by rapamycin (Fig. 5A, S4). On the other hand, when starved for nitrogen, the clg1Δ pcl1Δ pcl2Δ pcl9Δ quadruple mutant displayed a significantly lower Pho8Δ60 activity than the single clg1Δ mutant, suggesting that Pcl1, Pcl2 and Pcl9 might have some redundant role with Clg1 in autophagy regulation. Moreover, deletion of CLG1 significantly inhibited, but did not completely abolish the induction of autophagy by Pho85 overexpression (Fig. 5B, 4B), suggesting that Clg1 is not the sole cyclin that targets Pho85 to positively regulate autophagy. Since Pho85 promotes the induction of autophagy through destabilizing Sic1, then the corresponding cyclin(s) must also be involved in regulation of Sic1 stability. Sic1 must be phosphorylated to be targeted for degradation; accordingly, we then asked which cyclin(s) is responsible for Pho85-associated kinase activity toward Sic1. We found that Clg1 was the primary autophagy-regulating cyclin partner of Pho85 to phosphorylate Sic1 in vitro (Fig. 6A). In addition, we confirmed the previous result from Nishizawa et al. (1998) that the Pcl1-Pho85 (but not Pcl2-Pho85) complex phosphorylates Sic1 in vitro. Intriguingly, the stress responsive Pho80-Pho85 complex strongly phosphorylated Sic1 in vitro. We further investigated the mutant version of Sic1, Sic1-Δ3P, which contains T5A, T33V and S76A mutations. Sic1-Δ3P was phosphorylated to a significantly lower extent than wild-type Sic1 (Fig. 6A), suggesting at least T5, T33 and/or S76 were phosphorylated in vitro. To gain more insight into the physiological relevance of these phosphorylation events in vivo, we further examined the effect of overexpression of these cyclins on the regulation of Sic1 stability and the induction of autophagy. Indeed, overexpression of Clg1, Pcl1 or Pho80 significantly reduced the level of wild-type Sic1, but not Sic1-Δ3P, and overcame the inhibition of autophagy caused by overexpression of wild-type Sic1, but not Sic1-Δ3P (Fig. 6C and 6D). Taken together, these data support our model that Pho85 forms complexes with cyclins Clg1, Pcl1 and Pho80 to directly phosphorylate T5, T33 and/or S76 of Sic1, and hence promote autophagy by targeting Sic1 for degradation. The observations that Pho80-Pho85 negatively regulated autophagy through inactivating Rim15 and Pho4, and also positively regulated autophagy through downregulating Sic1, led us to propose that it has dual roles in autophagy regulation (Fig. 1A); however, further studies are clearly needed to clarify the relationship between Clg1, Pcl1 and Pho80 in Sic1 and autophagy regulation. The observation that single clg1Δ cells, but not other single pcl mutants, showed an autophagic defect (Fig. 5A, S4), indicated that Clg1 might play a primary role. Pho80-Pho85 is localized exclusively to the nucleus (Kaffman et al., 1998); Clg1 is localized primarily to the nucleus (our unpublished data), whereas Pcl1 localizes partially to the nucleus as well as sites of polarized cell growth (Moffat and Andrews, 2004). Moreover, transcript levels for CLG1 and PHO80 are constant throughout the cell cycle, whereas PCL1 is specifically expressed during the late G1 phase (Measday et al., 1997). Thus, it is likely that subcellular localization of Pcl-Pho85 complexes and/or the timing of expression of these cyclin genes contributes to the selection of when and where to phosphorylate Sic1 and promote its degradation.
In mammalian cells, the CDK inhibitor p27 functions as a positive regulator of autophagy (Liang et al., 2007). In the present study, however, we found that the yeast CDK inhibitor Sic1 is a negative regulator of autophagy, based on the observations that overexpression of Sic1 or Sic1-Δ3P, a degradation-resistant mutant, significantly inhibited autophagy, and that loss of Sic1 dramatically upregulated autophagy. Moreover, autophagy was inhibited in the cdc28-4ts and cdc34-2ts mutants, in which Sic1 accumulates (Schwob et al., 1994), whereas deletion of SIC1 in the cdc28-4ts mutant partially suppressed the defect in autophagy induction. These observations fit well with the view that hyper-accumulation of Sic1 inhibits autophagy. Cells that overexpress the Clb-Cdc28 inhibitor Sic1 accumulate in G1 phase, and the cdc34-2ts, cdc28-4ts and cdc28-13ts mutants also display a G1 cell cycle arrest after incubation at nonpermissive temperature (Nugroho and Mendenhall, 1994; Schwob et al., 1994; Verma et al., 1997). Thus, we cannot rule out the possibility that the defect in autophagy caused by hyper-accumulation of Sic1 might be due to cell cycle arrest at the G1 phase. On the other hand, induction of autophagy under conditions of nutrient limitation often accompanies cell cycle arrest at the G1 phase. Therefore, it is also possible that cell cycle arrest may not be directly linked to autophagy regulation, and both may occur in parallel. Further analysis is needed to identify the precise correlation between cell cycle arrest and autophagy.
One obvious question is how Sic1 negatively regulates autophagy. Our data suggest that Rim15 acts as a downstream target of Sic1. First, deletion of RIM15 completely suppressed the upregulation of autophagy in sic1Δ cells (Fig. 7A). Second, inhibition of autophagy caused by Sic1 overexpression did not occur in rim15Δ cells (Fig. 7B and 7C). Third, Rim15 overexpression partially suppressed the inhibition of autophagy caused by Sic1 overexpression (Fig. 7D and 7E). Since Sic1 is a stoichiometric inhibitor of Clb-Cdc28 kinases, we asked whether inhibition of Rim15 kinase activity occurs through a physical interaction between Rim15 and Sic1. However, only a weak interaction was detected in co-immunoprecipitation experiments (data not shown). Thus, further study will be needed to elucidate the mechanism by which Sic1 inhibits Rim15. Notably, our data suggest that Rim15 integrates signals from both the negative and positive regulatory pathways of Pho85 to properly control autophagy.
In conclusion, we have elucidated the regulatory mechanism by which Pho85 and its corresponding cyclin complexes modulate autophagy in yeast. We propose that Pho85, not only integrates nutrient signals via the Pho80-Pho85 and Pcl5-Pho85 cyclin-CDK complexes to negatively regulate autophagy, but that it also integrates information from the cell cycle inhibitor Sic1 via the Clg1-, Pcl1-, and Pho80-Pho85 cyclin-CDK complexes to promote autophagy. Although Pho85 does not play a major role in regulating autophagy compared to the TORC1 complex, the multifunctional CDK Pho85 is critical to ensure appropriate autophagy activity during various extracellular and intracellular stress conditions.
Experimental Procedures
Strains, Plasmids and Media
Yeast strains and plasmids used in this study are listed in Table S1 and S2.
Strains were grown at 30°C in standard rich medium with 2% glucose (YPD), or 2% galactose plus 2% raffinose (YPGal) as carbon source, or synthetic medium with 2% glucose (SMD), or 2% galactose plus 2% raffinose (SMGal) as carbon source. Rapamycin (dissolved in 90% ethanol, 10% Tween-20) was added to the media at a final concentration of 2 μg/ml. For starvation conditions, cells were shifted to SD-N or SG-N medium. 1-Na-PP1 was added to the media at a final concentration of 20 mM for the analysis of the ATP analogue-sensitive mutant PHO85F82G.
Additional Experimental Procedures can be found in the Supplemental Data available online.
Supplementary Material
Footnotes
- Pho80- and Pcl5-associated Pho85 complexes exert an inhibitory effect on autophagy.
- Sic1 functions as a negative regulator of autophagy.
- Clg1, Pcl1 and Pho80 target Pho85 to promote autophagy via destabilizing Sic1.
- Rim15 acts as a downstream target of Sic1.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol. 2007;8:149–160. doi: 10.1038/nrm2105. [DOI] [PubMed] [Google Scholar]
- Budovskaya YV, Stephan JS, Reggiori F, Klionsky DJ, Herman PK. The Ras/cAMP-dependent protein kinase signaling pathway regulates an early step of the autophagy process in Saccharomyces cerevisiae. J Biol Chem. 2004;279:20663–20671. doi: 10.1074/jbc.M400272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll AS, Bishop AC, DeRisi JL, Shokat KM, O'Shea EK. Chemical inhibition of the Pho85 cyclin-dependent kinase reveals a role in the environmental stress response. Proc Natl Acad Sci U S A. 2001;98:12578–12583. doi: 10.1073/pnas.211195798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll AS, O'Shea EK. Pho85 and signaling environmental conditions. Trends Biochem Sci. 2002;27:87–93. doi: 10.1016/s0968-0004(01)02040-0. [DOI] [PubMed] [Google Scholar]
- Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, Corral LG, Krenitsky VP, Xu W, Moutouh-de Parseval L, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood. 2008;111:4690–4699. doi: 10.1182/blood-2007-09-112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Ferrell JE., Jr Multisite phosphorylation and the countdown to S phase. Cell. 2001;107:819–822. doi: 10.1016/s0092-8674(01)00620-1. [DOI] [PubMed] [Google Scholar]
- Feldman RM, Correll CC, Kaplan KB, Deshaies RJ. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- Huang D, Friesen H, Andrews B. Pho85, a multifunctional cyclin-dependent protein kinase in budding yeast. Mol Microbiol. 2007;66:303–314. doi: 10.1111/j.1365-2958.2007.05914.x. [DOI] [PubMed] [Google Scholar]
- Huang J, Klionsky DJ. Autophagy and human disease. Cell Cycle. 2007;6:1837–1849. doi: 10.4161/cc.6.15.4511. [DOI] [PubMed] [Google Scholar]
- Kaffman A, Rank NM, O'Neill EM, Huang LS, O'Shea EK. The receptor Msn5 exports the phosphorylated transcription factor Pho4 out of the nucleus. Nature. 1998;396:482–486. doi: 10.1038/24898. [DOI] [PubMed] [Google Scholar]
- Komata T, Kanzawa T, Takeuchi H, Germano IM, Schreiber M, Kondo Y, Kondo S. Antitumour effect of cyclin-dependent kinase inhibitors (p16(INK4A), p18(INK4C), p19(INK4D), p21(WAF1/CIP1) and p27(KIP1)) on malignant glioma cells. Br J Cancer. 2003;88:1277–1280. doi: 10.1038/sj.bjc.6600862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Measday V, Moore L, Retnakaran R, Lee J, Donoviel M, Neiman AM, Andrews B. A family of cyclin-like proteins that interact with the Pho85 cyclin-dependent kinase. Mol Cell Biol. 1997;17:1212–1223. doi: 10.1128/mcb.17.3.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Andrews B. Late-G1 cyclin-CDK activity is essential for control of cell morphogenesis in budding yeast. Nat Cell Biol. 2004;6:59–66. doi: 10.1038/ncb1078. [DOI] [PubMed] [Google Scholar]
- Nishizawa M, Kawasumi M, Fujino M, Toh-e A. Phosphorylation of sic1, a cyclin-dependent kinase (Cdk) inhibitor, by Cdk including Pho85 kinase is required for its prompt degradation. Mol Biol Cell. 1998;9:2393–2405. doi: 10.1091/mbc.9.9.2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Matsuura A, Wada Y, Ohsumi Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1995;210:126–132. doi: 10.1006/bbrc.1995.1636. [DOI] [PubMed] [Google Scholar]
- Nugroho TT, Mendenhall MD. An inhibitor of yeast cyclin-dependent protein kinase plays an important role in ensuring the genomic integrity of daughter cells. Mol Cell Biol. 1994;14:3320–3328. doi: 10.1128/mcb.14.5.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzle T, Beck T, Martin DE, Hall MN. Activation of the RAS/cyclic AMP pathway suppresses a TOR deficiency in yeast. Mol Cell Biol. 2004;24:338–351. doi: 10.1128/MCB.24.1.338-351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider BL, Patton EE, Lanker S, Mendenhall MD, Wittenberg C, Futcher B, Tyers M. Yeast G1 cyclins are unstable in G1 phase. Nature. 1998;395:86–89. doi: 10.1038/25774. [DOI] [PubMed] [Google Scholar]
- Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- Shemer R, Meimoun A, Holtzman T, Kornitzer D. Regulation of the transcription factor Gcn4 by Pho85 cyclin PCL5. Mol Cell Biol. 2002;22:5395–5404. doi: 10.1128/MCB.22.15.5395-5404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Cargo proteins facilitate the formation of transport vesicles in the cytoplasm to vacuole targeting pathway. J Biol Chem. 2004;279:29889–29894. doi: 10.1074/jbc.M404399200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen E, Wanke V, Roosen J, Smets B, Dubouloz F, Pedruzzi I, Cameroni E, De Virgilio C, Winderickx J. Rim15 and the crossroads of nutrient signalling pathways in Saccharomyces cerevisiae. Cell Div. 2006;1:3. doi: 10.1186/1747-1028-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallóczy Z, Jiang W, Virgin HW, IV, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science. 1997;278:455–460. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wilson WA, Fujino MA, Roach PJ. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol Cell Biol. 2001a;21:5742–5752. doi: 10.1128/MCB.21.17.5742-5752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wilson WA, Fujino MA, Roach PJ. The yeast cyclins Pc16p and Pc17p are involved in the control of glycogen storage by the cyclin-dependent protein kinase Pho85p. FEBS Lett. 2001b;506:277–280. doi: 10.1016/s0014-5793(01)02914-3. [DOI] [PubMed] [Google Scholar]
- Wanke V, Pedruzzi I, Cameroni E, Dubouloz F, De Virgilio C. Regulation of G0 entry by the Pho80-Pho85 cyclin-CDK complex. EMBO J. 2005;24:4271–4278. doi: 10.1038/sj.emboj.7600889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Wysocki R, Javaheri A, Kristjansdottir K, Sha F, Kron SJ. CDK Pho85 targets CDK inhibitor Sic1 to relieve yeast G1 checkpoint arrest after DNA damage. Nat Struct Mol Biol. 2006;13:908–914. doi: 10.1038/nsmb1139. [DOI] [PubMed] [Google Scholar]
- Yang Z, Huang J, Geng J, Nair U, Klionsky DJ. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell. 2006;17:5094–5104. doi: 10.1091/mbc.E06-06-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, Zaman S, Broach JR, Klionsky DJ. Protein kinase A and Sch9 cooperatively regulate induction of autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2007;18:4180–4189. doi: 10.1091/mbc.E07-05-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.