

Summary
A combination of genetic and immunological features is useful for prediction of autoimmune diabetes. Patterns of immune response correspond to the progression from a pre-clinical phase of disease to end-stage islet damage, with biomarkers indicating transition from susceptibility to active autoimmunity, and to a final loss of immune regulation. Here we review the markers that provide evidence for immunological checkpoint failure and that also provide tools for assessment of individualized disease risk. When viewed in the context of genetic variation that influences immune response thresholds, progression from susceptibility to overt disease displays predictable modalities of clinical presentation resulting from a sequential series of failed homeostatic checkpoints for selection and activation of immunity.
Introduction
Progressive immune-mediated loss of insulin-secreting islet cells leads to type 1 diabetes (T1D). It also leaves a trail marked by characteristic immunological signs that provide well validated predictive markers of disease. The analysis of these immunological markers of autoimmune progression, combined with measurement of genetic susceptibility traits, leads to a comprehensive model of disease pathogenesis. In this model, T1D is the end result of a sequential series of failed homeostatic checkpoints for selection and activation of immunity. Several core concepts in human autoimmunity are illustrated by these checkpoints, which control the likelihood of disease-initiating events, the transition from autoimmune susceptibility to autoimmune progression, and ultimately a failure of peripheral immune regulation.
In this article, we review the immunological markers used for disease prediction, their interaction with underlying genetic susceptibility traits, and variables that influence clinical outcomes. We discuss how these markers provide evidence for immunological checkpoint failure and how they provide tools for assessment of individualized disease risk. Some words of caution are required up front. Current risk estimates represent extrapolation of findings from model cohorts and studies. Thus there are a number of assumptions and caveats in what is presented. First, much of the data obtained until now has been on studies of familial cases of T1D, which represent a minority of all cases. Although there is no evidence that T1D occurring in the absence of a family history differs with regard to prediction and pathogenesis, the possibility must be acknowledged. Second, much of what we discuss is derived from what we know about T1D in children and young adults. A substantial proportion of patients develops T1D in adulthood, where the relationships of disease with genes and immune markers are much less concrete, and where we have few biomarkers and know little about pathogenesis. Third, there are critical gaps in our knowledge. We know the major genes and a number of autoantibody biomarkers, but other biomarkers that could directly reflect pathogenesis, such as autoantigen-specific CD4+ and CD8+ effector, memory, and regulatory T cells, are still unknown or sparsely applied. In particular, we have limited knowledge in human subjects about the accuracy of findings in peripheral blood with respect to the pathology at the pancreas. Despite these caveats, we hope that the reader will gain an appreciation of what we believe is an advanced capability to predict T1D, how current biomarkers relate to models of pathogenesis, and what gaps and opportunities for further understanding remain.
Biomarkers of susceptibility: Tools for disease prediction
A portion of estimable genetic risk can be quantified from family history information and the presence of particular alleles of the genes that contribute to the familial risk. This risk estimate can already be determined at or close to birth, both for individuals with and without a family history of disease. Around 400 in every 100,000 US children will be born into an already affected family (Dabelea et al., 2007). These 400 children will have a T1D risk that exceeds 5%, as compared to around 0.4% in the remaining children. Risk in the 400 children can be further stratified on the basis of which affected family member has T1D (3%, 5%, and 8% if they have an affected mother, father, or sibling, respectively) (Bonifacio et al., 2004; Hemminki et al., 2009;). Moreover, a minority of such children will have two affected first degree relatives and will have a 20% risk.
Much of the familial risk is provided by Human Leukocyte Antigen (HLA) genotypes. In view of the multiple roles of HLA genes in T cell selection, antigen presentation, and immune response, there are many opportunities for HLA-mediated influences on disease risk and progression. HLA genetic susceptibility clearly influences the types of specificities recognized by autoreactive T cells, which can be considered the first checkpoint in the selection and activation of autoimmunity.
Alleles at the HLA DR and HLA DQ class II loci are the most useful determinants of inherited risk. T1D risk in a child who has a T1D sibling can be stratified from 0.3% up to 30% depending on his or her HLA class II genotype (Schenker et al., 1999; Aly et al., 2006). Importantly, T1D risk in the children without a family history of T1D can be stratified from around 0.01% to over 5% (Emery et al., 2005). Risk can be estimated empirically on the basis of the frequency of the HLA genotype of the child in the non-diabetic population and in those who have T1D. For example, the HLA DRB1*03,*04; DQB1*0302 genotype, which confers the highest T1D risk, is present in 2.3% of US-born Caucasian children and 39% of patients who develop T1D before age 20 (Odds Ratio 17), providing a T1D risk of 6.8% (Lambert et al., 2004). The same genotype is present in 7% of the 400 children born with an affected family member, and therefore risk in the 40 children who have a T1D relative and have the HLA DRB1*03,*04; DQB1*0302 genotype will be around 25%. Extreme T1D risk (up to 50%) will be present in children with the HLA DRB1*03,*04; DQB1*0302 genotype born into a family with two or more affected family members (Bonifacio et al., 2004). Similar extreme risks were reported for children who are HLA DRB1*03,*04; DQB1*0302 and are identical by descent to their affected sibling at these loci (Aly et al., 2006). Finally, T1D is special with respect to genetic susceptibility in that there are HLA genotypes that confer extreme protection (Baisch et al., 1990). Thus, T1D risk in a child with a T1D family history and with protective HLA-DQB alleles, such as HLA DQB1*0602 is reduced to approximately 1% of the risk in children with similar family history but without this allele. The major classification categories for T1D genetic risk that are currently clinically practicable are shown in Table 1, and they illustrate the disparity between risk for those with and without a family history of T1D. Alleles at HLA DP class II loci and class I loci, such as HLA A*24, B*38, and B*39, also contribute to T1D risk, but they have not been incorporated into risk prediction models. Other genes or chromosome regions have been shown to confer T1D risk, but as discussed below, their utility in T1D prediction may not be high.
Table 1.
Type 1 Diabetes risk stratification by T1D family history and HLA genotyping
| Population | Type 1 diabetes risk (%) |
|---|---|
| Low risk | |
| No affected FDR plus HLA protective genes | 0.01 |
| No affected FDR | 0.4 |
| Affected FDR plus HLA protective genes | 0.3 |
| Intermediate risk | |
| No affected FDR plus HLA risk genes | 4 |
| One affected FDR | 5 |
| • mother with T1D | 3 |
| • father with T1D | 5 |
| • sibling with T1D | 8 |
| High risk | |
| One affected FDR plus HLA high risk genes | 10-20 |
| Multiple affected FDRs | 20-25 |
| Very high risk | |
| Identical twin affected | 30-70 |
| Multiple affected FDRs plus HLA risk genes | 50 |
| Sibling affected plus HLA risk genes, identical by decent | 30-70 |
Abbreviations: FDR,first-degree relative; HLA risk genes: HLA DRB1*03,*04;DQB1*0302; HLA protective genes, HLA DQB1*0602.
The most important change in the T1D risk status of a child occurs when islet autoantibodies develop. Apart from autoantibodies that have been acquired through placental transfer, islet autoantibodies rarely appear prior to age 6 months (Ziegler et al., 1999; Naserke et al., 2001). Exceptions include cases of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome where neonates can develop insulin or glutamic acid decarboxylase (GAD) antibodies in the first months of life (Rubio-Cabezas et al., 2009). Autoantibodies to four islet antigen groups have so far been identified: insulin or proinsulin, GAD65 or GAD67, IA-2 (or ICA512 or IA-2β or PHOGRIN), and ZnT8 (Palmer et al., 1983; Baekkeskov et al., 1990; Rabin et al., 1994; Lu et al., 1996; Wenzlau et al., 2007). Antibodies to other islet antigens exist and contribute to the islet cell antibody (ICA) staining of the pancreas, but for the purposes of prediction, the four above mentioned autoantibodies are currently adequate for T1D that develops in children and young adults (Kulmala et al., 1998; Bingley et al., 2001; LaGasse et al., 2002). It is noteworthy, however, that these markers are useful, but insufficient for sensitive prediction of T1D in adults (Bottazzo et al., 2005; Lampasona et al., 2010). Unlike inherited traits, islet autoantibodies are biomarkers measured by immunoassay and are therefore defined by a quantifiable signal. Thus, our accuracy in identifying true disease-relevant signals will depend upon the quality of the assay. Because of the consequences associated with labeling a child as “islet autoantibody positive,” substantial efforts in standardizing and harmonizing assays for islet autoantibody measurement have occurred (Bingley et al., 2003; Achenbach et al., 2007). In this regard, T1D risk estimation is advanced in comparison to other autoimmune diseases.
The presence of autoantibodies to just one of the four antigen groups alone is associated with only a marginal increase in risk, both in subjects with and without a family history of T1D, indicating that with current assays and threshold for positivity, autoantibodies to single islet antigens are not rare. Thus, current assays probably include a variable component of non-specific binding, and an important checkpoint in disease is progression to the multiple islet autoantibody stage. T1D risk is, indeed, markedly increased when islet autoantibodies to two or more of the antigen groups are found in a child (Bingley et al., 1994; Verge et al., 1996; Achenbach et al., 2004b). Risk is incremental in relation to whether antibodies are against two, three, or four of the antigen groups and amongst those without the full complement of four islet autoantibodies. T1D risk can vary in relation to which of the islet autoantibodies is present (Achenbach et al., 2004b; Achenbach et al., 2006; Achenbach et al., 2009). In particular, the presence of antibodies to IA-2 (or ICA512 or IA-2β or PHOGRIN) is associated with highest risk. Similar to the number of islet autoantibodies, greater titer, affinity, and broadness of epitope reactivity are features of islet autoantibodies that are associated with high T1D risk. In other words, the more islet autoantibodies one has and the stronger they are, the higher the T1D risk.
Islet autoantibodies can be transient (Yu et al., 2000), but high titer islet autoantibodies to multiple islet antigens rarely if ever completely disappear prior to diabetes onset. Nevertheless, repeated testing of islet autoantibodies in positive cases is valuable in order to determine whether more antibodies have developed.
T1D risk is a combination of the likelihood of disease development and the rate at which it will develop. Importantly, it is an average probability, meaning that some will develop diabetes within days of being identified as islet autoantibody positive while others will take decades to develop disease and some not at all.
Considerable effort has been made to determine risk on the basis of genes, autoantibodies, and age, and to stage the prediabetes period using markers of beta cell function, as illustrated in Figure 1. Various combinations of the risk markers can give similar overall risk, and for most combinations the risk can be calculated empirically. Risk can be stratified from <1% to >70% (Figure 2). Current approaches use a stepwise decision tree. Genetic risk is usually the first marker applied in the form of family history and/or HLA class II genotype. Autoantibodies are selectively measured in those individuals who are considered to have sufficient genetic risk to warrant autoantibody testing. Islet autoantibody screening in genetically at-risk children is worthwhile from the age of about one year. Further genetic typing may be applied in autoantibody-positive individuals to exclude those with protective HLA class II genotypes. Finally, beta cell function is measured in islet autoantibody-positive individuals using either the ability of the beta cell to secrete insulin in response to an intravenous glucose challenge or the ability of the individual to clear glucose after a meal challenge (Srikanta et al., 1985). More recently it has also been recognized that insulin demand from the body is also likely to affect the timing of clinical diabetes (Fourlanos et al., 2004), and a global measure of insulin production together with insulin sensitivity may be a better measure of beta cell function for the purposes of T1D prediction.
Figure 1.
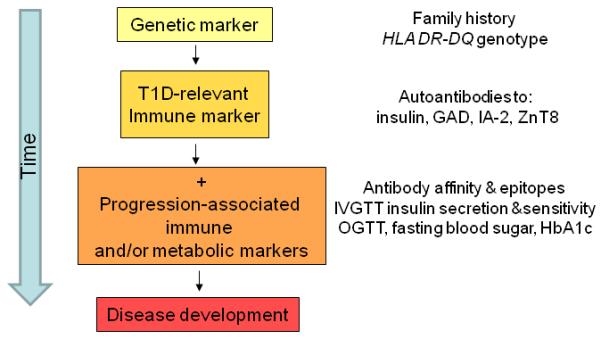
Individual stratification for diabetes risk reflects stages of disease progression. (GAD, glutamic acid decarboxylase; IA-2, islet-associated autoantibody 2; ZnT8, zinc transporter 8; IVGTT, intravenous glucose tolerance test; OGTT, oral glucose tolerance test; HbA1c, hemoglobin A1c).
Figure 2.
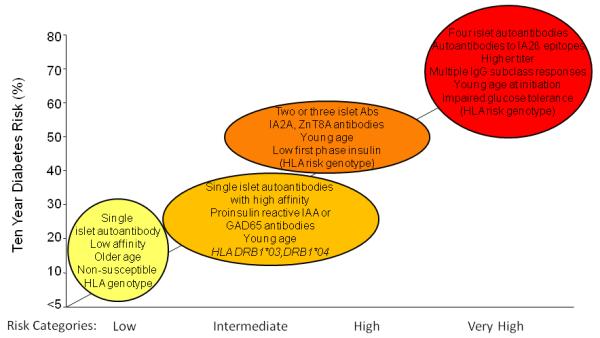
Type 1 diabetes risk stratification by islet autoantibody properties: Increase in T1D risk is associated with progression of islet autoantibodies from single to multiple autoantibodies. Characteristics of the initial antibody response can help predict disease progression. (IAA, insulin autoantibodies; GAD65, glutamic acid decarboxylase 65; IA-2, islet-associated autoantibody 2; ZnT8, zinc transporter 8;
Working our way down this decision tree provides a manner to select individuals who have sufficient risk to warrant inclusion into intervention trials. The risk that is considered sufficient will depend upon the toxicity and invasiveness of the intervention therapy applied. For the future, it could be expected that once effective preventative therapies become available, the decision tree approach will be replaced by the population-wide application of all the above markers in a public health prevention manner. In such an approach the stepwise algorithms are likely to be replaced by a risk score, based on the combination of all markers, representing a paradigm shift after years of increasingly complex layers of decisions in screening.
Because the determination of T1D risk of an individual depends on family history and on autoantibody status, it necessarily follows that risk estimates can change throughout life. For example, predictive risk for a child who has no family history of T1D at birth increases by a factor of ten if his or her sibling develops T1D, and if the child has an identical twin who develops T1D, predictive risk will increase dramatically to around 50% (Redondo et al., 2008). Risk calculated from the autoantibody status will usually increase over time as autoantibodies appear and their number rises. The relationship between age and T1D risk is complex. The risk of developing islet autoantibodies is high early in life as compared to late (Bingley, 1996). Moreover, the earlier autoantibodies appear, the faster the rate of progression to diabetes (Hummel et al., 2004). This may reflect a more aggressive or less regulated immune response, or it may be partially related to beta cell function and insulin demand of the body.
Genotypic variation influences immunological thresholds for selection and activation
The current technologies for identifying T1D risk are generally highly specific for the high risk categories of individuals, but have low overall sensitivity in the general population, since most cases of T1D arise in populations with low to moderate levels of overall risk. Prospects for filling some of the gaps rely on the development and appropriate use of additional genetic and immunological markers that involve alterations in immune response pathways implicated in disease. These additional biomarkers may be either dependent or independent of HLA-associated events, and others may target general pathways of immune activation.
There are two additional genetic loci commonly associated with a relative risk for T1D >2.0, INS and PTPN22 (Concannon et al., 2009). Recent studies now provide insight into how these genes influence the underlying HLA-associated disease risk.
Allelic variation at INS, the gene encoding proinsulin, confers differential susceptibility to T1D: variation in INS is categorized by polymorphisms in a VNTR element, associated with an Odds Ratio for T1D susceptibility of 2.2 in INS VNTR I subjects relative to the heterozygous or “protective” INS VNTR III genotypes (Bennett et al., 1995). The INS VNTR III genotype is associated with high expression of thymic proinsulin in thymic medullary epithelial cells, suggesting a correlation between antigen level and negative selection of high avidity autoreactive T cells, consistent with a prominent role for central tolerance in protection from insulin autoimmunity (Pugliese et al., 1997; Vafiadis et al., 1997). This hypothesis has been directly tested using proinsulin tetramers to profile T cells from genetically disparate subjects, and have shown that the expression of high avidity T cell receptor (TCR) specific for the dominant proinsulin epitope PI76-90 is under genetic control. Comparison of peripheral blood lymphocytes in HLA DRB1*04 subjects showed that 79% of INS VNTR I,I subjects had detectable proinsulin tetramer-positive T cells, compared with 29% of the INS VNTR III subjects (p < 0.0007) (Durinovic-Bello et al., 2010).
Consistent with prior reports from T cell function and cloning assays (Durinovic-Bello et al., 2002), proinsulin autoantigen-specific CD4+ T cells are present in both normal and T1D subjects (Yang et al., 2008). The frequency of these cells is low, estimated to range from 1:30,000 to 1:200,000 in peripheral blood samples. To distinguish between high avidity and low avidity T cell receptors specific for proinsulin, tetramers were created using agonist peptides containing amino acid substitutions in the PI76-90 epitope, which improve binding of the tetramer to the antigen-specific TCR; substitution of lysine to serine at peptide position p9 generates superagonist variant PI76-90S88, resulting in enhanced detection of a low avidity proinsulin-positive CD4+ T cell population (Yang et al., 2008). In contrast to the results cited above, when the same T cell samples were analyzed using the variant PI76-90S88 tetramer, there was no difference in tetramer-positive cells detected between the INS VNTR groups (Durinovic-Bello et al., 2010). Thus, low-avidity anti-proinsulin T cells are present independent of genotype, whereas the presence of anti-proinsulin T cells with higher avidity is under INS genetic control. This illustrates an early and primary checkpoint for autoreactivity that influences the frequency and avidity of the T cell repertoire. The genetic association between INS polymorphisms and T1D disease frequency, even among individuals identical for high risk HLA genes, suggests that the threshold for autoimmune predisposition is sensitive to modulation and is likely to be an important determinant of subsequent disease progression.
Lessons from PTPN22: Variation in immune activation checkpoints
Genetic variation in the protein tyrosine phosphatase N22 (PTPN22) is associated with T1D, as well as with other autoimmune diseases, including rheumatoid arthritis, systemic lupus erythematosus, Graves disease, and myasthenia gravis (Begovich et al., 2004; Bottini et al., 2004; Kyogoku et al., 2004; Onengut-Gumuscu et al., 2004; Smyth et al., 2004; Velaga et al., 2004; 2007; Ladner et al., 2005; Vang et al., 2005; Harley et al., 2008). Biochemical studies of the PTPN22-encoded protein variants have demonstrated an increase in phosphatase function associated with the disease-associated sequence, which, in the context of TCR activation, decreased signal transduction. Direct studies of human peripheral blood mononuclear cells (PBMC) from subjects differing at PTPN22 have confirmed that the disease-associated variant is a gain-of-function variant that leads to a blunting of both the BCR and the TCR signal upon stimulation (Rieck et al., 2007). In CD4+ T cells, this phenotype is characterized by diminished calcium flux upon TCR stimulation and diminished downstream expression of activation markers and cytokines. Interestingly, there is also an alteration in the percent of memory CD4+ T cell composition of the T cell compartment, suggesting increased survival of effector memory cells in the context of the disease-associated variant, perhaps as a result of impaired activation-induced cell death (AICD) signaling. Similar functional variation is seen in human B cells. Calcium flux after B cell receptor (BCR) stimulation was reported to be diminished in the B cells of individuals with the PTPN22 variant, as was phosphorylation of Syk kinase, phospholipase Cγ (PLCγ), and AKT kinase (Arechiga et al., 2009). B cell maturation involves progression through a series of checkpoint controls, so it is possible that the impaired signaling allows for persistence or redirection of less mature, more polyreactive phenotypes associated with autoimmunity.
The HLA, INS, and PTPN22 genetic associations with T1D illustrate the cardinal features of autoimmune predisposition: establishing a lowered threshold for selection and activation of autoreactive T cells that varies among individuals, based on genotype. As demonstrated by the proinsulin tetramer studies cited above, one consequence of this genetic programming is manifest through the level of high avidity autoreactive cells in the peripheral circulating immune repertoire. As indicated by the PTPN22 studies, disease pathogenesis is then influenced by variable activation thresholds related to autoimmune progression. There are important implications of these observations for T1D prediction because it may be possible to determine the likelihood of disease progression, separate from the primary biomarkers that are used to predict initial risk. Examples that shine a light on this gray area of disease prediction and suggest pathogenic mechanisms include the following:
T1D autoantigen-specific T cells from normal subjects are predominantly CD45RA+, indicating a naïve phenotype, for both GAD65 (Danke et al., 2005; Monti et al., 2007) and for proinsulin (Yang et al., 2008). In other words, these cells are inexperienced with respect to antigen and likely represent a potential autoimmune repertoire, which has not been activated in vivo. In contrast, the autoantigen-specific T cells from T1D subjects and at-risk relatives are both CD45RA+ and CD45RA-RO+, the latter representing a memory phenotype indicating prior antigen experience (Danke et al., 2005; Monti et al., 2007). This is consistent with the presence of islet autoantibodies in most such subjects, driven by T-dependent help.
When memory T cells are chronically activated, they are known to undergo maturation of several functional markers; one of these, Kv1.3, is a potassium channel upregulated in effector memory responses. GAD65-specific CD4+ T cells from T1D subjects, but not from non-diabetic individuals, display a Kv1.3-high phenotype (Beeton et al., 2006).
Another feature, which is an indicator of recurrent in vivo stimulation, is evidence of avidity maturation of the T cell response. Avidity maturation is the transition of a population of polyclonal T cells into a mature set of oligoclonal T cells, which have high avidity for a specific target antigen. This occurs in vivo in the setting of chronic exposure to low doses of antigen, and has been seen in GAD-specific CD4+ T cells in association with T1D (Bielekova et al., 2004; Standifer et al., 2009). This has been more extensively studied in the context of multiple sclerosis, where Martin et al. have shown that high avidity autoantigen-specific T cells are enriched for previously in vivo, activated cells and are significantly skewed toward a proinflammatory phenotype (Muraro et al., 2000; Bielekova et al., 2004).
Overall, then, the number and ratio of high to low avidity autoreactive cells in the developing immune system is a function of genes that control TCR engagement and lymphocyte maturation, most notably HLA molecules, but also including modifiers of T cell activation. Subsequent events that result in chronic antigen exposure—either damage to the islet cells from pathogens or toxins, or genetically influenced beta cell death from natural causes—may very well be the source of the antigen-specific stimuli, which trigger the transition seen when T cells evolve from naïve to memory markers. In this scenario, a second checkpoint regulates the persistence and maturation of autoreactive cells (a marker of disease progression), subsequent to the first checkpoint that establishes the autoreactive repertoire (a marker of underlying disease risk). People with a high genetic load subsequently carry a higher frequency of autoreactive T cells, and therefore they have a lower threshold for autoimmune activation when islet cell damage and antigen exposure occurs. Clinically this may account for earlier age of disease onset or for a higher overall prevalence of disease, and is reflected in the measurement of high titer serum autoantibodies. Conversely, people with a low genetic load and low numbers of autoreactive T cells have a higher threshold, and delay the transition to effector memory responses or require larger amounts of antigenic challenge to trigger disease progression. The phenotypic properties of the effector memory population outlined above suggest that after a particular frequency of autoreactive T cells is established, the transition from a highly susceptible stage of preclinical risk to a progressive disease stage leading to T1D in this model is represented by the accumulation of high avidity memory effector cells with autoantigen specificity. Whether measuring this transition will be useful in predicting disease in the low-to-moderate risk HLA cohorts remains to be determined.
Additionally, the link between these phenotypic properties of peripheral lymphocytes and the activity of autoreactive cells in the local environment of pancreatic islets and draining lymph nodes is unclear. This question currently represents a deep knowledge gap between the biomarker studies and the presumed role of tissue damage and antigen exposure in pathogenesis.
Other potential biomarkers forT1D prediction
There are complex interconnections of innate and adaptive immunity and with the tissue response to autoimmune attack, which undoubtedly changes during the progression from disease risk to overt diabetes. In an attempt to detect these changes, a number of “next generation” technologies are being explored for their capacity to find biomarkers that will improve disease prediction. Due to the large number of unknowns associated with the complex immunobiology of progressive autoimmunity, these new approaches have used unfocused screening methods to uncover potential molecular candidates.
One example is the use of transcript array analysis of whole blood samples or of circulating PBMC. This work is based on prior success with another autoimmune disease of children, systemic onset juvenile idiopathic arthritis (SOJIA). In SOJIA, a prominent set of transcripts collectively induced by interleukin-1 (IL-1) was identified as a biomarker of active disease (Allantaz et al., 2007). Based on the array findings, a clinical trial of IL-1 blockade was initiated and rapidly led to its adoption as a therapy of choice for this patient group (Pascual et al., 2008). A similar pilot study in T1D found a set of transcript markers associated with inflammation and hyperglycemia, but did not pinpoint any T1D-specific expression signatures (Chaussabel et al., 2008). A second example used serum from T1D subjects to elicit novel transcript responses from a standardized cell line, detected on large scale arrays. Again, there was prior precedent for this type of study, which had been used successfully to identify type 1 interferons as a prominent bioactive mediator in systemic lupus erythematosus (SLE) (Chaussabel et al., 2008). In these SLE studies, serum from patients taken prior to therapy elicited a type I interferon (IFN) transcript profile, an effect neutralized by adding anti-IFN Ab or by treatment of the patients to induce clinical remission. Although an initial report using a similar approach in T1D suggested the possibility of an IL-1-associated profile (Wang et al., 2008), this is currently a disputed area of investigation without a clear outcome (Jackson et al., 2008; Bergholdt et al., 2009). A third example is the application of a metabolomic analysis. Metabolite biology in the serum reflects a network of interactions, manifest as a complex and heterogeneous chemistry. A preliminary report of a potential marker for early T1D disease risk has been published, in which lipid mediators associated with inflammation were reportedly present prior to the detection of specific anti-islet autoantibodies (Oresic et al., 2008).
Pathogenesis and Disease Progression
T1D can occur in the context of widespread autoimmune dysregulation, such as within the IPEX or APS1 syndromes as a result of mutations in the forkhead box P3 (FOXP3) and autoimmune regulator (AIRE) genes, respectively (Wildin et al., 2001; Turunen et al., 2006), but the vast majority of patients with autoimmune diabetes do not have globally dysregulated immunity, and instead follow a fairly restricted and targeted immunological path. There are two characteristics of type 1 diabetes present in most patients, namely at least one susceptible HLA class II haplotype and islet autoantibodies. Thus, regardless of the etiological factor(s) that may favor the development of islet autoimmunity in a child, it remains highly probable that the initiation of the disease process is the effector immune response to islet beta cell antigens and that the appearance of islet autoantibodies is our first detectable sign of this process.
Islet autoantibodies rarely appear prior to around age 6 months, and amongst children with a family history of T1D there is a peak incidence at around 1 to 2 years of age ( Naserke et al., 1999; Hummel et al., 2004; Bonifacio et al., 2008). This is a relatively important observation because, in terms of the disease process, it suggests that the events leading to islet autoimmunity are encountered after 6 months of age and potentially at increased frequency in early infancy and/or that neonatal immune mechanisms are relatively protective from disease during the first six months of life. Islet autoantibodies that appear in the 1- to 2-year age period have certain characteristics. Antibodies to the insulin or proinsulin antigen group are often amongst the first to appear (Hummel et al., 2004), and within pre-type 1 diabetes children, they have a strong association with HLA DRB1*04; DQB1*0302 (Ziegler et al., 1991). Thus, HLA DRB1*04; DQB1*0302 associated immune activation of insulin reactive B and T cells is a frequent characteristic of the initial disease process of childhood diabetes. The insulin autoantibodies (IAA) are of high affinity IgG1 class already at first detection, suggesting rapid affinity maturation (Castano et al., 1993; Bonifacio et al., 1999; Achenbach et al., 2004a). IgM-IAA is rarely observed (unpublished). Spreading of the immune response to other islet antigens is frequent. The high affinity IAA almost always bind strongly to both insulin and proinsulin (Achenbach et al., 2004a), and can also bind the intermediate products desmin (DES)-(31-32)-proinsulin and DES-(64-65)-proinsulin that are found within insulin secretory granules. These IAA probably recognize a common epitope. While this insulin or proinsulin start and spread of autoimmunity to disease is common in this 1-to 2-year appearance group, other immunization profiles occur. There is an acute, “explosive” antibody response to all antigen groups with rapid progression to disease, suggesting uncontrolled rapid beta cell destruction. In addition, some children progress to diabetes after developing a high affinity autoantibody response to GAD65 prior to IAA. In contrast, antibodies to IA-2, IA-2b and ZnT8 antigen groups very rarely appear on their own or prior to IAA or GAD antibodies (Achenbach et al., 2009). Whether the “insulin,” “GAD,” and “explosive” antibody phenotypes of pre-type 1 diabetes simply represent immune response geneassociated variants or are consequences of different etiology is unknown.
The relatively homogeneous autoantibody profile found in a sizable proportion of children who develop diabetes in childhood is potentially informative with respect to pathogenesis. First, the major targets are preferentially or exclusively expressed in the beta cell. Second, the ubiquitous beta cell proteins, such as actin and nuclear proteins often targeted in systemic autoimmune diseases, are not prominent autoantigens in T1D. Third, there are particular HLA associations seen with specific autoantibody responses (Graham et al., 2002; Mayr et al., 2007; Achenbach et al., 2009). Fourth, in the case of the beta cell-specific ZnT8 antigen, there is an exquisite specificity of autoantibodies to self-polymorphic variants of the protein. ZnT8 residue 325 lies within a major epitope of ZnT8 autoantibodies (Wenzlau et al., 2007; Achenbach et al., 2009). Children who are homozygous for the 325R variant make antibodies to the epitope that expresses 325R and not to the epitope expressing 325W, and vice versa, suggesting autoimmunization as a result of physiological beta cell death or an event associated with induced beta cell death, e.g., cytopathic virus. In either case, the precise target specificity of the response is likely to be favored by protein abundance in the beta cell, location within secretory structures, and preferential presentation of its peptides by the T1D-associated HLA class molecules. The right constellation to reach the threshold needed for a sustained effector immune response is likely to also include alleles at other immune response genes that lower immune activation threshold and an inflammatory cytokine milieu.
Although childhood diabetes is characterized by early appearance of autoantibodies, autoimmunization can occur at any time during life. Observations from the German BABYDIAB cohort (children of parents with T1D) show a second peak incidence period around puberty (Figure 3). The characteristics of the islet autoantibody profiles seen in “late” autoantibody-positive children are heterogeneous, and the typical profile is different compared to that seen in the 1-to 2-year olds. Autoantibodies to single antigen groups, typically, insulin or GAD65, without spreading to other islet proteins, is common. Lower affinity IAA or GAD65 antibodies are also more common, as are antibodies directed against atypical epitopes. Whereas early autoantibody development is strongly linked to T1D–associated HLA class II genotypes, the distribution of HLA class II genotypes in children who develop islet autoantibodies late is less dominated by HLA DRB1*04; DQB1*0302 genotypes. One interpretation of the differences associated with age of islet autoantibody appearance is that etiology and immunization are truly different; i.e., events that lead to an insulin-dominant spreading autoimmunity at age one year are different from those that lead to a GAD65-restricted autoimmunity at age 11 years. Alternatively, immune activation thresholds differ with age, perhaps influenced by genetic variation, as a result of environmental exposures that condition and program memory and regulatory immune responses. This trend towards less HLA dominance and more heterogeneous antibody profiles is also pronounced in older individuals developing autoantibodies who lack a family history of T1D.
Figure 3.
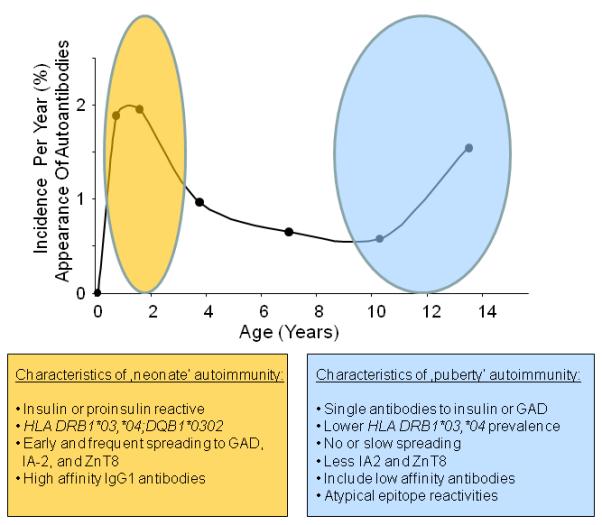
Incidence of islet autoantibodies (cases with at least one Ab of IAA, GADA, IA2A, or ZnT8A per year, expressed as a % of children with a family history of diabetes ascertained in the BABYDIAB study population); abbreviations as in legend to Figure 2). Two waves of islet autoimmunity with an increased incidence at around one year of age (“neonate” autoimmunity) and at around puberty are observed with distinct characteristics.
Assigning etiological causation to environmental triggering factors for islet autoimmunity remains a challenge. A major histocompatibility complex (MHC)-dominated process appears to be operating at very young age, so that environmental agents that favor (or protect from) T1D do so in an MHC-dependent manner. There are no consistent findings of single agents that account for the majority of children in which early islet autoimmunity occurs, and proposed mechanisms are speculative and not entirely consistent with the autoantibody appearance findings described above. An intriguing possibility is that immune response thresholds for islet autoimmunity during childhood are programmed during fetal life. Evidence in type 1 diabetes includes the observations that exposure to elevated blood glucose levels and maternal islet autoantibodies during a diabetic pregnancy are associated with reduced risk of islet autoantibodies in the child (Bonifacio et al., 2008). An ongoing study, called The Environmental Determinants of Diabetes in the Young study (Hagopian et al., 2006) is designed to address some of the proposed environmental associations.
There is no typical progression from the appearance of islet autoantibodies to clinical diabetes. It can be a matter of weeks to decades. By analogy with other autoimmune disorders, a shorter preclinical course most likely indicates aggressive, sustained islet beta cell destruction, whereas decades could represent a series of waxing and waning episodes of beta cell destruction or a constant slow beta cell loss. Notably, the observation that autoantibody responses “spread” to new islet antigens and epitopes at irregular intervals strongly suggests that active immunization is occurring at several occasions during the prediabetic period (Bonifacio et al., 1999). Finally, although measureable loss of beta cell function is a biomarker of impending clinical disease, some islet autoantibody-positive subjects can stay at a low level of insulin production for years prior to clinical disease. These observations are most consistent with the notion that immunologically mediated beta cell destruction is under some sort of regulatory control, as discussed below.
The final checkpoint: Regulatory failure in T1D
Many individuals with the immunological markers of pre-clinical T1D do not progress to clinical disease. An attractive hypothesis is that individuals who have islet autoantibodies and susceptibility genes, but who do not progress to T1D, may be the beneficiaries of a successful regulatory immune response. A great deal of attention has therefore been focused on the analysis of immunoregulatory function associated with T1D autoimmunity, based on the likelihood that a failure of immune regulation is required for complete immune-mediated beta cell loss.
A number of contradictory observations confound this field, however. Regulatory T cell numbers have variously been reported to be decreased or normal in T1D (Lindley et al., 2005; Putnam et al., 2005; Tree et al., 2006; Brusko et al., 2007; Oling et al., 2007; Yang et al., 2007; Link et al., 2008; Vrabelova et al., 2008; Grant et al., 2009; Luczynski et al., 2009), and functional assays have similarly described low, slightly decreased, or normal regulatory activity (Lawson et al., 2008; Schneider et al., 2008; Jin et al., 2009; Long et al., 2009a; 2009b; Putnam et al., 2009; Ryden et al., 2009). Much of this muddle is likely due to methodological differences in various laboratories, since the precise definition of human regulatory T cells has changed several times in recent years, and most of these studies have not distinguished between potentially distinct regulatory T cell subsets.
Nevertheless, a consensus is developing regarding a few key points: first, there is clear evidence for the existence of natural regulatory CD4+CD25+FOXP3+ T cells in both pre-T1D and post-diagnosis T1D subjects. Similarly, precursor cells are present, which can readily be induced to form CD4+FOXP3+ T cells with in vitro regulatory function (Putnam et al., 2009). Second, some of these induced Treg cells display antigen specificity for islet autoantigens and appear indistinguishable from similar cells from non-T1D subjects (Long et al., 2009b). Third, various protocols to expand CD4+CD25+FOXP3+ cells have been successful both in vitro and in vivo, particularly aided by the use of rapamycin, which, in addition to its therapeutic implications, unequivocally demonstrates the presence of these cells in T1D subjects (Battaglia et al., 2006; Monti et al., 2008). An analogous situation may exist with CD8+ regulatory T cells as well, unmasked after therapy of T1D subjects with anti-CD3 mAb (Bisikirska et al., 2005).
Alternatively, when natural or induced regulatory T cells are tested for suppressive function on autologous CD4+ effector T cells, T1D subjects generally show diminished regulatory activity compared with non-T1D controls (Lindley et al., 2005; Schneider et al., 2008). This now appears to be, at least in part, due to a distinct phenotype ascribed to the effector T cell population in T1D—namely, that these cells are somewhat refractory to the regulatory control of CD4+CD25+FOXP3+ Treg cells. In a study of HLA-matched control and T1D subjects, regulatory cells from T1D subjects or those from control subjects showed comparable suppression of effector T cells from the controls, whereas neither was effective at suppression of effector T cells from the T1D subjects (Schneider et al., 2008). The molecular mechanism accounting for this refractory effector cell phenotype is unknown.
Two biomarkers have been proposed as potential surrogates for predicting immune regulatory status in the context of disease prediction. HLA-DQB1*0602 is an MHC class II allele, which is negatively associated with T1D in population studies, even in individuals heterozygous for one of the major HLA T1D-associated genes (Baisch et al., 1990). Further, the rare T1D subject who does carry DQB1*0602 tends to develop disease at a later age, even in subjects who are positive for T1D autoantibodies (Pugliese et al., 1995; Sanjeevi et al., 1995). This dominant protection is strongly suggestive of a regulatory protection mechanism favoring disease remission, although the molecular and cellular mechanisms remain to be defined. The other potential regulatory biomarker is also a genetic trait, a polymorphism in the PTPN2 gene. This gene encodes a phosphatase that has widespread functions in many cell types, but in particular is critically involved in signaling of cytokines, such as IL-2 and IL-15. The T1D-associated variant of PTPN2 leads to diminished phosphorylation of STAT5 in T cells, interfering with cytokine signaling, raising the possibility that this variant may interfere with Treg cell survival, which is highly dependent on IL-2 activity (Long et al., 2009a).
Future directions
More than any other common autoimmune disease, childhood and early adult T1D is now partially predictable in genetically susceptible individuals by careful use and analysis of genetic and immunological biomarkers. Utilizing these predictive tools on a broad scale requires two major advances: first, the development of simple, inexpensive, point-of-use technologies suitable for widespread clinical use; and, second, the clinical rationale for intervening therapeutically in individuals who are at high diabetes risk. This latter clinical impetus will come when treatments capable of halting disease progression are shown to be safe and effective, a goal that currently drives many ongoing clinical trials in T1D. In the meantime, T1D prediction studies in large populations can help us refine our view of pathogenesis, by linking genetic variation with particular molecular and cellular mechanisms and perhaps by identifying individuals, who, in cases of slow disease progression, help us identify physiological mechanisms of successful immunoregulation.
Highlights.
Genetic and antibody markers provide a hierarchical stratification of diabetes risk;
Clinical phenotypes are shaped by selection and activation checkpoints in at-risk individuals;
Autoantibodies inform pathogenic mechanisms and classify pre-clinical disease progression;
Disease can be arrested in a pre-clinical phase, likely due to successful immunoregulation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achenbach P, Koczwara K, Knopff A, Naserke H, Ziegler AG, Bonifacio E. Mature high-affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. J. Clin. Invest. 2004a;114:589–597. doi: 10.1172/JCI21307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achenbach P, Lampasona V, Landherr U, Koczwara K, Krause S, Grallert H, Winkler C, Pfluger M, Illig T, Bonifacio E, Ziegler AG. Autoantibodies to zinc transporter 8 and SLC30A8 genotype stratify type 1 diabetes risk. Diabetologia. 2009;52:1881–1888. doi: 10.1007/s00125-009-1438-0. [DOI] [PubMed] [Google Scholar]
- Achenbach P, Schlosser M, Williams AJ, Yu L, Mueller PW, Bingley PJ, Bonifacio E. Combined testing of antibody titer and affinity improves insulin autoantibody measurement: Diabetes Antibody Standardization Program. Clin. Immunol. 2007;122:85–90. doi: 10.1016/j.clim.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Achenbach P, Warncke K, Reiter J, Naserke HE, Williams AJ, Bingley PJ, Bonifacio E, Ziegler AG. Stratification of type 1 diabetes risk on the basis of islet autoantibody characteristics. Diabetes. 2004b;53:384–392. doi: 10.2337/diabetes.53.2.384. [DOI] [PubMed] [Google Scholar]
- Achenbach P, Warncke K, Reiter J, Williams AJ, Ziegler AG, Bingley PJ, Bonifacio E. Type 1 diabetes risk assessment: improvement by follow-up measurements in young islet autoantibody-positive relatives. Diabetologia. 2006;49:2969–2976. doi: 10.1007/s00125-006-0451-9. [DOI] [PubMed] [Google Scholar]
- Allantaz F, Chaussabel D, Stichweh D, Bennett L, Allman W, Mejias A, Ardura M, Chung W, Smith E, Wise C, Palucka K, Ramilo O, Punaro M, Banchereau J, Pascual V. Blood leukocyte microarrays to diagnose systemic onset juvenile idiopathic arthritis and follow the response to IL-1 blockade. J. Exp. Med. 2007;204:2131–2144. doi: 10.1084/jem.20070070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aly TA, Ide A, Jahromi MM, Barker JM, Fernando MS, Babu SR, Yu L, Miao D, Erlich HA, Fain PR, Barriga KJ, Norris JM, Rewers MJ, Eisenbarth GS. Extreme genetic risk for type 1A diabetes. Proc. Natl. Acad. Sci. U. S. A. 2006;103:14074–14079. doi: 10.1073/pnas.0606349103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arechiga AF, Habib T, He Y, Zhang X, Zhang ZY, Funk A, Buckner JH. Cutting edge: the PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J. Immunol. 2009;182:3343–3347. doi: 10.4049/jimmunol.0713370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M, Folli F, Richter-Olesen H, De Camilli P. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 1990;347:151–156. doi: 10.1038/347151a0. [DOI] [PubMed] [Google Scholar]
- Baisch JM, Weeks T, Giles R, Hoover M, Stastny P, Capra JD. Analysis of HLA-DQ genotypes and susceptibility in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1990;322:1836–1841. doi: 10.1056/NEJM199006283222602. [DOI] [PubMed] [Google Scholar]
- Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J. Immunol. 2006;177:8338–8347. doi: 10.4049/jimmunol.177.12.8338. [DOI] [PubMed] [Google Scholar]
- Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW, Kolski-Andreaco A, Wei E, Grino A, Counts DR, Wang PH, LeeHealey CJ, Andrews S, Sankaranarayanan A, Homerick D, Roeck WW, Tehranzadeh J, Stanhope KL, Zimin P, Havel PJ, Griffey S, Knaus HG, Nepom GT, Gutman GA, Calabresi PA, Chandy KG. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc. Natl. Acad. Sci. U. S. A. 2006;103:17414–17419. doi: 10.1073/pnas.0605136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ST, Lucassen AM, Gough SC, Powell EE, Undlien DE, Pritchard LE, Merriman ME, Kawaguchi Y, Dronsfield MJ, Pociot F. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat. Genet. 1995;9:284–292. doi: 10.1038/ng0395-284. [DOI] [PubMed] [Google Scholar]
- Bergholdt R, Brorsson C, Lage K, Nielsen JH, Brunak S, Pociot F. Expression profiling of human genetic and protein interaction networks in type 1 diabetes. PLoS. One. 2009;4:e6250. doi: 10.1371/journal.pone.0006250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J. Immunol. 2004;172:3893–3904. doi: 10.4049/jimmunol.172.6.3893. [DOI] [PubMed] [Google Scholar]
- Bingley PJ. Interactions of age, islet cell antibodies, insulin autoantibodies, and first-phase insulin response in predicting risk of progression to IDDM in ICA+ relatives: the ICARUS data set. Islet Cell Antibody Register Users Study. Diabetes. 1996;45:1720–1728. doi: 10.2337/diab.45.12.1720. [DOI] [PubMed] [Google Scholar]
- Bingley PJ, Bonifacio E, Mueller PW. Diabetes Antibody Standardization Program: first assay proficiency evaluation. Diabetes. 2003;52:1128–1136. doi: 10.2337/diabetes.52.5.1128. [DOI] [PubMed] [Google Scholar]
- Bingley PJ, Bonifacio E, Ziegler AG, Schatz DA, Atkinson MA, Eisenbarth GS. Proposed guidelines on screening for risk of type 1 diabetes. Diabetes Care. 2001;24:398. doi: 10.2337/diacare.24.2.398. [DOI] [PubMed] [Google Scholar]
- Bingley PJ, Christie MR, Bonifacio E, Bonfanti R, Shattock M, Fonte MT, Bottazzo GF, Gale EA. Combined analysis of autoantibodies improves prediction of IDDM in islet cell antibody-positive relatives. Diabetes. 1994;43:1304–1310. doi: 10.2337/diab.43.11.1304. [DOI] [PubMed] [Google Scholar]
- Bisikirska B, Colgan J, Luban J, Bluestone JA, Herold KC. TCR stimulation with modified anti-CD3 mAb expands CD8+ T cell population and induces CD8+CD25+ Tregs. J. Clin. Invest. 2005;115:2904–2913. doi: 10.1172/JCI23961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacio E, Hummel M, Walter M, Schmid S, Ziegler AG. IDDM1 and multiple family history of type 1 diabetes combine to identify neonates at high risk for type 1 diabetes. Diabetes Care. 2004;27:2695–2700. doi: 10.2337/diacare.27.11.2695. [DOI] [PubMed] [Google Scholar]
- Bonifacio E, Pfluger M, Marienfeld S, Winkler C, Hummel M, Ziegler AG. Maternal type 1 diabetes reduces the risk of islet autoantibodies: relationships with birthweight and maternal HbA(1c) Diabetologia. 2008;51:1245–1252. doi: 10.1007/s00125-008-1022-z. [DOI] [PubMed] [Google Scholar]
- Bonifacio E, Scirpoli M, Kredel K, Fuchtenbusch M, Ziegler AG. Early autoantibody responses in prediabetes are IgG1 dominated and suggest antigen-specific regulation. J. Immunol. 1999;163:525–532. [PubMed] [Google Scholar]
- Bottazzo GF, Bosi E, Cull CA, Bonifacio E, Locatelli M, Zimmet P, Mackay IR, Holman RR. IA-2 antibody prevalence and risk assessment of early insulin requirement in subjects presenting with type 2 diabetes (UKPDS 71) Diabetologia. 2005;48:703–708. doi: 10.1007/s00125-005-1691-9. [DOI] [PubMed] [Google Scholar]
- Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, Eisenbarth GS, Comings D, Mustelin T. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 2004;36:337–338. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- Brusko T, Wasserfall C, McGrail K, Schatz R, Viener HL, Schatz D, Haller M, Rockell J, Gottlieb P, Clare-Salzler M, Atkinson M. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes. 2007;56:604–612. doi: 10.2337/db06-1248. [DOI] [PubMed] [Google Scholar]
- Castano L, Ziegler AG, Ziegler R, Shoelson S, Eisenbarth GS. Characterization of insulin autoantibodies in relatives of patients with type I diabetes. Diabetes. 1993;42:1202–1209. doi: 10.2337/diab.42.8.1202. [DOI] [PubMed] [Google Scholar]
- Chaussabel D, Quinn C, Shen J, Patel P, Glaser C, Baldwin N, Stichweh D, Blankenship D, Li L, Munagala I, Bennett L, Allantaz F, Mejias A, Ardura M, Kaizer E, Monnet L, Allman W, Randall H, Johnson D, Lanier A, Punaro M, Wittkowski KM, White P, Fay J, Klintmalm G, Ramilo O, Palucka AK, Banchereau J, Pascual V. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity. 2008;29:150–164. doi: 10.1016/j.immuni.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N. Engl. J. Med. 2009;360:1646–1654. doi: 10.1056/NEJMra0808284. [DOI] [PubMed] [Google Scholar]
- Dabelea D, Bell RA, D’Agostino RB, Jr., Imperatore G, Johansen JM, Linder B, Liu LL, Loots B, Marcovina S, Mayer-Davis EJ, Pettitt DJ, Waitzfelder B. Incidence of diabetes in youth in the United States. JAMA. 2007;297:2716–2724. doi: 10.1001/jama.297.24.2716. [DOI] [PubMed] [Google Scholar]
- Danke NA, Yang J, Greenbaum C, Kwok WW. Comparative study of GAD65-specific CD4+ T cells in healthy and type 1 diabetic subjects. J. Autoimmun. 2005;25:303–311. doi: 10.1016/j.jaut.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Durinovic-Bello I, Boehm BO, Ziegler AG. Predominantly recognized proinsulin T helper cell epitopes in individuals with and without islet cell autoimmunity. J. Autoimmun. 2002;18:55–66. doi: 10.1006/jaut.2001.0566. [DOI] [PubMed] [Google Scholar]
- Durinovic-Bello I, Wu RP, Gersuk VH, Sanda S, Shilling HG, Nepom GT. Insulin gene VNTR genotype associates with frequency and phenotype of the autoimmune response to proinsulin. Genes Immun. 2010 doi: 10.1038/gene.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery LM, Babu S, Bugawan TL, Norris JM, Erlich HA, Eisenbarth GS, Rewers M. Newborn HLA-DR,DQ genotype screening: age- and ethnicity-specific type 1 diabetes risk estimates. Pediatr. Diabetes. 2005;6:136–144. doi: 10.1111/j.1399-543X.2005.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourlanos S, Narendran P, Byrnes GB, Colman PG, Harrison LC. Insulin resistance is a risk factor for progression to type 1 diabetes. Diabetologia. 2004;47:1661–1667. doi: 10.1007/s00125-004-1507-3. [DOI] [PubMed] [Google Scholar]
- Graham J, Hagopian WA, Kockum I, Li LS, Sanjeevi CB, Lowe RM, Schaefer JB, Zarghami M, Day HL, Landin-Olsson M, Palmer JP, Janer-Villanueva M, Hood L, Sundkvist G, Lernmark A, Breslow N, Dahlquist G, Blohme G. Genetic effects on age-dependent onset and islet cell autoantibody markers in type 1 diabetes. Diabetes. 2002;51:1346–1355. doi: 10.2337/diabetes.51.5.1346. [DOI] [PubMed] [Google Scholar]
- Grant J, Bourcier K, Wallace S, Pan D, Conway A, Seyfert-Margolis V, Wallace PK. Validated protocol for FoxP3 reveals increased expression in type 1 diabetes patients. Cytometry B Clin. Cytom. 2009;76:69–78. doi: 10.1002/cyto.b.20446. [DOI] [PubMed] [Google Scholar]
- Hagopian WA, Lernmark A, Rewers MJ, Simell OG, She JX, Ziegler AG, Krischer JP, Akolkar B. TEDDY--The Environmental Determinants of Diabetes in the Young: an observational clinical trial. Ann. N. Y. Acad. Sci. 2006;1079:320–326. doi: 10.1196/annals.1375.049. [DOI] [PubMed] [Google Scholar]
- Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK, Guthridge JM, Cobb BL, Mirel DB, Marion MC, Williams AH, Divers J, Wang W, Frank SG, Namjou B, Gabriel SB, Lee AT, Gregersen PK, Behrens TW, Taylor KE, Fernando M, Zidovetzki R, Gaffney PM, Edberg JC, Rioux JD, Ojwang JO, James JA, Merrill JT, Gilkeson GS, Seldin MF, Yin H, Baechler EC, Li QZ, Wakeland EK, Bruner GR, Kaufman KM, Kelly JA. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 2008;40:204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki K, Li X, Sundquist J, Sundquist K. Familial association between type 1 diabetes and other autoimmune and related diseases. Diabetologia. 2009;52:1820–1828. doi: 10.1007/s00125-009-1427-3. [DOI] [PubMed] [Google Scholar]
- Hummel M, Bonifacio E, Schmid S, Walter M, Knopff A, Ziegler AG. Brief communication: early appearance of islet autoantibodies predicts childhood type 1 diabetes in offspring of diabetic parents. Ann. Intern. Med. 2004;140:882–886. doi: 10.7326/0003-4819-140-11-200406010-00009. [DOI] [PubMed] [Google Scholar]
- Jackson A, McWilliams C, Kaizer E, Chaussabel D, Glaser C, Noguchi H, Matsumoto S, Levy MF, Naziruddin B. Gene expression profiling of human pancreatic islets undergoing a simulated process of instant blood-mediated inflammatory reaction. Transplant. Proc. 2008;40:430–432. doi: 10.1016/j.transproceed.2008.01.021. [DOI] [PubMed] [Google Scholar]
- Jin Y, Chen X, Podolsky R, Hopkins D, Makala LH, Muir A, She JX. APC dysfunction is correlated with defective suppression of T cell proliferation in human type 1 diabetes. Clin. Immunol. 2009;130:272–279. doi: 10.1016/j.clim.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmala P, Savola K, Petersen JS, Vahasalo P, Karjalainen J, Lopponen T, Dyrberg T, Akerblom HK, Knip M. Prediction of insulin-dependent diabetes mellitus in siblings of children with diabetes. A population-based study. The Childhood Diabetes in Finland Study Group. J. Clin. Invest. 1998;101:327–336. doi: 10.1172/JCI119879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, Chang M, Ramos P, Baechler EC, Batliwalla FM, Novitzke J, Williams AH, Gillett C, Rodine P, Graham RR, Ardlie KG, Gaffney PM, Moser KL, Petri M, Begovich AB, Gregersen PK, Behrens TW. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am. J. Hum. Genet. 2004;75:504–507. doi: 10.1086/423790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner MB, Bottini N, Valdes AM, Noble JA. Association of the single nucleotide polymorphism C1858T of the PTPN22 gene with type 1 diabetes. Hum. Immunol. 2005;66:60–64. doi: 10.1016/j.humimm.2004.09.016. [DOI] [PubMed] [Google Scholar]
- LaGasse JM, Brantley MS, Leech NJ, Rowe RE, Monks S, Palmer JP, Nepom GT, McCulloch DK, Hagopian WA. Successful prospective prediction of type 1 diabetes in schoolchildren through multiple defined autoantibodies: an 8-year follow-up of the Washington State Diabetes Prediction Study. Diabetes Care. 2002;25:505–511. doi: 10.2337/diacare.25.3.505. [DOI] [PubMed] [Google Scholar]
- Lambert AP, Gillespie KM, Thomson G, Cordell HJ, Todd JA, Gale EA, Bingley PJ. Absolute risk of childhood-onset type 1 diabetes defined by human leukocyte antigen class II genotype: a population-based study in the United Kingdom. J. Clin. Endocrinol. Metab. 2004;89:4037–4043. doi: 10.1210/jc.2003-032084. [DOI] [PubMed] [Google Scholar]
- Lampasona V, Petrone A, Tiberti C, Capizzi M, Spoletini M, di Pietro S, Songini M, Bonicchio S, Giorgino F, Bonifacio E, Bosi E, Buzzetti R. Zinc transporter 8 antibodies complement GAD and IA-2 antibodies in the identification and characterization of adult-onset autoimmune diabetes: Non Insulin Requiring Autoimmune Diabetes (NIRAD) 4. Diabetes Care. 2010;33:104–108. doi: 10.2337/dc08-2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson JM, Tremble J, Dayan C, Beyan H, Leslie RD, Peakman M, Tree TI. Increased resistance to CD4+CD25hi regulatory T cell-mediated suppression in patients with type 1 diabetes. Clin. Exp. Immunol. 2008;154:353–359. doi: 10.1111/j.1365-2249.2008.03810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes. 2005;54:92–99. doi: 10.2337/diabetes.54.1.92. [DOI] [PubMed] [Google Scholar]
- Link M, Salur L, Kisand K, Rajasalu T, Tillmann V, Uibo R. Higher FoxP3 mRNA expression in peripheral blood mononuclear cells of GAD65 or IA-2 autoantibody-positive compared with autoantibody-negative persons. APMIS. 2008;116:896–902. doi: 10.1111/j.1600-0463.2008.00889.x. [DOI] [PubMed] [Google Scholar]
- Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, Zhang ZY, Pihoker C, Sanda S, Greenbaum C, Buckner JH. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4+CD25+ regulatory T cells of T1D subjects. Diabetes. 2009a doi: 10.2337/db09-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SA, Walker MR, Rieck M, James E, Kwok WW, Sanda S, Pihoker C, Greenbaum C, Nepom GT, Buckner JH. Functional islet-specific Treg can be generated from CD4+ Eur. J. Immunol. 2009b;39:612–620. doi: 10.1002/eji.200838819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Li Q, Xie H, Chen ZJ, Borovitskaya AE, Maclaren NK, Notkins AL, Lan MS. Identification of a second transmembrane protein tyrosine phosphatase, IA-2beta, as an autoantigen in insulin-dependent diabetes mellitus: precursor of the 37-kDa tryptic fragment. Proc. Natl. Acad. Sci. U. S. A. 1996;93:2307–2311. doi: 10.1073/pnas.93.6.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luczynski W, Stasiak-Barmuta A, Urban R, Urban M, Florys B, Hryszko M. Lower percentages of T regulatory cells in children with type 1 diabetes - preliminary report. Pediatr. Endocrinol. Diabetes Metab. 2009;15:34–38. [PubMed] [Google Scholar]
- Mayr A, Schlosser M, Grober N, Kenk H, Ziegler AG, Bonifacio E, Achenbach P. GAD autoantibody affinity and epitope specificity identify distinct immunization profiles in children at risk for type 1 diabetes. Diabetes. 2007;56:1527–1533. doi: 10.2337/db06-1715. [DOI] [PubMed] [Google Scholar]
- Monti P, Scirpoli M, Maffi P, Piemonti L, Secchi A, Bonifacio E, Roncarolo MG, Battaglia M. Rapamycin monotherapy in patients with type 1 diabetes modifies CD4+CD25+FOXP3+ regulatory T-cells. Diabetes. 2008;57:2341–2347. doi: 10.2337/db08-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti P, Scirpoli M, Rigamonti A, Mayr A, Jaeger A, Bonfanti R, Chiumello G, Ziegler AG, Bonifacio E. Evidence for in vivo primed and expanded autoreactive T cells as a specific feature of patients with type 1 diabetes. J. Immunol. 2007;179:5785–5792. doi: 10.4049/jimmunol.179.9.5785. [DOI] [PubMed] [Google Scholar]
- Muraro PA, Pette M, Bielekova B, McFarland HF, Martin R. Human autoreactive CD4+ T cells from naive CD45RA+ and memory CD45RO+ subsets differ with respect to epitope specificity and functional antigen avidity. J. Immunol. 2000;164:5474–5481. doi: 10.4049/jimmunol.164.10.5474. [DOI] [PubMed] [Google Scholar]
- Naserke HE, Bonifacio E, Ziegler AG. Immunoglobulin G insulin autoantibodies in BABYDIAB offspring appear postnatally: sensitive early detection using a protein A/G-based radiobinding assay. J. Clin. Endocrinol. Metab. 1999;84:1239–1243. doi: 10.1210/jcem.84.4.5597. [DOI] [PubMed] [Google Scholar]
- Naserke HE, Bonifacio E, Ziegler AG. Prevalence, characteristics and diabetes risk associated with transient maternally acquired islet antibodies and persistent islet antibodies in offspring of parents with type 1 diabetes. J. Clin. Endocrinol. Metab. 2001;86:4826–4833. doi: 10.1210/jcem.86.10.7931. [DOI] [PubMed] [Google Scholar]
- Oling V, Marttila J, Knip M, Simell O, Ilonen J. Circulating CD4+CD25 high regulatory T cells and natural killer T cells in children with newly diagnosed type 1 diabetes or with diabetes-associated autoantibodies. Ann. N. Y. Acad. Sci. 2007;1107:363–372. doi: 10.1196/annals.1381.038. [DOI] [PubMed] [Google Scholar]
- Onengut-Gumuscu S, Ewens KG, Spielman RS, Concannon P. A functional polymorphism (1858C/T) in the PTPN22 gene is linked and associated with type I diabetes in multiplex families. Genes Immun. 2004;5:678–680. doi: 10.1038/sj.gene.6364138. [DOI] [PubMed] [Google Scholar]
- Oresic M, Simell S, Sysi-Aho M, Nanto-Salonen K, Seppanen-Laakso T, Parikka V, Katajamaa M, Hekkala A, Mattila I, Keskinen P, Yetukuri L, Reinikainen A, Lahde J, Suortti T, Hakalax J, Simell T, Hyoty H, Veijola R, Ilonen J, Lahesmaa R, Knip M, Simell O. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J. Exp. Med. 2008;205:2975–2984. doi: 10.1084/jem.20081800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK, Paquette TL. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science. 1983;222:1337–1339. doi: 10.1126/science.6362005. [DOI] [PubMed] [Google Scholar]
- Pascual V, Allantaz F, Patel P, Palucka AK, Chaussabel D, Banchereau J. How the study of children with rheumatic diseases identified interferon-alpha and interleukin-1 as novel therapeutic targets. Immunol. Rev. 2008;223:39–59. doi: 10.1111/j.1600-065X.2008.00643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese A, Gianani R, Moromisato R, Awdeh ZL, Alper CA, Erlich HA, Jackson RA, Eisenbarth GS. HLA-DQB1*0602 is associated with dominant protection from diabetes even among islet cell antibody-positive first-degree relatives of patients with IDDM. Diabetes. 1995;44:608–613. doi: 10.2337/diab.44.6.608. [DOI] [PubMed] [Google Scholar]
- Pugliese A, Zeller M, Fernandez A, Jr., Zalcberg LJ, Bartlett RJ, Ricordi C, Pietropaolo M, Eisenbarth GS, Bennett ST, Patel DD. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat. Genet. 1997;15:293–297. doi: 10.1038/ng0397-293. [DOI] [PubMed] [Google Scholar]
- Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, Atkinson MA, Bluestone JA. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes. 2009;58:652–662. doi: 10.2337/db08-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam AL, Vendrame F, Dotta F, Gottlieb PA. CD4+CD25high regulatory T cells in human autoimmune diabetes. J. Autoimmun. 2005;24:55–62. doi: 10.1016/j.jaut.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Rabin DU, Pleasic SM, Shapiro JA, Yoo-Warren H, Oles J, Hicks JM, Goldstein DE, Rae PM. Islet cell antigen 512 is a diabetes-specific islet autoantigen related to protein tyrosine phosphatases. J. Immunol. 1994;152:3183–3188. [PubMed] [Google Scholar]
- Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic twins. N. Engl. J. Med. 2008;359:2849–2850. doi: 10.1056/NEJMc0805398. [DOI] [PubMed] [Google Scholar]
- Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J. Immunol. 2007;179:4704–4710. doi: 10.4049/jimmunol.179.7.4704. [DOI] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Minton JA, Caswell R, Shield JP, Deiss D, Sumnik Z, Cayssials A, Herr M, Loew A, Lewis V, Ellard S, Hattersley AT. Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care. 2009;32:111–116. doi: 10.2337/dc08-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryden A, Stechova K, Durilova M, Faresjo M. Switch from a dominant Th1-associated immune profile during the pre-diabetic phase in favour of a temporary increase of a Th3-associated and inflammatory immune profile at the onset of type 1 diabetes. Diabetes Metab Res. Rev. 2009;25:335–343. doi: 10.1002/dmrr.958. [DOI] [PubMed] [Google Scholar]
- Sanjeevi CB, Landin-Olsson M, Kockum I, Dahlquist G, Lernmark A. Effects of the second HLA-DQ haplotype on the association with childhood insulin-dependent diabetes mellitus. Tissue Antigens. 1995;45:148–152. doi: 10.1111/j.1399-0039.1995.tb02434.x. [DOI] [PubMed] [Google Scholar]
- Schenker M, Hummel M, Ferber K, Walter M, Keller E, Albert ED, Janka HU, Kastendiek C, Sorger M, Louwen F, Ziegler AG. Early expression and high prevalence of islet autoantibodies for DR3/4 heterozygous and DR4/4 homozygous offspring of parents with Type I diabetes: the German BABYDIAB study. Diabetologia. 1999;42:671–677. doi: 10.1007/s001250051214. [DOI] [PubMed] [Google Scholar]
- Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J. Immunol. 2008;181:7350–7355. doi: 10.4049/jimmunol.181.10.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JM, Vella A, Nutland S, Rance HE, Maier L, Barratt BJ, Guja C, Ionescu-Tirgoviste C, Savage DA, Dunger DB, Widmer B, Strachan DP, Ring SM, Walker N, Clayton DG, Twells RC, Gough SC, Todd JA. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes. 2004;53:3020–3023. doi: 10.2337/diabetes.53.11.3020. [DOI] [PubMed] [Google Scholar]
- Srikanta S, Ganda OP, Rabizadeh A, Soeldner JS, Eisenbarth GS. First-degree relatives of patients with type I diabetes mellitus. Islet-cell antibodies and abnormal insulin secretion. N. Engl. J. Med. 1985;313:461–464. doi: 10.1056/NEJM198508223130801. [DOI] [PubMed] [Google Scholar]
- Standifer NE, Burwell EA, Gersuk VH, Greenbaum CJ, Nepom GT. Changes in autoreactive T cell avidity during type 1 diabetes development. Clin. Immunol. 2009;132:312–320. doi: 10.1016/j.clim.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tree TI, Roep BO, Peakman M. A mini meta-analysis of studies on CD4+CD25+ T cells in human type 1 diabetes: report of the Immunology of Diabetes Society T Cell Workshop. Ann. N. Y. Acad. Sci. 2006;1079:9–18. doi: 10.1196/annals.1375.002. [DOI] [PubMed] [Google Scholar]
- Turunen JA, Wessman M, Forsblom C, Kilpikari R, Parkkonen M, Pontynen N, Ilmarinen T, Ulmanen I, Peltonen L, Groop PH. Association analysis of the AIRE and insulin genes in Finnish type 1 diabetic patients. Immunogenetics. 2006;58:331–338. doi: 10.1007/s00251-006-0088-3. [DOI] [PubMed] [Google Scholar]
- Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, Wickramasinghe S, Colle E, Polychronakos C. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat. Genet. 1997;15:289–292. doi: 10.1038/ng0397-289. [DOI] [PubMed] [Google Scholar]
- Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, Nika K, Tautz L, Tasken K, Cucca F, Mustelin T, Bottini N. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat. Genet. 2005;37:1317–1319. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- Velaga MR, Wilson V, Jennings CE, Owen CJ, Herington S, Donaldson PT, Ball SG, James RA, Quinton R, Perros P, Pearce SH. The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves’ disease. J. Clin. Endocrinol. Metab. 2004;89:5862–5865. doi: 10.1210/jc.2004-1108. [DOI] [PubMed] [Google Scholar]
- Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA, Chase HP, Eisenbarth GS. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes. 1996;45:926–933. doi: 10.2337/diab.45.7.926. [DOI] [PubMed] [Google Scholar]
- Vrabelova Z, Hrotekova Z, Hladikova Z, Bohmova K, Stechova K, Michalek J. CD 127- and FoxP3+ expression on CD25+CD4+ T regulatory cells upon specific diabetogeneic stimulation in high-risk relatives of type 1 diabetes mellitus patients. Scand. J. Immunol. 2008;67:404–410. doi: 10.1111/j.1365-3083.2008.02074.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Jia S, Geoffrey R, Alemzadeh R, Ghosh S, Hessner MJ. Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. J. Immunol. 2008;180:1929–1937. doi: 10.4049/jimmunol.180.3.1929. [DOI] [PubMed] [Google Scholar]
- Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, Rewers M, Eisenbarth GS, Jensen J, Davidson HW, Hutton JC. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc. Natl. Acad. Sci. U. S. A. 2007;104:17040–17045. doi: 10.1073/pnas.0705894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- Yang J, Danke N, Roti M, Huston L, Greenbaum C, Pihoker C, James E, Kwok WW. CD4+ T cells from type 1 diabetic and healthy subjects exhibit different thresholds of activation to a naturally processed proinsulin epitope. J. Autoimmun. 2008;31:30–41. doi: 10.1016/j.jaut.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Yang Z, Zhou Z, Huang G, Ling H, Yan X, Peng J, Li X. The CD4(+) regulatory T-cells is decreased in adults with latent autoimmune diabetes. Diabetes Res. Clin. Pract. 2007;76:126–131. doi: 10.1016/j.diabres.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Yu J, Yu L, Bugawan TL, Erlich HA, Barriga K, Hoffman M, Rewers M, Eisenbarth GS. Transient antiislet autoantibodies: infrequent occurrence and lack of association with “genetic” risk factors. J. Clin. Endocrinol. Metab. 2000;85:2421–2428. doi: 10.1210/jcem.85.7.6670. [DOI] [PubMed] [Google Scholar]
- Ziegler AG, Hummel M, Schenker M, Bonifacio E. Autoantibody appearance and risk for development of childhood diabetes in offspring of parents with type 1 diabetes: the 2-year analysis of the German BABYDIAB Study. Diabetes. 1999;48:460–468. doi: 10.2337/diabetes.48.3.460. [DOI] [PubMed] [Google Scholar]
- Ziegler AG, Standl E, Albert E, Mehnert H. HLA-associated insulin autoantibody formation in newly diagnosed type I diabetic patients. Diabetes. 1991;40:1146–1149. doi: 10.2337/diab.40.9.1146. [DOI] [PubMed] [Google Scholar]