

Abstract
Hypoxic microenvironment supports cancer stem cell survival, causes poor response to anticancer therapy and tumor recurrence. Inhibition of Notch-1 signaling in adenocarcinoma of the lung (ACL) cells causes apoptosis specifically under hypoxia. Here we found that Akt-1 activation is a key mediator of Notch-1 pro-survival effects under hypoxia. Notch-1 activates Akt-1 through repression of phosphatase and tensin homolog (PTEN) expression and induction of the Insulin-like Growth Factor 1 Receptor (IGF-1R). The latter seems to be the major determinant of Akt-1 stimulation, since Notch-1 signaling affects Akt-1 activation in PTEN−/− ACL cells. Both downregulation of Insulin Receptor Substrate 1 (IRS-1) and dominant-negative IGF-1R sensitized ACL cells to γ-secretase inhibitor (GSI)-induced apoptosis. Conversely, overexpression of IGF-1R protected ACL cells from GSI toxicity. Inhibition of Notch-1 caused reduced IGF-1R expression, while forced Notch-1 expression yielded opposite effects. ChIP experiments suggested Notch-1 direct regulation of the IGF-1R promoter. Experiments in which human ACL cells were injected in mice confirmed elevated and specific co-expression of Notch-1IC, IGF-1R and pAkt-1 in hypoxic tumor areas.
Our data provide a mechanistic explanation for Notch-1 mediated pro-survival function in hypoxic ACL tumor microenvironment. The results identify additional targets that may synergize with Notch-1 inhibition for ACL treatment.
Keywords: Notch signaling, lung cancer, hypoxia, IGF-1R, cancer cell survival
Introduction
IGF-1R signaling plays a central role in human embryonic stem cell survival and expansion (Bendal et al., 2007). It is also a pivotal pathway in oncogenesis, as fibroblast cell cultures established from Igf-1r−/− mice cannot be transformed by a variety of viral and cellular oncogenes (Sell et al., 1994; Baserga et al. 1997). A link between tissue low oxygen concentrations and IGF-1 signaling has been proposed (Moromisato et al., 1996; Treins et al., 2005).
Hypoxia is a major challenge for anticancer therapy. A growing body of evidence supports the notion that standard anticancer therapy fails because it is ineffective in targeting hypoxic tumor microenvironment (Milas and Hittelman, 2009; Schuurbiers et al., 2009). In addition, this latter environment appears to provide a niche for solid cancers’ tumor stem cells (Keith and Simon, 2007).
The cellular response to low oxygen concentrations is mostly mediated by the transcription factor Hypoxia Inducible Factor 1α (HIF-1α) (Keith and Simon, 2007). A cooperative function between HIF-1α and Notch-1 has been demonstrated (Gustafsson et al., 2005).
Notch signaling is an evolutionarily conserved pathway regulating critical cell fate decision during development and post-natal life (Artavanis-Tsakonas et al., 1999). Upon binding to their ligands, Notch receptors undergo a number of proteolitic cleavages, including a final cleavage operated by a γ-secretase complex, which releases the active forms of Notch receptors, or NotchIC (Artavanis-Tsakonas et al., 1999). NotchIC isoforms translocate to the nucleus where they regulate transcription of a number of downstream genes, including the transcriptional repressor family of Enhancer of Split in Drosophila (HES and HEY genes in humans) (Artavanis-Tsakonas et al., 1999). Therefore, inhibition of γ-secretase activity in turn causes inhibition of Notch signaling.
Notch receptors (Notch-1 through -4) and ligands have been linked to cancer, although the exact role that each isoform plays seems to be tissue- and context-dependent (Miele et al., 2006). Notch’s role in non-small cell lung cancer (NSCLC) still awaits a better understanding. A pro-oncogenic role for Notch-3 has been proposed in a subset of NSCLCs (Dang et al., 2000; Haruki et al., 2005; Konishi et al., 2007). We showed that targeting Notch-1, either using shRNA or a γ-secretase inhibitor (MRK-003), caused ACL cells to undergo apoptosis specifically under hypoxia (Chen et al., 2007), a condition typical of ACL in vivo (Chen and Dehdashti, 2005). Re-expression of intracellular (active) Notch-1 (Notch-1IC) rescued the pro-apoptotic effects of MRK-003 (Chen et al., 2007). On the other hand, Notch-1 inhibition in normoxic ACL cells had no effect on ACL cells survival (Chen et al., 2007). Here we studied the mechanisms leading to Notch-1-dependent pro-survival signals to ACL cells under hypoxia.
Results
Unless otherwise specified, all experiments were performed in 1% O2, 5%CO2, 94% N2 (hypoxia). The concentrations of gasses remained constant throughout the experiments (see Materials and Methods).
Notch-1 activates Akt-1 in ACL cells
Notch-1 activation in 1% oxygen appeared to be Hypoxia Inducible Factor-1α (HIF-1α) dependent, because HIF-1α siRNA reduced Notch-1IC expression and the Notch downstream target HES-1 (Figure 1a-b), confirming previous results (Gustafsson et al., 2005). ACL cells express HIF-2α. However, this protein does not seem to affect Notch-1 signaling (Supplementary Figure S1).
Figure 1.

Notch-1 signaling is dependent on HIF-1α and negatively regulates PTEN expression in ACL cells. (a) Representative Western blot analysis of A549 cells transfected with either a control siRNA (cont) or with a siRNA targeting the HIF-1α mRNA (siHIF-1α). Note decreased Notch-1IC expression when HIF-1α was artificially downregulated. (b) Quantitative RT-PCR of the HES-1 mRNA in A549 transfected with either a control siRNA (cont, black column) or with a siRNA targeting the HIF-1α mRNA (siHIF-1α, gray column). mRNA levels were normalized for 18S rRNA expression levels. Columns represent the average of three independent experiments. Error bars represent SD.
(c) Western blot analysis of the indicated proteins in H1299 cells transfected with a control siRNA (cont, left), with a siRNA targeting the Notch-1 mRNA (siN1), with a control plasmid (cont, right) or with a plasmid expressing Notch-1IC (N1). (d) Both siRNA to Notch-1 and GSI treatment increase the PTEN mRNA expression levels. H1299 cells were transfected with either a control siRNA (cont, left) or with a siRNA targeting the Notch-1 mRNA (siN1). Alternatively, H1299 cells were treated with either DMSO (cont, right) or with 20 μM MRK-003 for 48 hr (GSI). The mRNA levels were measured using Q-RT-PCR and normalized for 18S rRNA content. Note that both genetic and chemical inhibition of Notch-1 causes increased PTEN mRNA expression. Columns represent average of three independent experiments. Error bars represent SD. We obtained similar results in A549 and H1755 cells (one experiment for each cell line).
In other systems Notch-1 positively regulates Akt-1 activation by suppressing PTEN transcription (Palomero et al., 2007; Graziani et al., 2008). We asked whether Notch-1 influenced PTEN expression in ACL cells. We manipulated Notch-1 expression in these cells and we determined that Notch-1 negatively regulates PTEN expression at both the protein and mRNA levels (Figure 1c–d). In parallel, we found that Notch-1 stimulated Akt-1, its upstream activator phosphoinositide-dependent kinase-1 (PDK-1) (Alessi et al., 1997) and downstream effector mammalian target of rapamycin (Ruggero and Pandolfi, 2003) (mTOR; Figure 2a–c). Forced expression of Notch-1IC caused increased phosphorylation of PDK-1, Akt-1 and mTOR (Figure 2a-c), while siRNA to Notch-1 caused reduced phosporylation of these proteins (Figure 2d). Conversely, Notch-1IC induction in the same cells in normoxia did not cause PDK-1/Akt-1/mTOR activation (Supplementary Figure S2), confirming previous results indicating different biologic outcomes following Notch-1 activation in ACL cells in different oxygen concentrations (Chen et al., 2007).
Figure 2.

Notch-1 regulates Akt-1 phosphorylation in ACL cells under hypoxia; Akt-1 activation protects ACL cells from apoptosis triggered by Notch inhibition under hypoxia. (a) A549 cells were transduced with an empty lentiviral vector (A549-TR-D) or with a lentivirus in which Notch-1IC was under the control of a doxycycline-inducible promoter (A549-TR-N1). Upon doxycycline induction of Notch-1IC, increased phosphorylation of Akt-1, PDK-1 and mTOR was observed (compare lanes 2 and 4). (b) Transient transfection of ACL cells with a plasmid expressing Notch-1IC causes Akt-1 phosphorylation in ACL cells. Western blot analysis of the indicated proteins; cont, ACL cells transfected with pCDNA3; N1, ACL cells transfected with a plasmid expressing Notch-1IC. (c) A549-TR-N1 cells were treated with the indicated concentrations of doxycycline. Notch-1 appeared to stimulate Akt-1 phosphorylation in a dose-dependent fashion. (d) Notch-1 artificial downregulation obtained using a siRNA to Notch-1 caused decreased activation of PDK-1, Akt-1 and mTOR. A549 cells were transfected with either a control siRNA (cont) or a siRNA targeting the Notch-1 mRNA (siN1). 48 hr after transfection cells were assayed by Western blot. Similar results were obtained in H1299 and H1755 cells (not shown).
Constitutively active Akt-1 protects ACL cells from GSI-induced cell death under hypoxia. (e) Western blot analysis of the indicated proteins in A549 cells transfected with either a control vector (cont) or with a vector expressing an NH2-terminal myristoylatable Akt-1 (constitutively active Akt, or aAkt). (f) Annexin V/7-AAD staining of A549 cells transfected with either a control plasmid (cont) or with aAkt, and exposed to 20 μM MRK-003 for 48 hr. The histogram represents the average of three independent experiments. Error bars represent SD. Black columns: alive cells; gray columns: dead cells (annexin V positive). Of note, A549 cells grown under hypoxia (with no additional manipulations) have 6.42 ± 2.11% annexin V positive cells. This number is not significantly different from cells transfected with aAkt-1 and treated with MRK-003.
Activated Akt-1 plays a major role in Notch-mediated protection from apoptosis under hypoxia, since transient transfection of ACL cells with an NH2-terminal myristoylatable Akt-1 (constitutively active Akt, or aAkt) rescued 71.59 ± 2.18% cells from MRK-003-induced apoptosis (Figure 2f). High concentrations (100 μM) of MRK-003 caused nearly complete cell death 48 hr after exposure. Even under these conditions, aAkt-1 kept alive about 50% of transfected cells (Supplementary Figure S3). Taken together, these observations suggest that Akt-1 activation could be a major target of Notch-1 induced ACL cells’ resistance to apoptosis under hypoxia.
Through its regulation of Akt-1, Notch-1 indirectly regulated the expression of Bcl2-A1 and caspase-1, two proteins involved in apoptosis response (Supplementary Figure S4).
To test whether Notch-1 regulation of Akt-1 activation was mainly a result of Notch-1 control of PTEN expression, we modulated the expression of Notch-1 in PTEN−/− ACL cell line H1650 (Guo et al., 2008). Even in these cells, downregulation of endogenous Notch-1 through siRNA led to decreased phosphorylation of PDK-1, Akt-1 and mTOR, while forced expression of Notch-1IC led to opposite effects (Figure 3).
Figure 3.

Notch-1 modulates the activation levels of PDK-1, Akt-1 and mTOR in PTEN null ACL cells. Western blot analysis of the indicated proteins. H1650 cells were transfected with either a control siRNA (cont), with a siRNA targeting the Notch-1 mRNA (siN1), with a control plasmid (pcDNA) or with a plasmid expressing Notch-1IC (N1IC). Cells were analyzed 48 hr after transfection. Note that artificial downregulation of Notch-1 depresses the activation of the PDK-1/Akt-1/mTOR axis, while forced expression of Notch-1IC elicits opposite effects in ACL cells lacking PTEN.
These results suggest that factors other than PTEN are responsible for Notch-1-mediated Akt-1 activation in ACL cells under hypoxia.
Notch-1 regulates IGF-1R expression
In several models, IGF-1R and IRS-1 mediate the majority of PI3 kinase/Akt-1 induction (Adams et al., 2000). We hypothesized a possible cross-talk between Notch-1 and IGF-1 signaling because IGF-1 autocrine signaling can regulate HIF-1α expression (Treins et al., 2005). Furthermore, hypoxia increases the expression of IGF-1 and IGF-1R (Moromisato et al., 1996) (and Figure 4a). Both transient siRNA to IRS-1 and dominant-negative IGF-1R sensitized ACL cells to GSI treatment (Figure 4b–c), reinforcing the possible link between IGF-1R and Notch-1 signaling. On the other hand, expression of IGF-1R under the regulation of a CMV promoter protected ACL cells from GSI-induced apoptosis (Figure 4d). We then asked if Notch-1 could regulate the expression of IGF-1R. We determined that either siRNA to Notch-1 or MRK-003 treatment reduced expression of IGF-1R protein (Figure 5a;d) and mRNA (Figure 5b). Conversely, forced expression of Notch-1IC increased IGF-1R protein (Figure 5a,c) and mRNA expression (Figure 5b). The MRK-003-induced repression of IGF-1R expression was reversed after induction of Notch-1IC (Figure 5e). Collectively, this data suggest that Notch-1 regulates the IGF-1R expression under hypoxia. To explore the contribution of HIF-1α to this phenomenon we downregulated HIF-1α using siRNA to HIF-1α. ACL cells with downregulated HIF-1α showed reduced IGF-1R expression (Figure 5f), further confirming the cooperation between HIF-1α and Notch-1 signaling (Gustafsson et al., 2005; Chen et al., 2007). Notch-1 regulation of IGF-1R expression was not restricted to ACL cells, since siRNA to Notch-1 depressed the IGF-1R expression in cell lines of different tissue derivation (Supplementary Figure S5). To rule out possible off-target effects, we used a different siRNA to Notch-1 (Qiagene). Artificial downregulation of Notch-1 using this alternative siRNA also caused reduced IGF-1R expression in two separate experiments (Supplementary Figure S6).
Figure 4.

IGF-1R signaling affects ACL susceptibility to GSI treatment. (a) A549 cells cultured in a standard incubator or under hypoxia. Top, representative Western blot analysis of the indicated proteins; bottom, quantitative RT-PCR of the IGF-1R mRNA in A549 grown either in normoxia (21% O2, black column) or under hypoxia (1% O2, gray columns). Messenger RNA levels were normalized for 18S rRNA content. Columns represent the average of three independent experiments. Error bars represent SD. (b) siRNA to IRS-1 sensitizes ACL cells to MRK-003 induced cell death under hypoxia. A549 cells were transfected with either a control siRNA (cont) or with a siRNA targeting the IRS-1 mRNA. Top, representative Western blot analysis of the indicated proteins; bottom, annexin V/7AAD staining of cells transfected with siRNA and exposed to 20 μM MRK-003 under hypoxia for 48 hr. Black column, alive cells; gray column, annexin V positive cells. Columns represent the average of three independent experiments. Error bars representing SD. P value is indicated. (c) Dominant negative IGF-1R sensitizes ACL cells to MRK-003 induced cell death under hypoxia. A549 cells were transfected with either a control plasmid (cont) or with a plasmid expressing a dominant-negative IGF-1R. Top, representative Western blot analysis of the indicated proteins; bottom, annexin V/7AAD staining of cells transfected with plasmids and exposed to 20 μM MRK-003 under hypoxia for 48 hr. Black column, alive cells; gray column, annexin V positive cells. Columns represent the average of three independent experiments. Error bars represent SD. P value is indicated. (d) Transfection of IGF-1R under the control of a CMV promoter protects ACL cells from MRK-003 induced cell death under hypoxia. A549 cells were transfected with either a control plasmid (cont) or with a plasmid expressing full-length IGF-1R. Top, representative Western blot analysis of the indicated proteins; bottom, annexin V/7AAD staining of cells transfected with plasmids and exposed to 20 μM MRK-003 under hypoxia for 48 hr. Black column, alive cells; gray column, annexin V positive cells. Columns represent the average of three independent experiments. Error bars represent SD. P value is indicated. As all experiments where the oxygen concentration is not indicated, the analyses in (b), (c) and (d) were performed under hypoxia.
Figure 5.

Notch-1 regulates IGF-1R expression. (a) Representative Western blot of the indicated proteins in A549 cells trasfected with a control plasmid (cont, left) or with a plasmid expressing Notch-1IC (N1IC), or transfected with a control siRNA (cont, right) or with a siRNA targeting the Notch-1 mRNA (siN1). (b) quantitative RT-PCR of the IGF-1R mRNA in A549 transfected with the nucleic acids described above. Additionally, A549 were exposed to DMSO (C) or with 20 μM MRK-003 (GSI) for 48 hr under hypoxia. mRNA values were normalized for 18S rRNA expression levels. *: P< 0.001; **: P< 0.001; ***: P< 0.01. Columns represent the average of three independent experiments. Error bars represent SD. (c) Western blot analysis of the indicated proteins. H1755 and H1299 cells were either transfected with a control plasmid (N1IC −) or with a plasmid expressing Notch-1IC (N1IC +). The two panels for IGF-1R represent two different exposure times of the same gel. Note increased IGF-1R expression when Notch-1IC was overexpressed. Additional abbreviations: TM, transmembrane, uncleaved Notch-1; IC, Notch-1IC. (d) Western blot analysis of the indicated proteins of different ACL lines (indicated) exposed to 20 μM MRK-003 (GSI+) or to vehicle alone (GSI−) for 48 hr. (e) A549-TR-DEST (lanes 1 and 2) and A549-TR-N1 (lanes 3 and 4) were exposed to either 1 μg/ml doxycycline (DOX), 20 μM MRK-003 (GSI) or both for 48 hr. Note that doxycycline treatment alone reduced the IGF-1R expression levels. A more marked effect was elicited by MRK-003 treatment on the expression of the IGF-1R. However, induction (obtained through doxycycline treatment) of Notch-1IC led to a 4.8-fold increase in the IGF-1R expression levels (compare lanes 3 and 4). Notch-1IC failed to restore the IGF-1R expression levels completely possibly because of the compounding doxycycline effects (compare lanes 1 and 2). (f) Artificial downregulation of HIF-1α causes reduced expression of the IGF-1R in ACL cells under hypoxia. A549 cells were transfected with either a control siRNA or with a siRNA targeting the HIF-1α mRNA. 48 hr after transfection cells were assayed by Western blot analysis for the indicated proteins.
Interestingly, hypoxia induced the both the IGF-1 and IGF-2 expression levels in ACL cells. However, IGF-2 did not appear to be regulated by Notch-1, since artificial downregulation of Notch-1 did not affect the IGF-2 expression levels (Supplementary Figure S7). Instead, siRNA to Notch-1 decreased the IGF-1 mRNA expression levels of ~58% (Supplementary Figure S7).
To investigate the mechanisms through which Notch-1 regulates IGF-1R expression we transfected ACL cells with known Notch downstream targets (namely, HES-1, HES-5, HEY-1, HEY-L and c-MYC) (Klinakis et al., 2006; Palomero et al., 2006). None of these proteins significantly affected IGF-1R expression levels, neither at the protein nor at the mRNA level (not shown). For this reason we hypothesized a Notch-1 direct regulation of the IGF-1R promoter. There are two canonical CBF-1 consensus sequences in the DNA region surrounding the IGF-1R initiation codon (position +1): one at position −612 and a second at position +1478 (Figure 6a). We analyzed the association of Notch-1IC with these DNA regions using chromatin immunoprecipitation (ChIP). We found no association of Notch-1IC with the −612 site in 7 independent experiments. Instead, Notch-1IC reproducibly associated with the +1478 site (Supplementary Figure S8 and Figure 6b). We sought additional evidence that Notch-1IC stimulates transcription of the IGF-1R gene when associated with the +1478 region. To this end, we immunoprecipitated the Notch transcriptional coactivators MAML-1 (Nam et al., 2006) and p300 (Fryer et al., 2002); the +1478 DNA region coimmunoprecipitated with both proteins (Figure 6b). Transfection of ACL cells with a dominant-negative MAML-1 (dnMAML-1) prevented the immunoprecipitation of the +1478 region with Notch-1IC, dnMAML-1 and p300, and significantly decreased the amount of acetylated histone H3 at this site (Figure 6b). This result can be explained with the requirement of a complete protein complex for stable Notch-1 association with the +1478 region. Dominant negative MAML-1 lacks its central domain responsible for p300 recruitment, which is replaced by green fluorescence protein (GFP) (Weng et al., 2003). Alternatively, the absence of p300 activity in this site may decrease the overall acetylation of histone H3 (Figure 6b) leading to a local rearrangement of the chromatin structure that could render this region less accessible to transcription factors/coactivators. Furthermore, transfection of ACL cells with dnMAML-1 reduced IGF-1R expression (Figure 6c). Collectively, the above data suggest that Notch-1 directly regulates IGF-1R transcription through its association with the +1478 DNA region.
Figure 6.
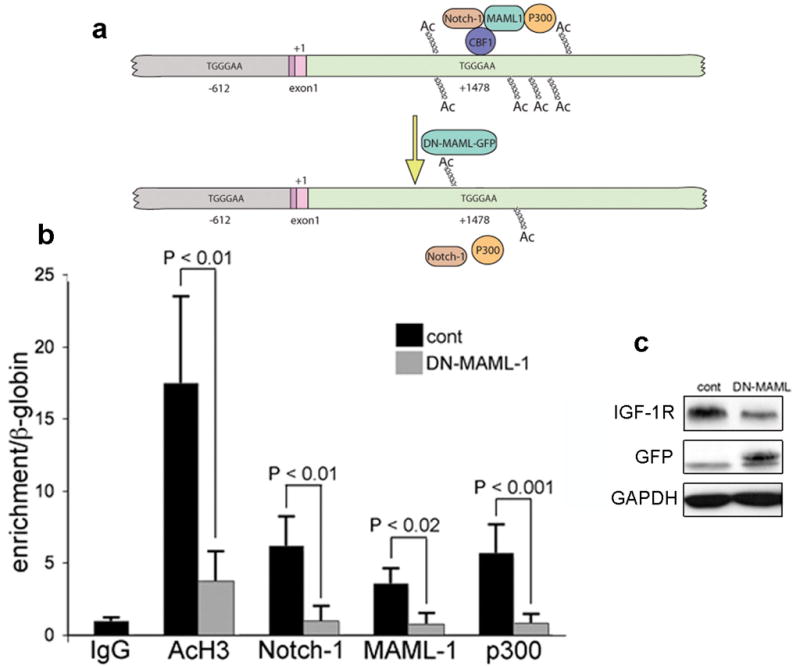
Notch-1 associates with the +1478 region of the IGF-1R gene alongside MAML-1 and p300; transfection of ACL cells with dominant negative MAML-1 disrupts such association. (a) Schematic representation of the region of the IGF-1R that we analyzed by ChIP for the indicated proteins. We focused our attention to the two DNA fragments containing canonical CBF-1 binding sites (indicated). Gray: region upstream of the initiation of transcription; darker pink: 5′ UTR; lighter pink: first fragment of the CDS (+1, initiation of translation); green: 5′ region of the first intron. Top; at the +1478 region we detected association of Notch-1, MAML-1 and p300. Upon transfection with dominant negative MAML-1 (DN-MAML-GFP) (bottom) these proteins are no longer associated to this DNA region, and the amount of acetylated histone H3 is significantly reduced (see panel b). (b) Quantitative ChIP of the +1478 region (62 bp-long TaqMan fragment) after immunoprecipitation of the indicated proteins in A549 cells transfected with a control plasmid (black columns) or with a plasmid coding for dominant negative MAML-1 (gray columns). The IgG column represents the average of three different pre-immune IgGs. Each column represents the average of four (black columns) or five (gray columns) independent experiments (IgG colum represents 9 independent immunoprecipitations). Error bars represent SD. (c) Representative Western blot analysis of A549 cells transfected with either control plasmid or the plasmid expressing dominant negative (dn) MAML-1 (visualized with an antibody against GFP). Note that upon dnMAML-1 expression (24 hr after transfection), the expression levels of IGF-1R are substantially decreased. All the experiments shown in this figure were conducted 24 hr after transfection. This was because dnMAML-1 appeared to be toxic to ACL cells for longer time-points after transfection.
The hypoxia/Notch-1IC/IGF-1R overexpression/Akt-1 activation circuitry takes place in vivo
We then asked if the HIF-1α/Notch-1/IGF-1R/Akt-1 circuitry would be active in vivo in experimentally induced, human metastatic ACL in mice (Supplementary Figure S9). To this end, we performed a number of immunofluorescence experiments on snap frozen, 8 μm thick tissue slides obtained from a total of 6 mice (see Materials and Methods for details). We used GLUT-1 as a marker of hypoxia (Ebert et al., 1995; Behrooz and Ismail-Beigi, 1999) because it reliably evidenced hypoxia in ACL cells (Supplementary Figure S10), and because it was co-expressed with HIF-1α in tumor samples (Figure 7a). Strikingly, only hypoxic tumor areas expressed Notch-1IC, maximally expressed IGF-1R, and appeared to be the only tumor areas where Akt-1 phosphorylation was detectable (Figure 7b–d). On the contrary, PTEN was never expressed in tumor areas that expressed hypoxic marker GLUT-1 (Figure 7e). Noteworthy, in these experiments we detected co-expression rather than co-localization because the different proteins assayed here have different cellular localization (e.g., cell surface receptors, cytoplasmic proteins, nuclear transcription factors).
Figure 7.

Notch-1IC is coexpressed in hypoxic tumor cells together with IGF-1R and phosphorylated Akt-1. Coimmunofluorescence stainings of the indicated proteins on 8 μm-thick slides obtained from tumor-bearing lungs of SCID mice injected with A549 cells through the tail vein. (a) Co-expression of GLUT-1 and HIF-1α. (b) Co-expression of GLUT-1 and Notch-1IC. (c) Expression of IGF-1R is maximal in GLUT-1 expressing tumor cells. (d) Phosphorylated (S473) Akt-1 is detected in GLUT-1 expressing (hypoxic) tumor cell only. (e) PTEN is expressed in GLUT-1 negative tumor cell only. Note that GLUT-1 and PTEN positive cells appear to be contiguous. This is likely an event due to the lateral, asymmetric nature of Notch signaling. Bar, 20 μm. Similar results were obtained in mice injected with H1299 and H1755.
Discussion
The results presented here confirm that hypoxia (through HIF-1α) is a major determinant of Notch-1 activation. This leads to strong Akt-1 activation in part through PTEN repression but prevalently because Notch-1 regulates IGF-1R expression (a schematic is reported in Supplementary Figure S11). To the best of our knowledge, this is the first study describing Notch-1 regulation of IGF-1R. This regulation appears to be direct, because Notch-1 downstream targets (HES-1, HES-5, HEY-1, HEY-L and c-MYC) failed to modify the IGF-1R expression levels. Furthermore, Notch-1 directly associated to the 5′ region of the IGF-1R first intron alongside MAML-1 and p300. Transfection of ACL cells with dominant negative MAML-1 disrupted this association and dramatically reduced the amounts of acetylated histone H3 in that DNA region. Regulatory regions are not uncommonly located within the first intron. For example, the IGF-1 promoter comprises regions that extend well into the second exon (Shemer et al., 1992; McLellan et al., 2006).
In coimmunoflurescence experiments, IGF-1R appears expressed also in non-hypoxic tumor areas. This suggests that Notch-1 must not be the sole regulator of IGF-1R expression. Upon stimulation, IGF-1R undergoes internalization, degradation and recycling (Carelli et al., 2006; Romanelli et al., 2007). These phenomena may artificially reduce the level of co-expression that we detected in our immunofluorescence experiments. Furthermore, Akt activation appears to be maximally sustained in cells actively recycling IGF-1R (Romanelli et al., 2007). Notch regulation of IGF-1 signaling may be evolutionarily ancient. In C. elegans, recovery from dauer stage (which is a diapause-like stage that allows this nematode to survive unfavorable environmental conditions) requires both Notch and insulin-like signaling, with Notch suggested to be upstream from IGF-1 (Ouellet et al., 2008).
Since IGF-1R signaling critically provides pro-survival signals in normal and cancer cells (Baserga et al., 1997; Adams et al., 2000), it is possible that depressed IGF-1R signaling may contribute to the Notch-1 knockout mice phenotype. Notch-1−/− mice die at around day 11 of gestation (Swiatek et al., 1994). Histological evaluation of these mutant embryos showed that, although patterning and somite formation appeared normal, the most noticeable tissue abnormality detected at day 9 of gestation was “widespread cell death” (Swiatek et al., 1994).
Notch-1 appears to affect the PDK-1/Akt-1/mTOR pathways of ACL cells in hypoxia only. It is well known that HIF controls 1 to 2% of the human genome (Manalo et al., 2005). It is therefore possible that under normoxia the IGF-1R promoter undergoes a number of reorganization rendering it inaccessible to Notch-1.
The IGF-1R activation may be the result of autocrine stimulation in vitro (ACL cells express both IGF-1 and IGF-2 (Supplementary Figure S6), while in vivo circulating IGFs levels may contribute to such activation.
For our understanding of solid tumors biology, Notch-1 regulation of the IGF-1R signaling pathway in hypoxic tumor microenvironment has potentially far-reaching implications. Hypoxic tumor areas are likely to be most resistant to apoptosis because of exacerbated Akt-1 activation, as a result of the HIF-1α/Notch-1/IGF-1R circuitry. Our data suggest that chemotherapeutic intervention that ultimately reduces the oxygen concentration within the tumor might inadvertently activate Notch in hypoxic tumor tissue and promote the survival of tumor cells. This would explain recent data indicating that angiogenesis inhibitors, although reducing the size of the primary tumors, appear to promote tumor invasiveness and metastasis (Pàez-Ribes et al., 2009; Ebos et al., 2009). This effect could be mediated by Notch-1 promotion of metastasization observed under hypoxia (Sahlgren et al., 2008). Markedly, hypoxia also promotes the “stem-like” phenotype of putative tumor-initiating cells (Keith and Simon, 2007). In other models, such as breast, these cells are highly Notch signaling dependent (Korkaya and Wicha, 2009). While such cells in NSCLC haven’t been definitively identified, it stands to reason that if a slowly proliferating, highly drug-resistant (Milas and Hittelman, 2009) cell population exists within hypoxic tumor areas, the HIF-1α-Notch-IGF-1R-Akt pathway may be involved in their maintenance. Inhibiting Notch-1 through a GSI or other pharmacological interventions in NSCLC may preferentially target hypoxic areas and the tumor-initiating cells within them. For this reason, the use of Notch inhibitors in combinations with inhibitors of the IGF-1/Akt pathway and/or cytotoxic chemotherapy may result in synergistic anti-tumor effects in ACL.
Conclusions and future directions
Here we provided evidence that Notch-1 regulates a pivotal pathway controlling growth (at the whole organism and cellular levels) and survival (at the cellular level). The latter seems to play a primary role in hypoxic tumor environment. Insulin-like and insulin signaling likely diverged from an ancestral pathway regulating growth according to nutrients’ availability (Yoyoshima et al., 2008), to differentiate into a branch specialized in regulating growth and survival (IGFs, their receptors and a number of IGF binding proteins of IGFBPs) versus a signaling mostly devoted to energy metabolism (insulin and its receptor, or IR). Cancer cells express IR (Belfiore, 2007; Frasca et al., 2008), and there is a growing body of evidence linking high circulating insulin levels, dietary habits, cancer incidence and prognosis (Heuson and Legros, 1972; Venkateswaran et al., 2007). The role of IR in cancer is not completely elucidated. However, there is circumstantial evidence that IR and IGF-1R signaling could be deeply intertwined in regulating cancer cell growth, survival and metabolism (reviewed in Pollak, 2008). The possibility that Notch-1 could regulate IR alongside IGF-1R deserves future investigations.
Similarly, the key mediators downstream of these receptors should be analyzed in more detail. Here we focused on Akt-1 because of its critical role in cell growth and survival (Chen et al., 2001; Cho et al., 2001). Akt-2 seems to primarily function in muscle and liver by regulating glucose metabolism (Cho et al., 2001B), while Akt-3 activity seems to play an important role in the central nervous system development (Tschopp et al., 2005). However, it is possible that more Akt isoforms could play distinct, yet essential roles, possibly in relation with activation of different receptors in ACL.
Materials and methods
Cells grown in hypoxia (1% O2, 5%CO2, 94% N2) were maintained in chambers (Stem Cell Technologies, Vancouver, BC, Canada) filled with a certified, aforementioned mixture of gases (Airgas North Central, West Chicago, IL, USA) at 37 °C. MiniOX1 oxygen meters (Mine Safety Appliances Company, Pittsburgh, PA, USA) were used to measure oxygen concentration. The experiments shown here were performed on ACL lines A549, H1299 and H1755 (ATCC, Manassas, VA, USA). Some experiments were performed in ACL cell line H1650, while other cell lines are specified in Supplementary Information. All cell lines were fingerprinted using the GenePrint fluorescent STR system (Promega, Madison, WI, USA).
GSI toxicity experiments were performed in RPMI 1640 supplemented with 10% fetal bovine serum and 5 nmol/l of purified IGF-1 (Sigma-Aldrich, Saint Louis, MO).
Oligonucleotides, antibodies, plasmids, retroviral vectors and siRNAs
A list of all antibodies used in this study is reported in Supplementary Table T1. A list of all oligonucleotides used here is reported in Supplementary Table T2. Plasmids encoding aAkt-1 (myr-Akt1) was from Upstate Biotechnology (Billerica, MA, USA). The plasmid encoding full-length IGF-1R was previously described (Bocchetta et al., 2008), the plasmid encoding dominant-negative IGF-1R was a gift from Dr Hua Zhang (University of Minnesota), the plasmid encoding dominant-negative MAML-1 was previously described (Weng et al., 2003). The plasmids encoding HES-1 was from Invitrogen (Carlsbad, CA, USA). The plasmid encoding HES-5 was from OriGene (Rockville, MD, USA). The plasmid encoding c-myc was from AddGene. The plasmid encoding HEY-L was obtained by amplifying the HEY-L cDNA from a liver cDNA library. This cDNA was subsequently cloned in the Xho I/Bam HI sites of pCDNA3 (Invitrogen). The Bcl2-A1 expressing plasmid was constructed following a similar strategy: the complete CDS was PCR amplified from a lung cDNA library and cloned into the Bam HI/Xho I sites of pCDNA4 (Invitrogen). The development of stably transducted, tetracycline-inducible ACL cells expressing Notch-1IC within the pLenti4/TO/V5-DEST (Invitrogen) backbone has been previously described (Chen et al., 2007). The plasmid encoding Notch-1IC has been previously described (Chen et al., 2007). siRNA to Notch-1 and IRS-1 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); an additional siRNA to Notch-1 was from Qiagen; siRNAs to HIF-1α and HIF-2α were from Qiagen (Germantown, MD, USA), while siRNA negative control was the Allstars negative control (Qiagen). Transient transfections were done using an electroporator (GenePulserII, Bio-Rad, Hercules, CA, USA) under the following parameters: 300 kV, 975 μF; 1 μg of plasmid DNA/106 cells or 20 pmol siRNA/106 cells. Efficiency of transfection was >95%.
Microscope image acquisition
We used an AX80 microscope (Olympus, Center Valley, PA, USA). Lenses were UPlanApo (Olympus) with a numerical aperture of 10x. Images were acquired at room temperature. The imaging medium was a fluorescence mounting medium (Dako, Glostrup, Denmark). We used as flourochromes Alexa Fluor 488 and Alexa Fluor 568. The camera was RETIGA 4000R (QImaging, Surrey, BC, Canada). The software for image acquisition was Photoshop 6.0 (Adobe Systems Inc., San Jose, CA, USA). Images were processed (overlaid) using Illustrator CS software (Adobe).
Mice and mouse procedures
We injected 6 weeks old female NOD.CB17-Prkdcscid/J mice (Jackson Laboratories, Bar Harbor, ME, USA) in the tail vein with 2.5 × 106 cells (A549, H1299, H1755 in separate experiments) in 100 μl of sterile saline solution. Mice were housed in a pathogen-free animal facility at Loyola University Medical Center. All procedures were done in accordance with the Institutional Animal Care and Use Committee of the Loyola University Chicago Medical Center. Animals were monitored until they reached the endpoint (dyspnea). At euthanasia, human cells comprised 93±0.8% of the total lungs of mice. Tumor burden quantification was performed using quantitative PCR measuring human versus mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene. After sequence alignments of the human and mouse GAPDH genomic regions (Clustalw software), we designed primers discriminating the two genes (Supplementary Table T2). Primers were carefully validated, and calibration curves to match Real Time PCR results to cell number were developed. We extracted DNA from the whole left lung, the whole left liver lobe, and the whole left cerebral hemisphere of each mouse. The remaining organs were excised and flash frozen. 8μm-thick slides were used for standard immunofluorescence experiments.
Cell viability studies and gene and protein expression analysis
We performed annexin V/7-AAD staining (cell viability) assays, quantitative (Q) RT-PCR (using SYBR green incorporation), Western blot and immunohistochemistry assays as previously described (Chen et al., 2007). As an additional control for equal loading, after ECL development Western blot membranes were stained with Ponceau S solution (Sigma-Aldrich). Bands in Western blot experiments were quantitatively evaluated using a FujiFilm LAS-3000 imaging system (FujiFilm, Tokyo, Japan). The working dilutions for each primary antibody were those recommended by the various manufacturers.
Pathway specific arrays based on quantitative RT-PCR were from SuperArray (Frederick, MD, USA). Results were confirmed in targeted Q-RT-PCR experiments. The GSI MRK-003 (Merck Inc., Whitehouse Station, NJ, USA) was dissolved in DMSO to 40 mM final concentration. The working concentrations and the duration of exposure used in each experiment are specified in figure legends. The percentage of GSI induced cell death is dependent on GSI concentration but also on cell confluency. In this study we used an MRK-003 concentration of 20 μM and a cell confluency of 70–80% in most experiments. In these conditions a 48 hr exposure to MRK-003 under hypoxia causes a 23±3% rate of cell death.
Chromatine Immunoprecipitation (ChIP) experiments
We performed end-point or Q-PCR based ChIP assay as previously described (Graziani et al., 2008) with a few modifications. CT values obtained in association with each immunoprecipitation reaction were normalized for β-globin. The average value of three pre-immune IgG was arbitrarily set to 1, and the β-globin normalized values of each immunoprecipitation reaction was expressed as fold enrichment over the values obtained for the pre-immune IgGs. Real time PCR was performed using TaqMan (Applied Biosystems, Foster City, CA, USA) primers and probes (Supplementary Table T2).
Statistical analyses
We performed statistical analysis by Student’s t-test. Values were considered statistically significant at P < 0.05 in two-tailed tests.
Supplementary Material
Acknowledgments
Financial support: American Cancer Society grant RSG-05-077-MBC, Award Number RO1 CA134503 from the National Cancer Institute, and by a grant by the Riviera Country Club and Sport Center (M. Bocchetta). Merck Inc. provided the drug MRK-003.
We thank Patricia Simms for help with FACS analyses. Merck & CO., Inc. (Whitehouse Station, N.J.) kindly provided the MRK-003 compound. We thank Dr Michele Carbone for critical review of this manuscript. This work was supported by grants from the American Cancer Society RSG-05-077-MBC, Award Number RO1 CA134503 from the National Cancer Institute, and by a grant from the Riviera Country Club and Sport Center.
Footnotes
Conflict of interest. The authors declare no conflict of interest.
References
- Adams TE, Epa VC, Garrett TP, Ward CW. Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol Life Sci. 2000;57:1050–1093. doi: 10.1007/PL00000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Baserga R, Hongo A, Rubini M, Prisco M, Valentinis B. The IGF-I receptor in cell growth, transformation and apoptosis. Biochim Biophys Acta. 1997;1332:F105–F126. doi: 10.1016/s0304-419x(97)00007-3. [DOI] [PubMed] [Google Scholar]
- Behrooz A, Ismail-Beigi F. Stimulation of Glucose Transport by Hypoxia: Signals and Mechanisms. News Physiol Sci. 1999;14:105–110. doi: 10.1152/physiologyonline.1999.14.3.105. [DOI] [PubMed] [Google Scholar]
- Belfiore A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr Pharm Des. 2007;13:671–686. doi: 10.2174/138161207780249173. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Stewart MH, Menendez P, George D, Vijayaragavan K, Werbowetski-Ogilvie T, et al. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature. 2007;448:1015–1021. doi: 10.1038/nature06027. [DOI] [PubMed] [Google Scholar]
- Bocchetta M, Eliasz S, De Marco MA, Rudzinski J, Zhang L, Carbone M. The SV40 large T antigen-p53 complexes bind and activate the insulin-like growth factor-I promoter stimulating cell growth. Cancer Res. 2008;68:1022–1029. doi: 10.1158/0008-5472.CAN-07-5203. [DOI] [PubMed] [Google Scholar]
- Carelli S, Di Giulio AM, Paratore S, Bosari S, Gorio A. Degradation of insulin-like growth factor-I receptor occurs via ubiquitin-proteasome pathway in human lung cancer cells. J Cell Physiol. 2006;208:354–362. doi: 10.1002/jcp.20670. [DOI] [PubMed] [Google Scholar]
- Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DL, Dehdashti F. Advances in positron emission tomographic imaging of lung cancer. Proc Am Thorac Soc. 2005;2:541–544. doi: 10.1513/pats.200507-075DS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, De Marco MA, Graziani I, Gazdar AF, Strack PR, Miele L, et al. Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Res. 2007;67:7954–7959. doi: 10.1158/0008-5472.CAN-07-1229. [DOI] [PubMed] [Google Scholar]
- Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001B;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- Dang TP, Gazdar AF, Virmani AK, Sepetavec T, Hande KR, Minna JD, et al. Chromosome 19 translocation, overexpression of Notch3, and human lung cancer. J Natl Cancer Inst. 2000;92:1355–1357. doi: 10.1093/jnci/92.16.1355. [DOI] [PubMed] [Google Scholar]
- Ebert BL, Firth JD, Ratcliffe PJ. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J Biol Chem. 1995;270:29083–29089. doi: 10.1074/jbc.270.49.29083. [DOI] [PubMed] [Google Scholar]
- Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA. Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev. 2002;16:1397–1411. doi: 10.1101/gad.991602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasca F, Pandini G, Sciacca L, Pezzino V, Squatrito S, Belfiore A, et al. The role of insulin receptors and IGF-I receptors in cancer and other diseases. Arch Physiol Biochem. 2008;114:23–37. doi: 10.1080/13813450801969715. [DOI] [PubMed] [Google Scholar]
- Graziani I, Eliasz S, De Marco MA, Chen Y, Pass HI, De May RM, et al. Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer Res. 2008;68:9678–9685. doi: 10.1158/0008-5472.CAN-08-0969. [DOI] [PubMed] [Google Scholar]
- Guo A, Villén J, Kornhauser J, Lee KA, Stokes MP, Rikova K, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A. 2008;105:692–697. doi: 10.1073/pnas.0707270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Haruki N, Kawaguchi KS, Eichenberger S, Massion PP, Olson S, Gonzalez A, et al. Dominant-negative Notch3 receptor inhibits mitogen-activated protein kinase pathway and the growth of human lung cancers. Cancer Res. 2005;65:3555–3561. doi: 10.1158/0008-5472.CAN-04-3132. [DOI] [PubMed] [Google Scholar]
- Heuson JC, Legros N. Influence of insulin deprivation on growth of the 7,12-dimethylbenz(a)anthracene-induced mammary carcinoma in rats subjected to alloxan diabetes and food restriction. Cancer Res. 1972;32:226–232. [PubMed] [Google Scholar]
- Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinakis A, Szabolcs M, Politi K, Kiaris H, Artavanis-Tsakonas S, Efstratiadis A. Myc is a Notch1 transcriptional target and a requisite for Notch1-induced mammary tumorigenesis in mice. Proc Natl Acad Sci U S A. 2006;103:9262–9267. doi: 10.1073/pnas.0603371103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi J, Kawaguchi KS, Vo H, Haruki N, Gonzalez A, Carbone DP, et al. Gamma-secretase inhibitor prevents Notch3 activation and reduces proliferation in human lung cancers. Cancer Res. 2007;67:8051–8057. doi: 10.1158/0008-5472.CAN-07-1022. [DOI] [PubMed] [Google Scholar]
- Korkaya H, Wicha MS. HER-2, notch, and breast cancer stem cells: targeting an axis of evil. Clin Cancer Res. 2009;15:1845–1847. doi: 10.1158/1078-0432.CCR-08-3087. [DOI] [PubMed] [Google Scholar]
- Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- McLellan AS, Kealey T, Langlands K. An E box in the exon 1 promoter regulates insulin-like growth factor-I expression in differentiating muscle cells. Am J Physiol Cell Physiol. 2006;291:C300–C307. doi: 10.1152/ajpcell.00345.2005. [DOI] [PubMed] [Google Scholar]
- Miele L, Golde T, Osborne B. Notch signaling in cancer. Curr Mol Med. 2006;6:905–918. doi: 10.2174/156652406779010830. [DOI] [PubMed] [Google Scholar]
- Milas L, Hittelman WN. Cancer stem cells and tumor response to therapy: current problems and future prospects. Semin Radiat Oncol. 2009;19:96–105. doi: 10.1016/j.semradonc.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Moromisato DY, Moromisato MY, Zanconato S, Roberts CT., Jr Effect of hypoxia on lung, heart, and liver insulin-like growth factor-I gene and receptor expression in the newborn rat. Crit Care Med. 1996;24:919–924. doi: 10.1097/00003246-199606000-00008. [DOI] [PubMed] [Google Scholar]
- Nam Y, Sliz P, Song L, Aster JC, Blacklow SC. Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell. 2006;124:973–983. doi: 10.1016/j.cell.2005.12.037. [DOI] [PubMed] [Google Scholar]
- Ouellet J, Li S, Roy R. Notch signalling is required for both dauer maintenance and recovery in C. elegans. Development. 2008;135:2583–2592. doi: 10.1242/dev.012435. [DOI] [PubMed] [Google Scholar]
- Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- Romanelli RJ, LeBeau AP, Fulmer CG, Lazzarino DA, Hochberg A, Wood TL. Insulin-like growth factor type-I receptor internalization and recycling mediate the sustained phosphorylation of Akt. J Biol Chem. 2007;282:22513–22524. doi: 10.1074/jbc.M704309200. [DOI] [PubMed] [Google Scholar]
- Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A. 2008;105:6392–6397. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurbiers OC, Kaanders JH, van der Heijden HF, Dekhuijzen RP, Oyen WJ, Bussink J. The PI3-K/AKT-pathway and radiation resistance mechanisms in non-small cell lung cancer. J Thorac Oncol. 2009;4:761–767. doi: 10.1097/JTO.0b013e3181a1084f. [DOI] [PubMed] [Google Scholar]
- Sell C, Dumenil G, Deveaud C, Miura M, Coppola D, DeAngelis T, et al. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol Cell Biol. 1994;14:3604–3612. doi: 10.1128/mcb.14.6.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemer J, Adamo ML, Roberts CT, Jr, LeRoith D. Tissue-specific transcription start site usage in the leader exons of the rat insulin-like growth factor-I gene: evidence for differential regulation in the developing kidney. Endocrinology. 1992;131:2793–2799. doi: 10.1210/endo.131.6.1446616. [DOI] [PubMed] [Google Scholar]
- Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8:707–719. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- Treins C, Giorgetti-Peraldi S, Murdaca J, Monthouël-Kartmann MN, Van Obberghen E. Regulation of hypoxia-inducible factor (HIF)-1 activity and expression of HIF hydroxylases in response to insulin-like growth factor I. Mol Endocrinol. 2005;19:1304–1317. doi: 10.1210/me.2004-0239. [DOI] [PubMed] [Google Scholar]
- Tschopp O, Yang ZZ, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, et al. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development. 2005;132:2943–2954. doi: 10.1242/dev.01864. [DOI] [PubMed] [Google Scholar]
- Venkateswaran V, Haddad AQ, Fleshner NE, Fan R, Sugar LM, Nam R. Association of diet-induced hyperinsulinemia with accelerated growth of prostate cancer (LNCaP) xenografts. J Natl Cancer Inst. 2007;99:1793–1800. doi: 10.1093/jnci/djm231. [DOI] [PubMed] [Google Scholar]
- Weng AP, Nam Y, Wolfe MS, Pear WS, Griffin JD, Blacklow SC, et al. Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of notch signaling. Mol Cell Biol. 2003;23:655–664. doi: 10.1128/MCB.23.2.655-664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoyoshima Y, Monson C, Duan C, Wu Y, Gao C, Yakar S, et al. The role of insulin receptor signaling in zebrafish embryogenesis. Endocrinology. 2008;149:5996–6005. doi: 10.1210/en.2008-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.