

Abstract
Fibrocalcific aortic stenosis (AS) results from an active process similar to atherosclerosis that involves basement membrane disruption, lipid deposition, inflammatory cell infiltration, and calcification. Consequently, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) have been extensively studied as potential therapeutic agents capable of slowing the progression of AS. However, two randomized trials, SALTIRE and the SEAS study, showed no benefit with statin therapy for AS. These results have shed doubt over the efficacy of statin therapy for AS, although their potential efficacy at early stages of aortic valve disease remains possible. In this article, we review the pathophysiology of fibrocalcific AS and discuss future directions for its nonsurgical management in the post-SEAS era.
Keywords: Calcification, Aortic valve, Statin, Stenosis
Introduction
Fibrocalcific aortic stenosis (AS) was traditionally considered the consequence of a passive, degenerative process that occurs with aging. However, significant data now support the notion that fibrocalcific aortic valve remodeling results from an active process similar to atherosclerosis [1•, 2•, 3]. Observational studies have associated AS and aortic valve calcification (AVC) with several cardiovascular risk factors [4, 5]. Analyses of human explanted stenotic aortic valve specimens indicate an active process involving basement membrane disruption, lipid deposition, inflammatory cell infiltration, and in some instances calcification consisting of active bone remodeling [6]. Moreover, several experimental studies have shown that valve disease is inducible using hypercholesterolemic conditions [2•]. As such, much attention has been given to lipid-lowering therapies, specifically 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins), as potential therapeutic agents in managing fibrocalcific AS. Initial observational and nonrandomized studies were promising [1•, 7•]; however, two randomized trials, SALTIRE and more recently the SEAS study, have shown no benefit to statin therapy in slowing the progression of AS [8, 9••]. These results have shed doubt over the efficacy of statin therapy for AS. As such, surgical aortic valve replacement surgery remains most efficacious for managing patients with symptomatic severe AS. In this article, we review the pathophysiology of fibrocalcific AS and discuss future directions for its nonsurgical management in the post-SEAS era.
Epidemiology
In the United States alone, heart valve disease is estimated to afflict 4.2 to 5.6 million people and to contribute to more than 25,000 deaths annually [10]. Fibrocalcific aortic valve disease, which frequently culminates in severe calcification and stenosis, is the most common cause of valvular heart disease in the Western world, present in over 20% of older adults [4, 5]. Even in the absence of outflow obstruction, aortic valve disease is associated with a 50% increase in the risk of death from cardiovascular cause and of myocardial infarction [11].
Several population-based studies have established significant associations between cardiovascular risk factors and the development and progression of fibrocalcific aortic valve remodeling. A landmark analysis from the Cardiovascular Health Study cohort demonstrated higher risk of aortic sclerosis with increased age, male gender, present smoking, hypertension, and elevated lipoprotein(a) and low-density lipoprotein (LDL) cholesterol levels [5]. More recent data from the MESA revealed increased risk of AVC in subjects with diabetes, the metabolic syndrome, and renal dysfunction [12, 13]. Another study from MESA associated LDL cholesterol levels with calcific aortic valve disease only in participants younger than 65 years, although an elevated total cholesterol/high-density lipoprotein (HDL) cholesterol ratio was associated with increased risk in all age groups [14]. In the KORA/MONICA study, age, smoking status, and increased total cholesterol predicted aortic valve disease and smaller aortic valve area at echocardiographic assessment 10 years later [4]. Together, these studies suggest that risk factor modification may effectively prevent or delay the development of AS.
Pathogenesis of Fibrocalcific AS
No longer considered a benign consequence of aging, AS is the result of lipid deposition, inflammatory cell infiltration, renin-angiotensin system (RAS) activation, and an active calcification process. Valvular inflammation activates valve myofibroblasts to release tumor necrosis factor-α (TNF-α) and transforming growth factor-β1 (TGF-β1). These cytokines stimulate expression of matrix metalloproteinases and bone morphogenic protein (BMP), ultimately resulting in transdifferentiation of myofibroblasts into an osteoblast-like cell type and calcification (Fig. 1) [1•, 3, 15].
Fig. 1.
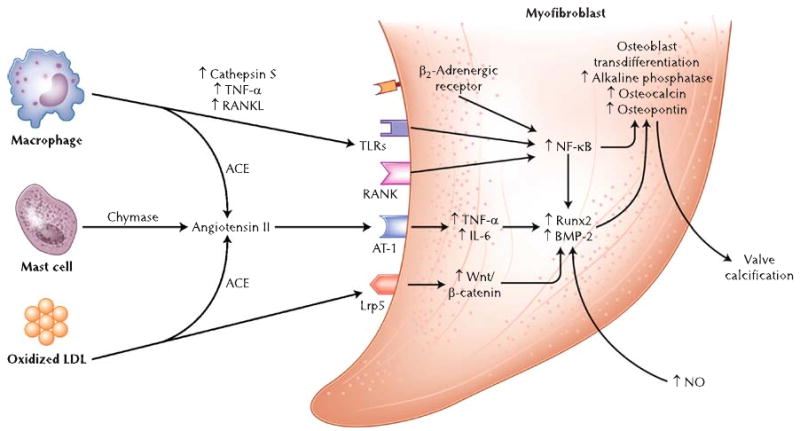
Pathogenesis of fibrocalcific aortic stenosis (AS). ACE angiotensin-converting enzyme, AT-1 angiotensin II type 1 receptor, BMP-2 bone morphogenic protein-2, IL-6 interleukin-6, LDL low-density lipoprotein, Lrp5 LDL receptor-related protein 5, NF-κB nuclear factor-κB, NO nitric oxide, RANK receptor activator of NF-κB, RANKL receptor activator of NF-κB ligand, TLRs toll-like receptors, TNF-α tumor necrosis factor-α
Because calcification is the final pathway leading to AS, much of the recent work in this field focused on delineating specific processes responsible for valve calcification. Data from several laboratories have demonstrated that the pathogenesis of valve calcification mirrors that of bone formation [6, 16, 17, 18••]. Calcified cardiac valves often possess elements of mature lamellar bone and increased expression of osteopontin, osteonectin, and the bone-specific transcription factor Runx2/Cbfa-1 [6, 16, 18••]. Rajamanan et al. [15] and Caira et al. [16] established that the development of an osteogenic phenotype in aortic valves is mediated at least in part by the Wnt/LDL receptor-related protein 5/β-catenin pathway (Fig. 1).
The evidence supporting inflammation as a trigger for valve calcification continues to increase. Aikawa et al. [18••] used near-infrared fluorescent nanoparticles to demonstrate the real-time association of macrophages with early osteogenic activity. They additionally demonstrated that proinflammatory cathepsin S, a potent elastase present in macrophages, accelerates valve and vascular calcification [19]. Nuclear factor (NF)-κB may serve as an added link between inflammation and calcification given the presence of inflammatory cytokines, oxidized LDL, and receptor activator of NF-κB ligand in valve lesions (Fig. 1) [3]. Stimulation of immune-modulating toll-like receptors two and four results in activation of NF-κB, increased valve myofibroblast inflammatory cytokine production, and increases BMP-2 and Runx2 expression [20, 21]. The sympathoadrenergic system may regulate AVC via activation of NF-κB as well; stimulation of β2-adrenergic receptors on aortic valve myofibroblasts results in dramatic reductions in the expression of several bone markers [22]. Recent data also suggest that the complement system may be active in stenotic aortic valves with upregulation of C3a and C5a receptors [23].
Levels of superoxide and hydrogen peroxide are increased in calcified aortic valves, specifically in areas of calcification, implicating oxidative stress in the calcification process [24–26]. Increased oxidative stress, which is noted early in the disease process, is related to hypercholesterolemia [26] and due to uncoupled nitric oxide synthase activity and reduced antioxidant activity within diseased valves (Fig. 1) [25]. Whether NADPH plays a role in aortic valves, as it does in atherosclerosis, remains controversial [24, 25].
Genetic evidence continues to add to our understanding of aortic valve remodeling and calcification. Mutations in the NOTCH1 gene have been associated with a spectrum of aortic valve disease, including heavy calcification [27]. NOTCH1 represses calcification pathways mediated by Runx2 and BMP-2 (Fig. 1) [27, 28]. Recent data suggest that periostin is a critical regulator of NOTCH1, the absence of which leads to decreased NOTCH1 and increased AVC [29]. Mutations in genes for the vitamin D receptor, estrogen receptor, several apolipoproteins, interleukin-10, and TGF-β1 also have been implicated in AS [30•]. These findings highlight the growing role genomics will play in furthering our understanding of valve remodeling and in identifying potential therapeutic targets.
Pharmacotherapy for Fibrocalcific AS
Lipid-Based Interventions
Significant evidence supports lipids as the primary offending agents triggering fibrocalcific aortic valve remodeling [1•, 2•, 3]. Lipid deposits are present in explanted stenotic aortic valve specimens, and several experimental studies have induced aortic valve remodeling and stenosis in hypercholesterolemic conditions [2•]. Consequently, statins have been extensively studied as a potential therapeutic agent for AS [1•, 2•]. Several retrospective studies have associated statin therapy with slowed progression of AS and with less AVC [1•]; however, results from prospective studies has been conflicting (Table 1).
Table 1.
Prospective trials testing statins for the management of AS
| Study | Trial | N | Study design | Patient characteristics | Intervention | End points | Conclusions |
|---|---|---|---|---|---|---|---|
| Cowell et al. [8] | SALTIRE | 155 | Randomized clinical trial | Calcified AS with aortic jet velocity > 2.5 m/s | Randomized to atorvastatin (80 mg) vs placebo daily | Primary: Change in aortic-jet velocity on echocardiogram and in AVC score on CT. Secondary: Composite of clinical end points, AVR, death from any cause, hospitalization for any cause and for CV causes | Atorvastatin did not significantly reduce the progression of AS or the rates of major clinical end points |
| Moura et al. [7•] | RAAVE | 121 | Open-label, prospective study | Asymptomatic moderate to severe AS | Rosuvastatin (20 mg) vs placebo based on baseline LDL | Primary: AS progression and improvement in LDL cholesterol. | Rosuvastatin significantly slowed the progression of AS and reduced serum LDL. Inflammatory markers were also reduced with rosuvastatin |
| Secondary: Improvement in inflammatory markers | |||||||
| Rossebø et al. [9••] | SEAS | 1873 | Randomized clinical trial | Asymptomatic mild to moderate AS | Randomized to simvastatin (40 mg) plus ezetimibe (10 mg) vs placebo daily | Primary: Composite of major CV events. | Simvastatin and ezetimibe did not reduce the composite end point or events related to AS, but did reduce ischemic CV events. No effect on echocardiographic progression of AS was seen |
| Secondary: Events related to AS and ischemic CV events |
AS aortic stenosis, AVC aortic valve calcification, AVR aortic valve replacement, CV cardiovascular, LDL low-density lipoprotein
In the RAAVE study, 121 patients with moderate to severe AS were treated with and without rosuvastatin according to the National Cholesterol Education Program Adult Treatment Panel III guidelines [7•]. Statin therapy in patients with increased LDL cholesterol levels slowed the echocardiographic progression of AS compared to patients with normal cholesterol not receiving rosuvastatin (Table 1). In contrast, two randomized trials failed to demonstrate clinical benefit in patients with AS with statin therapy. The SALTIRE randomly assigned 155 patients with calcific AS to atorvastatin (80 mg/d) or placebo. No benefit was seen in echocardiographic progression of AS or in AVC on CT (Table 1) [8]. The more recent SEAS study randomized 1,873 patients with mild to moderate AS to 40 mg of simvastatin with 10 mg of ezetimibe or placebo [9••]. Despite significant reductions in serum LDL cholesterol levels, no benefit was seen with aortic valve-related events or with the progression of AS, although lipid-lowering therapy significantly reduced the incidence of ischemic cardiovascular events (Table 1).
The definitive SEAS study appears to refute previous studies supporting lipid-lowering therapy for AS [9••]; however, even after SEAS, data in support of lipid-lowering therapies continue to mount (Table 2). In apolipoprotein E−/− mice fed a high-cholesterol diet, atorvastatin reduced macrophage burden and osteogenesis [18••]. Miller et al. [26] demonstrated that lowering serum LDL cholesterol levels in mice with early aortic valve disease arrested its progression. Several more observational clinical studies also reinforced the association of increased serum lipids with AS [14, 31, 32]. Importantly, each of these studies also may explain why the randomized statin trials have been negative in the face of such strong basic and observational clinical data.
Table 2.
Pharmacotherapy for fibrocalcific AS
| Agent | Mechanisms |
|---|---|
| Statins | ↓LDL |
| ↓ Inflammation | |
| ↓TGF-β | |
| ↓TNF-α | |
| ↓MMP | |
| ↓BMP-2 | |
| ↓Protein prenylation | |
| HDL/Apo A1–based interventions | ↑HDL |
| ↓LDL | |
| ↓Inflammation | |
| ↓NF-κB | |
| ↓MCP-1 | |
| RAS inhibition | ↓Inflammation |
| ↓Oxidative stress | |
| ↓Proteoglycan | |
| ↑Fibrinolysis | |
| Bisphosphonates | ↓Inflammation |
| ↓IL-1 | |
| ↓IL-6 | |
| ↓TNF-α | |
| ↓MMP | |
| ↓Protein prenylation | |
| ↓Bone resorption | |
| ↓Ca/PO4 | |
| ↓LDL | |
| ↑HDL | |
| Antiproliferative drugsa | ↓Smooth muscle cell/myofibroblast proliferation |
Rapamycin, paclitaxel
Apo apolipoprotein, BMP-2 bone morphogenic protein-2, HDL high-density lipoprotein, IL interleukin, LDL low-density lipoprotein, MCP-1 monocyte chemoattractant protein-1, MMP matrix metalloproteinase, NF-κB nuclear factor-κB, TGF-β transforming growth factor-β, TNF-α tumor necrosis factor-α
As previously mentioned, data from MESA demonstrate that age interacts with the association between serum LDL cholesterol levels and the risk of AVC such that LDL cholesterol levels are only predictive of AVC in those younger than 65 years of age [14]. A smaller study of patients undergoing aortic valve replacement surgery found that lipid profiles of elderly patients were less atherogenic than those of middle-aged patients [32]. In a large series of 1,046 patients with a mean follow-up time of 5.6±3.2 years, Antonini-Canterin et al. [31] showed that statin therapy effectively slowed the progression of aortic valve disease in those with aortic sclerosis or mild AS, but not in moderate AS. Together, these data suggest that randomized statin trials may have failed because statins were being administered too late in the disease progression. Perhaps moderate AS represents the point at which the calcification process is too far advanced for lipid-lowering therapies to modify its continued progression. This suggests that lipid-lowering therapy may be most effective as a preventative strategy targeted to those with aortic sclerosis or only mild AS. Clinical trials designed to test this hypothesis will be crucial for managing AS.
In addition to lowering serum LDL cholesterol levels, recent investigation is targeting HDL-based interventions for managing AS (Table 2). Epidemiologic data have associated accelerated AS progression with low HDL cholesterol levels and with high total cholesterol/HDL cholesterol ratio [2•, 3], suggesting impaired reverse cholesterol transport as a potential therapeutic target in the treatment of AS. Moreover, HDL cholesterol has been associated with significant pleiotropic effects including anti-inflammatory activities [33], which may be beneficial in AS. A recent study described reversal of experimental AS in a hypercholesterolemic rabbit model after administration of the apolipoprotein A-1 (apo A-1) mimetic peptide [34]. Similar results were observed with recombinant apo A-1 Milano, a mutant form of apo A-1 associated with low incidence of cardiovascular disease, via reduction of valve myofibroblast cholesterol content and downregulation of monocyte chemoattractant protein-1 and NF-κB [35]. Whether HDL-based interventions and reverse cholesterol transport will be effective at later stages of aortic valve disease remains to be determined.
Renin-Angiotensin System Inhibition
The RAS has been implicated in the development of AVC based in part on the presence of angiotensin-converting enzyme (ACE), angiotensin II, and angiotensin II type 1 receptor in diseased valves, but not normal ones [3]. Angiotensin II, which is known to play a role in increasing inflammation and oxidative stress and in decreasing fibrinolysis, is generated within valve lesions via LDL-associated ACE, macrophage-associated ACE, and mast cell chymase. These findings prompted clinical investigation into the utility of ACE inhibitors for managing fibrocalcific valve disease (Table 2). Retrospective data suggested that ACE inhibitor use is associated with slowed aortic valve calcium accumulation but not with slowed progression of AS [3]. Since these conflicting results, no further study has tested the clinical efficacy of RAS blockade in slowing the progression of AS; however, recent data continue to support RAS involvement in the development of fibrocalcific valve remodeling. ACE gene polymorphisms have been associated with AVC severity [36]. In a hyperlipidemic apo E knockout mouse, uremia-induced aortic valve changes were attenuated by ACE inhibition [37]. Additionally, in a study of explanted stenotic aortic valves, serum levels of angiotensin II were associated with increased aortic valve expression of TNF-α and interleukin-6 [38]. As with statin therapy, the conflicting clinical results with RAS inhibition are felt to be secondary to the late initiation of therapy in clinical studies and the short duration of follow-up [3].
Modulation of Bone Metabolism
Bisphosphonates are primarily used in the management of osteoporosis to prevent osteoclast-mediated bone resorption by binding to hydroxyapatite in bone. The potent nitrogen-containing bisphosphonates (NCBPs) inhibit farnesyl-pyrophosphate synthase, an enzyme in the cholesterol biosynthesis/mevalonate pathway distal to 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the site of statin action [39]. Consequently, NCBPs share several effects with statins, decreasing serum LDL cholesterol levels by approximately 5%, raising HDL cholesterol by 10% to 18% [40, 41], and reducing inflammation (Table 2) [39]. Price et al. [42] demonstrated that NCBP inhibit deposition of calcium in vascular and valvular tissues in animal models, possibly by preventing release of calcium phosphate particles from bone. Thus, NCBPs may provide a unique and novel means to slow AVC in addition to their statin-like effects. Additionally, pretreatment of bioprosthetic aortic valve cups with synthesized thiol bisphosphonate, 2-mercaptoethylidene-1, 1-bisphosphonic acid inhibited degenerative calcification [43]. These data suggest that NCBPs may serve as an effective and well-tolerated way to slow AVC, and clinical data supporting this theory are beginning to emerge. An analysis from MESA suggests that NCBP use is associated with less risk of AVC in older women; however, an increased risk of AVC was associated with NCBP use in those women younger than 65 years of age [44]. In another study, osteoporosis therapies, mostly bisphosphonates, were strongly associated with slowed progression of AS [45]. Further evaluation of NCBP is needed to determine whether potential benefits on the aortic valve are due to statin-like mechanisms or secondary to effects on bone metabolism, in which case other inhibitors of bone resorption such as calcitonin and estrogen-receptor modulators may also prove fruitful in treating AVC. It is also important to determine whether the increased risk of AVC in younger NCBP users in the MESA cohort reflects heightened cardiovascular risk associated with osteoporosis, a toxic effect of these drugs, or an epiphenomenon [44].
Antiproliferative Drugs
Antiproliferative drugs, such as rapamycin and paclitaxel, have revolutionized the field of interventional cardiology as a treatment of restenosis after coronary stenting. These agents act via inhibition of vascular smooth muscle cell proliferation and migration, steps critical for stent restenosis (Table 2) [46, 47]. Although human heart valves do not contain smooth muscle cells, the valve myofibroblasts share several properties with vascular smooth muscle cells. For example, both cell types transdifferentiate into osteoblast-like cells that lead to calcification [2•]. Additionally myofibroblasts express α-actin, vimentin, and desmin, as do smooth muscle cells [48]. Thus, it is conceivable that both cell types may react similarly to antiproliferative agents. Because systemic administration of these agents is often complicated by numerous adverse effects, a recent study evaluated the potential for local delivery of paclitaxel to the aortic valve using a paclitaxel-eluting valvuloplasty balloon in pigs and found that drug concentrations within the valve were at therapeutic levels [49]. These preliminary results are intriguing, but clearly further study is needed to evaluate the use of antiproliferative agents in managing fibrocalcific aortic valve disease.
Transcatheter Therapies for Fibrocalcific AS
To date, no pharmacologic intervention has definitively been shown to slow the progression of AS. Moreover, experimental and clinical data suggest that medical management may only interrupt valve remodeling if initiated early in the disease process. Surgical aortic valve replacement surgery consequently remains the standard therapy for patients with symptomatic severe AS [50•]. However, many patients with symptomatic severe AS are precluded from surgical intervention due to high operative risk because of advanced age, left ventricular dysfunction, poor functional capacity, porcelain aorta, past thoracic radiation, and lack of contractile reserve. Transcatheter aortic valve replacement (TAVR) is emerging as an alternative in these high-risk patients who would otherwise not receive aortic valve surgery. Two percutaneous heart valves, the Edwards SAPIEN valve (Edwards Lifescience, Irvine, CA) and the CoreValve ReValving system (CoreValve, Irvine, CA), have been used in over 4,000 cases worldwide [50]. Despite being in its infancy, TAVR has resulted in dramatic hemodynamic improvements, with 30-day mortality ranging from 6% to 18% depending on the technique and device being employed. Although high, these otherwise inoperable patients with symptomatic, severe AS possess 1- and 5-year survival of only 60% and 32%, respectively.
The techniques for TAVR have rapidly evolved from necessitating general anesthesia, extracorporeal circulatory support, and surgical vascular closure to a truly percutaneous technique that can be performed with no hemodynamic support. The retrograde femoral artery approach is most frequently used, although a transapical approach is feasible in those with poor vascular access or a tortuous or heavily calcified aorta. A multitude of TAVR devices are currently in development that will be repositionable and retrievable, offer thinner profiles, and possess self-positioning properties, features anticipated to minimize procedural morbidity while increasing procedural success. With continued technologic advancements, TAVR promises to significantly augment our ability to treat severe AS in ever-aging and more complex patient populations.
Within the United States, the Edwards SAPIEN valve is currently being tested in the PARTNER trial. Additionally, the US-based PAVIS trial is anticipated to further evaluate the CoreValve prosthesis once US Food and Drug Administration approval is obtained. These pivotal trials will randomize patients within two arms. The first will compare conventional, surgical aortic valve replacement to TAVR using the transapical or retrograde transfemoral approaches in higher-risk patients who remain surgical candidates, whereas the second will randomize patients who are deemed too high risk for surgical intervention to TAVR or routine medical management with balloon aortic valvuloplasty.
Conclusions
Fibrocalcific aortic valve disease is associated with significant morbidity, mortality, and health care expenditures, and the burden of disease is expected to rise as the population continues to age. Currently, surgical aortic valve replacement remains the standard treatment for symptomatic severe AS. Pharmacologic agents capable of slowing the progression of AS would transform the way in which this common disease process is managed. To date, statins have been extensively studied in this regard with disappointing results from two recent randomized trials; however, experimental data suggest that statins may effectively prevent or slow AS progression if initiated early in the disease process. Emerging data have identified HDL/ apo A-1, the RAS, and bone metabolism as potential targets for modulating aortic valve disease. Additionally, transcatheter valve replacement is gaining momentum as a reasonably safe option for high-risk patients with symptomatic severe AS. Beyond the SEAS, further understanding of aortic valve pathology and clinical research will likely yield fresh insight into pharmaceutical approaches. TAVR is poised to dramatically change the way aortic valve disease is treated in the clinic.
Acknowledgments
A portion of Dr. Mohler's salary is paid for by NIH grant number K12 HL83772-01A1.
Clinical Trial Acronyms
- MESA
Multi-Ethnic Study of Atherosclerosis
- PARTNER
Placement of Aortic Transcatheter
- PAVIS
Percutaneous Aortic Valve Intervention Study
- RAAVE
Rosuvastatin Affecting Aortic Valve Endothelium
- SALTIRE
Scottish Aortic Stenosis and Lipid Lowering Trial, Impact on Regression
- SEAS
Simvastatin and Ezetimibe in Aortic Stenosis
Footnotes
Disclosure Dr. Sammy Elmariah is supported in part by the GlaxoSmithKline Research and Education Foundation for Cardiovascular Disease. Dr. Emile R. Mohler III has received research support from GlaxoSmithKline and Bristol-Myers Squibb, and is on the speakers' bureau for Merck and Bristol-Myers Squibb-Sanofi Aventis.
References
Papers of particular interest, published recently, have been highlighted as follows:
• Of importance
•• Of major importance
- 1•.Goldbarg SH, Elmariah S, Miller MA, Fuster V. Insights into degenerative aortic valve disease. J Am Coll Cardiol. 2007;50:1205–1213. doi: 10.1016/j.jacc.2007.06.024. [DOI] [PubMed] [Google Scholar]; This is an informative review of degenerative aortic valve disease pathophysiology and therapeutic options.
- 2•.Rajamannan NM. Calcific aortic stenosis: lessons learned from experimental and clinical studies. Arterioscler Thromb Vasc Biol. 2009;29:162–168. doi: 10.1161/ATVBAHA.107.156752. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a thorough review highlighting all experimental models of AS.
- 3.O'Brien KD. Pathogenesis of calcific aortic valve disease: a disease process comes of age (and a good deal more) Arterioscler Thromb Vasc Biol. 2006;26:1721–1728. doi: 10.1161/01.ATV.0000227513.13697.ac. [DOI] [PubMed] [Google Scholar]
- 4.Stritzke J, Linsel-Nitschke P, Markus MR, et al. Association between degenerative aortic valve disease and long-term exposure to cardiovascular risk factors: results of the longitudinal population-based KORA/MONICA survey. Eur Heart J. 2009;30:2044–2053. doi: 10.1093/eurheartj/ehp287. [DOI] [PubMed] [Google Scholar]
- 5.Stewart BF, Siscovick D, Lind BK, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol. 1997;29:630–634. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 6.Mohler ER, 3rd, Gannon F, Reynolds C, et al. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 7•.Moura LM, Ramos SF, Zamorano JL, et al. Rosuvastatin affecting aortic valve endothelium to slow the progression of aortic stenosis. J Am Coll Cardiol. 2007;49:554–561. doi: 10.1016/j.jacc.2006.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the only prospective study to demonstrate beneficial effect of statins on AS progression.
- 8.Cowell SJ, Newby DE, Prescott RJ, et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- 9••.Rossebø AB, Pedersen TR, Boman K, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]; This is a large, randomized clinical trial that failed to show a benefit of statin therapy on AS progression.
- 10.Thom T, Haase N, Rosamond W, et al. Heart disease and stroke statistics-2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;113:e85–e151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- 11.Otto CM, Lind BK, Kitzman DW, et al. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. 1999;341:142–147. doi: 10.1056/NEJM199907153410302. [DOI] [PubMed] [Google Scholar]
- 12.Ix JH, Shlipak MG, Katz R, et al. Kidney function and aortic valve and mitral annular calcification in the Multi-Ethnic Study of Atherosclerosis (MESA) Am J Kidney Dis. 2007;50:412–420. doi: 10.1053/j.ajkd.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 13.Katz R, Wong ND, Kronmal R, et al. Features of the metabolic syndrome and diabetes mellitus as predictors of aortic valve calcification in the Multi-Ethnic Study of Atherosclerosis. Circulation. 2006;113:2113–2119. doi: 10.1161/CIRCULATIONAHA.105.598086. [DOI] [PubMed] [Google Scholar]
- 14.Owens DS, Katz R, Johnson E, et al. Interaction of age with lipoproteins as predictors of aortic valve calcification in the multiethnic study of atherosclerosis. Arch Intern Med. 2008;168:1200–1207. doi: 10.1001/archinte.168.11.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajamannan NM, Bonow RO, Rahimtoola SH. Calcific aortic stenosis: an update. Nat Clin Pract Cardiovasc Med. 2007;4:254–262. doi: 10.1038/ncpcardio0827. [DOI] [PubMed] [Google Scholar]
- 16.Caira FC, Stock SR, Gleason TG, et al. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. 2006;47:1707–1712. doi: 10.1016/j.jacc.2006.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaden JJ, Bickelhaupt S, Grobholz R, et al. Expression of bone sialoprotein and bone morphogenetic protein-2 in calcific aortic stenosis. J Heart Valve Dis. 2004;13:560–566. [PubMed] [Google Scholar]
- 18••.Aikawa E, Nahrendorf M, Figueiredo JL, et al. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850. doi: 10.1161/CIRCULATIONAHA.107.732867. [DOI] [PubMed] [Google Scholar]; This paper discussed using a novel near-infrared fluorescent nanoparticle to image early stages of vascular and valvular calcification.
- 19.Aikawa E, Aikawa M, Libby P, et al. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009;119:1785–1794. doi: 10.1161/CIRCULATIONAHA.108.827972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng X, Ao L, Song Y, et al. Expression of functional toll-like receptors 2 and 4 in human aortic valve interstitial cells: potential roles in aortic valve inflammation and stenosis. Am J Physiol Cell Physiol. 2008;294:C29–C35. doi: 10.1152/ajpcell.00137.2007. [DOI] [PubMed] [Google Scholar]
- 21.Yang X, Fullerton DA, Su X, et al. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol. 2009;53:491–500. doi: 10.1016/j.jacc.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 22.Osman L, Chester AH, Sarathchandra P, et al. A novel role of the sympatho-adrenergic system in regulating valve calcification. Circulation. 2007;116:I282–I287. doi: 10.1161/CIRCULATIONAHA.106.681072. [DOI] [PubMed] [Google Scholar]
- 23.Helske S, Oksjoki R, Lindstedt KA, et al. Complement system is activated in stenotic aortic valves. Atherosclerosis. 2008;196:190–200. doi: 10.1016/j.atherosclerosis.2007.03.040. [DOI] [PubMed] [Google Scholar]
- 24.Liberman M, Bassi E, Martinatti MK, et al. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol. 2008;28:463–470. doi: 10.1161/ATVBAHA.107.156745. [DOI] [PubMed] [Google Scholar]
- 25.Miller JD, Chu Y, Brooks RM, et al. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008;52:843–850. doi: 10.1016/j.jacc.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller JD, Weiss RM, Serrano KM, et al. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. 2009;119:2693–2701. doi: 10.1161/CIRCULATIONAHA.108.834614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garg V, Muth AN, Ransom JF, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 28.Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. 2009;47:828–834. doi: 10.1016/j.yjmcc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tkatchenko T, Moreno-Rodriguez R, Conway S, et al. Lack of periostin leads to suppression of Notch1 signaling and calcific aortic valve disease. Physiol Genomics. 2009;39:160–168. doi: 10.1152/physiolgenomics.00078.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30•.Bosse Y, Mathieu P, Pibarot P. Genomics: the next step to elucidate the etiology of calcific aortic valve stenosis. J Am Coll Cardiol. 2008;51:1327–1336. doi: 10.1016/j.jacc.2007.12.031. [DOI] [PubMed] [Google Scholar]; This is an informative review highlighting genetic mutations that have been associated with AS.
- 31.Antonini-Canterin F, Hirsu M, Popescu BA, et al. Stage-related effect of statin treatment on the progression of aortic valve sclerosis and stenosis. Am J Cardiol. 2008;102:738–742. doi: 10.1016/j.amjcard.2008.04.056. [DOI] [PubMed] [Google Scholar]
- 32.Mohty D, Pibarot P, Despres JP, et al. Age-related differences in the pathogenesis of calcific aortic stenosis: the potential role of resistin. Int J Cardiol. 2009 Jan 20; doi: 10.1016/j.ijcard.2008.12.068. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 33.Choi BG, Vilahur G, Yadegar D, et al. The role of high-density lipoprotein cholesterol in the prevention and possible treatment of cardiovascular diseases. Curr Mol Med. 2006;6:571–587. doi: 10.2174/156652406778018590. [DOI] [PubMed] [Google Scholar]
- 34.Busseuil D, Shi Y, Mecteau M, et al. Regression of aortic valve stenosis by ApoA-I mimetic peptide infusions in rabbits. Br J Pharmacol. 2008;154:765–773. doi: 10.1038/bjp.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cimmino G, Speidl WS, Ibanez B, et al. oxLDL Induces the expression of MCP-1 in aortic valve myofibroblasts via activation of NF-kappa B—inhibitory effects of recombinant Apo AI-Milano [abstract 1043–227] J Am Coll Cardiol. 2009;53:A415. [Google Scholar]
- 36.Ertas FS, Hasan T, Ozdol C, et al. Relationship between angiotensin-converting enzyme gene polymorphism and severity of aortic valve calcification. Mayo Clin Proc. 2007;82:944–950. doi: 10.4065/82.8.944. [DOI] [PubMed] [Google Scholar]
- 37.Simolin MA, Pedersen TX, Bro S, et al. ACE inhibition attenuates uremia-induced aortic valve thickening in a novel mouse model. BMC Cardiovasc Disord. 2009;9:10. doi: 10.1186/1471-2261-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cote N, Pibarot P, Pepin A, et al. Oxidized low-density lipoprotein, angiotensin II and increased waist circumference are associated with valve inflammation in prehypertensive patients with aortic stenosis. Int J Cardiol. 2009 Jun 12; doi: 10.1016/j.ijcard.2009.05.054. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 39.Corrado A, Santoro N, Cantatore FP. Extra-skeletal effects of bisphosphonates. Joint Bone Spine. 2007;74:32–38. doi: 10.1016/j.jbspin.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 40.Adami S, Braga V, Guidi G, et al. Chronic intravenous aminobisphosphonate therapy increases high-density lipoprotein cholesterol and decreases low-density lipoprotein cholesterol. J Bone Miner Res. 2000;15:599–604. doi: 10.1359/jbmr.2000.15.3.599. [DOI] [PubMed] [Google Scholar]
- 41.Montagnani A, Gonnelli S, Cepollaro C, et al. Changes in serum HDL and LDL cholesterol in patients with Paget's bone disease treated with pamidronate. Bone. 2003;32:15–19. doi: 10.1016/s8756-3282(02)00924-9. [DOI] [PubMed] [Google Scholar]
- 42.Price PA, Faus SA, Williamson MK. Bisphosphonates alendronate and ibandronate inhibit artery calcification at doses comparable to those that inhibit bone resorption. Arterioscler Thromb Vasc Biol. 2001;21:817–824. doi: 10.1161/01.atv.21.5.817. [DOI] [PubMed] [Google Scholar]
- 43.Rapoport HS, Connolly JM, Fulmer J, et al. Mechanisms of the in vivo inhibition of calcification of bioprosthetic porcine aortic valve cusps and aortic wall with triglycidylamine/mercapto bisphosphonate. Biomaterials. 2007;28:690–699. doi: 10.1016/j.biomaterials.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elmariah S, O'Brien KD, Budoff MJ, et al. The relationship of bisphosphonate use to the prevalence of aortic valve calcification in women. The Multi-Ethnic Study of Atherosclerosis [abstract 1001–299] J Am Coll Cardiol. 2009;53:A461. [Google Scholar]
- 45.Skolnick AH, Osranek M, Formica P, Kronzon I. Osteoporosis treatment and progression of aortic stenosis. Am J Cardiol. 2009;104:122–124. doi: 10.1016/j.amjcard.2009.02.051. [DOI] [PubMed] [Google Scholar]
- 46.Poon M, Badimon JJ, Fuster V. Overcoming restenosis with sirolimus: from alphabet soup to clinical reality. Lancet. 2002;359:619–622. doi: 10.1016/s0140-6736(02)07751-6. [DOI] [PubMed] [Google Scholar]
- 47.Wessely R, Schomig A, Kastrati A. Sirolimus and Paclitaxel on polymer-based drug-eluting stents: similar but different. J Am Coll Cardiol. 2006;47:708–714. doi: 10.1016/j.jacc.2005.09.047. [DOI] [PubMed] [Google Scholar]
- 48.Olsson M, Rosenqvist M, Nilsson J. Expression of HLA-DR antigen and smooth muscle cell differentiation markers by valvular fibroblasts in degenerative aortic stenosis. J Am Coll Cardiol. 1994;24:1664–1671. doi: 10.1016/0735-1097(94)90172-4. [DOI] [PubMed] [Google Scholar]
- 49.Spargias K, Milewski K, Debinski M, et al. Drug delivery at the aortic valve tissues of healthy domestic pigs with a Paclitaxel-eluting valvuloplasty balloon. J Interv Cardiol. 2009;22:291–298. doi: 10.1111/j.1540-8183.2009.00447.x. [DOI] [PubMed] [Google Scholar]
- 50•.Zajarias A, Cribier AG. Outcomes and safety of percutaneous aortic valve replacement. J Am Coll Cardiol. 2009;53:1829–1836. doi: 10.1016/j.jacc.2008.11.059. [DOI] [PubMed] [Google Scholar]; This is a recent review summarizing patient outcomes and safety with percutaneous aortic valve replacement.