

Abstract
We study Re analogues of 99mTc renal agents to interpret previous results at the 99mTc tracer level. The relative propensities of amine donors vs. carboxylate oxygen donors of four L = polyaminocarboxylate ligands to coordinate in fac-[ReI(CO)3L]n complexes were assessed by examining the reaction of fac-[ReI(CO)3(H2O)3]+ under conditions differing in acidity and temperature. All four L [N,N-bis-(2-aminoethyl)glycine (DTGH), N,N-ethylenediaminediacetic acid, diethylenetriamine-N-malonic acid, and diethylenetriamine-N-acetic acid] can coordinate as tridentate ligands while creating a dangling chain terminated in a carboxyl group. Dangling carboxyl groups facilitate renal clearance in fac-[99mTcI(CO)3L]n agents. Under neutral conditions, the four ligands each gave two fac-[ReI(CO)3L]n products with HPLC traces correlating well with known traces of the fac-[99mTcI(CO)3L]n mixtures. Such mixtures are common in renal agents because the needed dangling carboxyl group can compete for a coordination site. However, the HPLC separations needed to assess the biodistribution of a single tracer are impractical in a clinical setting. One goal in investigating this Re chemistry is to identify conditions for avoiding this problem of mixtures in preparations of fac-[99mTcI(CO)3L]n renal tracers. After separation and isolation of the fac-[ReI(CO)3L]n products, NMR analysis of all products and single crystal X-ray crystallographic analysis of both DTGH products as well as one product each from the other L allowed us to establish coordination mode unambiguously. The product favored in acidic conditions has a dangling amine chain and more bound oxygen. The product favored in basic conditions has a dangling carboxyl chain and more bound nitrogen. At the elevated temperatures used for simulating tracer preparation, equilibration was facile (ca. one hour or less), allowing selective formation of one product by utilizing acidic or basic conditions. The results of this fundamental study offer protocols and guidance useful for the design and preparation of fac-[99mTcI(CO)3L]n agents consisting of a single tracer.
Introduction
Tracers containing the 99mTc radionuclide are widely used in clinical nuclear medicine.1,2 Our goal of developing 99mTc radiopharmaceuticals with a rapid renal clearance has led us to seek 99mTc agents having ligands with a dangling (uncoordinated) carboxyl group,3-8 because the interaction of the renal receptor with the carboxyl group is important for clearance of small peptides.9-11 Many ligands have been investigated that form robust 99mTcVO anionic compounds with one or two dangling carboxyl groups.2,9,12-14 Such ligands pose a challenge because the carboxyl group can coordinate under some circumstances.15 The design of these 99mTcVO tracers has been aided by the evaluation of the inorganic chemistry of the ReVO analogues.9,15-24 The use of the Re analogue approach has provided well-defined chemical information that has facilitated the identification of 99mTcVO agents with excellent renal clearance in humans; however, 99mTcVO tracers do not have ideal properties.13,14,25-29
In recent years, technetium and rhenium complexes in low oxidation states have attracted significant attention.30-32 In particular, the fac-{MI(CO)3}+ core (M = Re, 99mTc) has been assessed because of its small size, its low-spin d6 electronic configuration, and the high chemical and kinetic stability of its complexes.30 These useful properties of the fac-{MI(CO)3}+ core allow the labeling of low-molecular-weight biomolecules attached through the three remaining facial sites available for substitution to produce agents having high specific activities.30,33-38 A convenient kit described by Alberto and colleagues for generating solutions of the fac-[99mTc(CO)3(H2O)3]+ cation is now commercially available.39,40 The uncharged aqua ligands in the fac-[99mTc(CO)3(H2O)3]+ precursor can be readily substituted by a wide variety of ligating groups, including amines, imines, thioethers, thiols, phosphines, and charged oxygen donors (carboxylates, phosphates, and phosphonates).4,7,41-51 Agents exhibiting the most favorable stability and pharmacokinetics have tridentate chelating ligands.42,44,52
We have been investigating the possibility that this relatively new core will allow us to develop renal agents with properties superior to those of the widely used agents with the 99mTcVO core.5-8,53 To explore the analogous Re chemistry, we recently developed a reliable and straightforward preparation of an aqueous solution of the fac-[Re(CO)3(H2O)3]+ cation.4 It is synthetically convenient to utilize polyaminocarboxylic acid ligands with acetate groups. Such ligands, including amino acid analogues, readily form stable complexes with the fac-{MI(CO)3}+ core.3,43,54,55 On the other hand, the catholic nature of the binding ability of this core raises concerns about the core’s coordination preferences for various donor atoms,43,47 especially the relative preference for amine groups vs. carboxylate groups.56,57
We initiated our studies with the new fac-{99mTcI(CO)3}+ core by using polyaminocarboxylate ligands (L).3,58,59 Treating fac-[99mTc(CO)3(H2O)3]+ with L formed robust fac-[99mTc(CO)3L]n anionic agents having a free carboxyl group useful for renal targeting, but often a mixture of two major anionic agents formed in a ratio dependent on preparative conditions.59 Therefore, we decided to utilize the Re analogue approach to investigate how temperature and acidic/neutral/basic conditions affected the formation and properties of fac-[Re(CO)3L]n analogues, primarily with N,N-bis(2-aminoethyl)glycine (DTGH, 1), but also with N,N-ethylenediaminediacetic acid (UEDDAH2, 2), diethylenetriamine-N-malonic acid (DTMH2, 3), and diethylenetriamine-N-acetic acid (DTAH, 4), Chart 1. Note that the L in Chart 1 exist as zwitterions, and the Hx in the names designates the number of dissociable protons. These L contain N and O donor groups appropriately oriented so as to allow facial, tridentate coordination, while at the same time having the potential to bind via different coordination modes (NNN vs NNO; NNO vs ONO). These ligands contain combinations of ethylene amino groups and acetate groups; the groups are attached to N donors in such a way that only five-membered chelate rings can form and such that any dangling group is anchored to Re via an N donor. Note that because all tricarbonyl complexes used or made here have a facial geometry, fac- is not used for specific tricarbonyl compounds but is used in general designations such as fac-[Re(CO)3L]n or fac-[99mTcI(CO)3L]n.
Chart 1.

Experimental Section
DTGH (1),60 UEDDAH2 (2),61 DTAH (4)60 and potassium bromomalonate62 were synthesized according to published methods. The trifluoroacetate of diprotonated DTGH (DTGH·2CF3CO2H) was obtained during the purification process and was used as a crude product for the next step. An aqueous solution of the precursor, [Re(CO)3(H2O)3]OTf (OTf = triflate anion), was prepared as reported elsewhere.4 All solvents were purchased from Aldrich and were used without purification. Elemental analyses were performed by Atlantic Microlabs, Atlanta, GA. 1H NMR spectra were recorded on Varian INOVA 400 or 600 MHz spectrometers and referenced to internal sodium 3-(trimethylsilyl)propionate-d4. HPLC analyses (monitored at 254 nm) were performed on a Waters Breeze system equipped with a Waters 2487 dual wavelength absorbance detector, Waters 1525 binary pump, and XTerra MS C18 column (5 μm; 4.6 × 250 mm). HPLC solvents consisted of the buffer [0.05 M TEAP (aqueous triethylammonium phosphate) at pH 2.5, solvent A] and methanol [solvent B]. The HPLC system started with 100% A from 0 to 3 min. The eluent switched at 3 min to 75% A/25% B, at 6 min to 66% A/34% B, and remained for 3 more min, followed by linear gradients: 66% A/34% B to 34% A/66% B from 9 to 20 min; and 34% A/66% B to 100% A from 20 to 30 min. The flow rate was 1 mL/min. An Orion Research Digital Ionalyzer/501 equipped with a glass/calomel ultrathin longstem combination pH electrode (Z11,343-3, Aldrich) was used to measure the pH of NMR solutions, including solutions being monitored over time.
Re(CO)3(DTG) Products (5 and 6)
A solution of [Re(CO)3(H2O)3]OTf (2.5 mL, 0.1 M) was added to a solution of DTGH·2CF3CO2H (0.097 g, 0.25 mmol) in 2 mL of water, and the pH of this equimolar reaction mixture was adjusted to 3.5 or (in a single experiment to assess pH effects) to pH 7.5. Normally for reaction mixtures, 1 N NaOH or HCl was used to adjust the pH, which was determined by using the Orion Research pH meter or EDM colorpHast pH test strips. The solution was heated at reflux, and the progress of the reaction was monitored by HPLC. Two products, 5 ([Re(CO)3(DTGH)-NNO]+) and 6 ([Re(CO)3(DTG)-NNN]), formed, having HPLC retention times (RT) of 3.8 min (5) and 7.6 min (6). Ratios of 5 : 6 of ~90 : 10 at pH 3.5 and ~45 : 55 at pH 7.5 were observed after one hour of heating. Reactions in both acidic and basic conditions were repeated several times under similar pH conditions (± 0.3 pH units) and the reported ratio of products was based on the average peak ratio, which did not change significantly with time.
5 (NNO Ligand Binding Mode)
Solid KPF6 (5 mg, 0.25 mmol) was added (in pH 3.5 reactions), and the mixture was stirred until the solid dissolved completely. After filtration, the solution was passed through a Sephadex G-15 column, and the products were eluted with deionized water. Eluted fractions containing only the NNO isomer were collected and concentrated to dryness by rotary evaporation, yielding a white solid that was recrystallized from water. Yield, 0.13 g (86%) of pure 5 as the [Re(CO)3(DTGH)-NNO]PF6 salt, designated hereafter as the 5 PF6 salt. Anal. Calcd for C9H15F6N3O5PRe·H2O: C, 18.17; H, 2.86; N, 7.06. Found: C, 18.39; H, 2.86; N, 7.12. 1H NMR [δ (ppm), DMSO-d6]: 2.47 (m, 1H), 2.65 (d, 1H), 2.90 (m, 1H), 3.05 (m, 1H), 3.16 (m, 2H), 3.43 (m, 1H), 3.64 (d, 2H), 3.66 (m, 1H), 4.91 (m, 1H), 5.14 (m, 1H) 7.74 (bs, 3H). [The 5 PF6 salt was obtained mainly for the X-ray crystal structure determination and elemental analyses. For other low-pH reactions 5 was isolated as a white solid (5 Cl salt) from low pH reaction mixtures (pH adjusted with 1 N HCl) in the same manner as described above but without the addition of KPF6.]
6 (NNN Ligand Binding Mode)
Pure 6 was isolated when the 5 Cl salt was dissolved in water at pH 12 and the solution was kept at room temperature overnight. White crystals of the neutral NNN product 6 ([Re(CO)3(DTG)-NNN], neutral solid) were collected, washed with a small amount of water, and vacuum dried. Anal. Calcd for C9H14N3O5Re·H2O: C, 24.10; H, 3.60; N, 9.37. Found: C, 24.06; H, 3.57; N, 9.39. 1H NMR [δ (ppm), D2O, pH 8.7]: 3.17 (m, 4H), 3.26 (m, 4H), 4.12 (s, 2H). Solids of 5 (PF6 and Cl forms) and 6 (neutral form) are slightly soluble in water.
Re(CO)3(UEDDA) Products (7 and 8)
8 (NNO Ligand Binding Mode)
[Re(CO)3(H2O)3]OTf (5 mL, 0.1 M) was added to a solution of UEDDAH2 (0.081 g, 0.50 mmol) in 5 mL of water, and the pH of the equimolar reaction mixture was adjusted to 7 with 1 N NaOH. The clear pH 7 solution was stirred at 30 to 40 °C. The reaction progress was monitored by HPLC, which showed that the precursor was consumed after 3 h, and one sharp peak was observed (RT = 14.5 min). The solution was concentrated to 2 to 3 mL by rotary evaporation and desalted on a Sephadex G-15 column (eluted with deionized water). The fractions collected were evaporated to dryness by rotary evaporation and then dried under vacuum to give 8, most probably as a sodium salt (Na[Re(CO)3(UEDDA)-NNO], NNO ligand binding mode), in 90% yield (0.210 g). A small amount of this white solid was dissolved in 2 mL of water/NaOH; the solution was filtered, the pH was adjusted to ~1 with 1 N HCl, and the solution was allowed to stand at 0 to 5 °C. Crystals of 8, as the neutral [Re(CO)3(UEDDAH)-NNO] zwitterion with a protonated carboxylic group, formed overnight and were suitable for X-ray crystallography. Anal. Calcd for C9H11N2O7Re: C, 24.27; H, 2.49; N, 6.29. Found: C, 24.35; H, 2.46; N, 6.27. 1H NMR [δ (ppm), D2O, pH 7.0]: 2.72 (m, 1H), 2.99 (m, 1H), 3.14 (m, 1H), 3.40 (m, 1H), 3.96 (d, 1H, J = 16.4 Hz), 4.02 (d, 1H, J = 17.6 Hz), 4.14 (mb, 1H), 4.18 (d, 1H, J = 17.6 Hz), 4.21 (d, 1H, J = 16.4 Hz), 4.88 (mb, 1H).
7 (ONO Ligand Binding Mode)
When the above preparative procedure was carried out at pH 3 to 4, instead of at pH 7, HPLC analysis showed that the precursor was consumed after ~4 h, and one sharp major peak (RT = 8.8 min, 96%) was observed. The white solid that precipitated from the reaction mixture overnight was collected by filtration, washed with a small amount of cold water, and dried under vacuum. Yield, 0.15 g (67%). Anal. Calcd for C9H11N2O7Re·H2O: C, 23.33; H, 2.83; N, 6.05. Found: C, 23.25; H, 2.83; N, 5.95. Elemental analysis suggests that 7 is the neutral [Re(CO)3(UEDDAH)-ONO)] zwitterion, with a protonated amino group, and an isomer of 8. 1H NMR (δ (ppm), D2O, pH 4.0) analysis showed that the product has an ONO ligand binding mode (7: 3.41 (m, 2H), 3.72 (overlapping d, J = 16.8 Hz, 2H and m, 2H), 3.93 (d, 2H, J = 16 Hz)). All attempts to obtain crystals of 7 suitable for X-ray crystallography were unsuccessful.
Re(CO)3(DTM) Products (9 and 10)
To prepare diethylenetriamine-N-malonic acid (DTMH2, 3), an aqueous solution of potassium bromomalonate (4.43 g, 17.1 mmol, 5 mL) was added dropwise to an aqueous solution (5 mL) of diethylenetriamine (1.85 mL, 17.1 mmol) at 25 °C. The pH of the reaction mixture was adjusted to 10 with 2.5 N HCl, and the reaction mixture was maintained at 60 °C and pH 10 for 2.5 h before it was cooled to 25 °C. The solution was acidified with concentrated HCl (pH ~ 4) and left overnight at 0 to 5 °C. After a white precipitate formed, the mixture was centrifuged, the supernatant liquid removed, and the precipitate was washed with cold water and vacuum dried; yield, 1.78 g (51%). The 1H NMR spectrum in D2O (pH 6) indicated that the solid contained ~80% of 3. Although an elaborate purification method gave product with much cleaner NMR spectra (1H NMR [δ (ppm), D2O, pH 3.5]: 3.39 (t, J = 6.6 Hz, 2H), 3.44 (m, 4H), 3.52 (t, J = 6.0 Hz, 2H); 13C NMR [δ (ppm), D2O, pH 4.3]: 41.3 (NCH2), 47.0 (NCH2), 47.9 (NCH2), 48.1 (NCH2), 69.4 (CH), 172.2 (CO2H)), the crude product was sufficiently pure for use in the next step.
[Re(CO)3(H2O)3]OTf (8.2 mL, 0.1 M) was added to a solution of DTMH2 (0.168 g, 0.82 mmol) in 15 mL of water and the pH of the reaction mixture was adjusted to 7 with 1 N NaOH. The solution was stirred at 50 to 60 °C for 2 h. HPLC analysis of the solution showed two peaks with RT = 3.5 min for 9 ([Re(CO)3(DTMH)-NNO]) and RT = 6.8 min for 10 ([Re(CO)3(DTM)-NNN]−). 1H NMR (D2O) analysis of the reaction mixture at pH 7 showed a 9 : 10 ratio of 45 : 55.
9 (NNO Ligand Binding Mode)
Pure 9 as the neutral [Re(CO)3(DTMH)-NNO] product (9 neutral) was obtained by adjusting the pH of the reaction mixture to 3.5 with 1 N HCl and stirring the mixture at room temperature for 30 min. The solution was then concentrated to 2-3 mL by rotary evaporation and desalted on a Sephadex G-15 column (eluted with deionized water). The product fractions collected were evaporated to dryness to give 9 neutral as a white solid; yield, 0.162 g (42%). Crystals suitable for X-ray crystallography were obtained by crystallization from water. Anal. Calcd for C10H14N3O7Re: C, 25.32; H, 2.97; N, 8.86. Found: C, 25.14; H, 3.30; N, 9.06. 1H NMR [δ (ppm), D2O, pH 6.0]: 2.34 (m, 1H), 3.09-3.26 (m, 6H), 3.43 (m, 1H), 4.01 (s, 1H), 5.75 (m, 1H), 6.15 (s, 1H).
10 (NNN Ligand Binding Mode)
Pure 10 was obtained by adjusting the pH of the reaction mixture to 10 with 1 N KOH and stirring at room temperature for 1 h. After the solution was concentrated to 2 to 3 mL and desalted on a Sephadex G-15 column (eluted with deionized water), the fractions containing product were collected and evaporated to dryness. The residue was treated with ethanol (10 mL) and the mixture was kept overnight at 0-5 °C. The white solid that formed was separated by decantation and dried; yield, 0.24 g (55%) as the K[Re(CO)3(DTM)-NNN] salt. Anal. Calcd for C10H13KN3O7Re·H2O: C, 22.63; H, 2.92; N, 7.92. Found: C, 22.93; H, 2.96; N, 7.90. 1H NMR [δ (ppm), D2O, pH 10.5]: 2.66 (m, 1H), 2.75 (m, 1H), 2.96 (m, 1H), 3.05 (m, 2H), 3.12 (dd, 1H), 3.24 (m, 1H), 3.45 (m, 1H), 4.12 (s, 1H). The spectrum lacked the very characteristic upfield signal of 9 at 2.34 ppm; thus, 9 (the product related to 10 but with the NNO coordination mode) was not formed under these conditions. Compound 10 is freely soluble in water. All attempts to obtain crystals of 10 suitable for X-ray crystallography were unsuccessful.
Re(CO)3(DTA) Products (11 and 12)
During the purification of 10 on a Sephadex G-15 column, several fractions collected contained a small amount of a product with the rhenium tricarbonyl moiety and having an HPLC RT of 7.8 min. Water was removed by rotary evaporation to give a white solid (12, [Re(CO)3(DTA)-NNN]. 1H NMR [δ (ppm), D2O, pH 7.0]: 2.66 (m, 1H), 2.81 (m, 1H), 2.96 (m, 3H), 3.08 (m, 1H), 3.24 (m, 1H), 3.37 (m, 1H), 3.77 (dd, 2H), 4.03 (m, 1H), 4.22 (m, 1H), 5.17 (m, 1H), 6.55 (s, 1H). Crystals suitable for X-ray structural determination were obtained by crystallizing a few milligrams of the crude product from methanol. An aqueous solution of 12 kept at pH ~2 overnight showed no 1H NMR spectral changes. When equimolar amounts (0.5 mmol) of the DTAH (4) ligand and [Re(CO)3(H2O)3]OTf in 5 mL of water were heated at pH ~ 6-7 and 70 °C for 2 h, the HPLC analysis of the solution showed two peaks with RT = 3.6 min and RT = 7.8 min in a 48 : 52 ratio. Preparative HPLC afforded only a few milligrams of each product. The 1H NMR spectrum of the product with RT = 7.8 min was identical with that of 12 ([Re(CO)3(DTA)-NNN]). The 1H NMR (D2O, pH 3.0) analysis of the product with RT = 3.6 min (11) showed that the ligand in this product has the NNO ligand binding mode; 1H NMR [δ (ppm), D2O, pH 3.0]: 2.36 (m, 1H), 2.98 (m, 1H), 3.08 (m, 4H), 3.28 (m, 2H), 3.53 (d, 1H, J = 17.6 Hz), 3.91 (dd, 1H, J = 17.6, 7.6 Hz), 5.75 (bs, 1H), 6.50 (bs, 1H)). We are confident that 11 is ([Re(CO)3(DTAH)-NNO]+. However, all attempts to isolate 11 in a pure form for the full characterization were unsuccessful.
X-ray Crystal Structural Determination
Suitable crystals of 5, 6, 8, 9, and 12 were coated with Paratone N oil, suspended in a small fiber loop, and placed in a cooled nitrogen gas stream at 100 K on a Bruker D8 SMART APEX CCD sealed tube diffractometer with graphite monochromated Cu Kα (1.54178 Å) radiation (for 5, 6, 8, 12) or Mo Kα (0.71073 Å) radiation (for 9). Data were measured by using a series of combinations of phi and omega scans with 10-second frame exposures and 0.5° frame widths. Data collection, indexing, and initial cell refinements were all carried out with APEX 263 software. SAINT64 software was used for frame integration and final cell refinements. The SADABS65 program was used to carry out absorption corrections.
The structures were solved by using direct methods and difference Fourier techniques (SHELXTL, V5.10).66 All non-hydrogen atoms were located in difference Fourier maps and refined anisotropically except for the N and O atoms in 5; the C and N atoms in 6; the C, O4 and O5 atoms in 8; C9 in 9; and C1s and O1s in 12. Hydrogen atoms were placed in their expected chemical positions by using the HFIX command and were included in the final cycles of least squares with isotropic Uij’s related to the atoms ridden upon. Scattering factors and anomalous dispersion corrections were obtained from the International Tables for X-ray Crystallography.67 Structure solution, refinement, graphics, and generation of publication materials were performed by using SHELXTL, V5.10 software. Crystal data and refinement parameters for 5, 6, 8, 9, and 12 are listed in Table 1. Selected bond distances and angles appear in Table 2.
Table 1.
Crystal Data and Structure Refinement for [Re(CO)3(DTGH)-NNO]PF6·H2O (5 PF6·H2O), [Re(CO)3(DTG)-NNN]·H2O (6·H2O), [Re(CO)3(UEDDAH)-NNO] (8), [Re(CO)3(DTMH)-NNO]·2H2O (9·2H2O) and [Re(CO)3(DTA)-NNN]·CH3OH (12·CH3OH)
| 5 PF6·H2O | 6·H2O | 8 | 9·2H2O | 12·CH3OH | |
|---|---|---|---|---|---|
| empirical formula | C9H15F6N3O6PRe | C9H14N3O6Re | C9H11N2O7Re | C10H18N3O9Re | C10H18N3O6Re |
| fw | 592.41 | 446.43 | 445.40 | 510.47 | 462.47 |
| T (K) | 173 (2) | 100 (2) | 173 (2) | 100 (2) | 173 (2) |
| λ (Å) | 1.54178 | 1.54178 | 1.54178 | 0.71073 | 1.54178 |
| crystal system | monoclinic | monoclinic | orthorhombic | monoclinic | monoclinic |
| space group | P21/c | P21/n | Pna21 | P21/n | P21/n |
| unit cell dimensions | |||||
| a (Å) | 9.7966 (4) | 7.8847 (10) | 11.8855 (5) | 10.241 (9) | 8.2324 (9) |
| b (Å) | 12.3907 (5) | 12.7776 (2) | 7.7212 (3) | 13.256 (11) | 12.6243 (13) |
| c (Å) | 14.4301 (5) | 13.4044 (2) | 13.6549 (6) | 11.602 (11) | 13.6879 (14) |
| β (°) | 107.363 (2) | 94.2740 (10) | 90 | 92.37 (2) | 96.321 (14) |
| V (Å3) | 1671.81 (11) | 1346.70 (3) | 1253.11 (9) | 1574 (2) | 1413.9 (3) |
| Z | 4 | 4 | 4 | 4 | 4 |
| ρ calc (mg/m3) | 2.354 | 2.202 | 2.361 | 2.155 | 2.173 |
| μ (mm−1) | 16.086 | 17.929 | 19.309 | 7.773 | 17.103 |
| R indices [I>4σ(I)] | R1 = 0.0633 | R1 = 0.0756 | R1 = 0.0230 | R1 = 0.0946 | R1 = 0.0252 |
| wR2 = 0.2006 | wR2 = 0.2629 | wR2 = 0.0583 | wR2 = 0.2390 | wR2 = 0.0661 | |
| R indices (all data) | R1 = 0.0690 | R1 = 0.0890 | R1 = 0.0230 | R1 = 0.1051 | R1 = 0.0257 |
| wR2 = 0.2197 | wR2 = 0.2966 | wR2 = 0.0583 | wR2 = 0.2491 | wR2 = 0.0665 | |
R1 = (Σ | |Fo| - |Fc| | )/Σ |Fo|; wR2 = [Σ[w(Fo2 – Fc2)2]/Σ[w(Fo2)2]]1/2 ; where w = 1/[σ2(Fo2) + (aP)2 + bP] and P = [(Max;0, Fo2) + 2 Fc2]/3
Table 2.
Selected Bond Distances (Å) and Bond Angles (°) of [Re(CO)3(DTGH)-NNO]PF6·H2O (5 PF6·H2O), [Re(CO)3(DTG)-NNN]·H2O (6·H2O), [Re(CO)3(UEDDAH)-NNO] (8), [Re(CO)3(DTMH)-NNO]·2H2O (9·2H2O) and [Re(CO)3(DTA)-NNN]·CH3OH (12·CH3OH)
| 5 PF6·H2O | 6·H2O | 8 | 9·2H2O | 12·CH3OH | |
|---|---|---|---|---|---|
| Re(1)–N(1) | 2.191(8) | 2.181(10) | 2.242(4) | 2.160(7) | 2.230(4) |
| Re(1)–N(2) | 2.237(8) | 2.252(11) | 2.203(5) | 2.234(19) | 2.235(4) |
| Re(1)–N(3) | - | 2.200(11) | - | - | 2.221(4) |
| Re(1)–O(4) | 2.150(6) | - | 2.151(4) | 2.119(14) | - |
| O(4)–Re(1)– C(1) | 173.9(3) | - | 170.6(3) | 174.1(8) | - |
| O(4)–Re(1)– C(2) | 96.1(4) | - | 96.79(17) | 96.1(8) | - |
| O(4)–Re(1)– C(3) | 95.2(3) | - | 98.4(3) | 96.7(11) | - |
| O(4)–Re(1)– N(1) | 80.9(3) | - | 76.2(3) | 76.1(6) | - |
| O(4)–Re(1)– N(2) | 78.5(3) | - | 78.61(15) | 83.0(8) | - |
| N(1)–Re(1)– C(3) | 173.0(3) | 175.5(5) | 173.9(2) | 171.7(10) | 89.77(17) |
| N(1)–Re(1)– C(1) | 94.8(4) | 92.0(5) | 95.1(2) | 98.0(9) | 98.88(17) |
| N(1)–Re(1)– C(2) | 94.6(4) | 95.8(5) | 98.3(2) | 98.5(9) | 174.21(18) |
| N(2)–Re(1)– C(2) | 172.3(4) | 167.4(5) | 175.05(18) | 178.2(8) | 97.66(19) |
| N(2)–Re(1)– C(1) | 96.5(4) | 98.7(5) | 96.5(2) | 95.6(10) | 172.94(17) |
| N(2)–Re(1)– C(3) | 94.3(3) | 98.4(5) | 97.4(2) | 95.4(9) | 96.45(17) |
| N(3)–Re(1)– C(1) | - | 176.5(5) | - | - | 96.18(17) |
| N(3)–Re(1)– C(2) | - | 90.4(5) | - | - | 93.24(17) |
| N(3)–Re(1)– C(3) | - | 93.9(5) | - | - | 173.81(16) |
| N(1)–Re(1)–N(2) | 79.2(3) | 77.1(4) | 78.86(18) | 79.8(7) | 76.55(14) |
| N(1)–Re(1)–N(3) | - | 85.0(4) | - | - | 85.89(14) |
| N(2)–Re(1)–N(3) | - | 78.7(4) | - | - | 78.25(15) |
Results and Discussion
Complexes 5 to 12 were prepared by treating each polyaminocarboxylic acid ligand with the fac-[Re(CO)3(H2O)3]+ precursor in aqueous solution (Scheme 1). For all ligands, two products with different coordination modes were formed and the ratio of the products depended on whether the reaction mixture was acidic, neutral or basic. As indicated in Scheme 1, under most conditions, the protonation state of the coordinated ligand is different in the two related complexes. Thus, the complexes in a pair have different charge and are not, strictly speaking, isomers. The fac-[Re(CO)3L]n complexes (n = +1, 0, or −1) were characterized by analytical and spectroscopic methods; the structures of several complexes (5, 6, 8, 9, and 12) were also confirmed by X-ray crystallographic methods.
Scheme 1.

Crystallographic Studies
All X-ray structural analyses (Figures 1 to 4, below) confirm that three CO ligands are coordinated to one face of the pseudo-octahedron, while the other face is occupied by tridentate coordinated ligands; donor sets are ONO in 7, NNO in 5, 8, and 9, and NNN in 6 and 12. In all cases, the ligands form two five-membered chelate rings. The structural parameters involving Re are similar and typical for all structures (Table 2), with unexceptional Re-C bond distances (1.900 to 1.924 Å; Table S1 in Supporting Information) consistent with values found in similar complexes.3,68-70 The trans angles fall between 170.6° and 178.2°, with the exception of the smaller N(2)–Re(1)–C(2) angle in 6 (167.4(5)°), in which the ligating tertiary nitrogen atom anchors two chelate rings and bears the dangling CH2CO2− group. The cis angles show large deviations from the idealized octahedral geometry (76.1-98.9°). The Re–O distances (2.119-2.151 Å) are close to those found in related complexes in which a carboxylate oxygen is coordinated trans to a carbonyl ligand.3,42,44,69,70 The Re–N bond distances of 2.160-2.252 Å are consistent with sp3-hybridized nitrogen donors. Projecting away from the Re center is the dangling side arm of the ligand, namely the acetate group in 6, 12 (-CH2CO2−; Figures 1 (right) and 4, respectively) and 8 (-CH2CO2H; Figure 2) and the ethylammonium group in 5 and 9 (-CH2CH2NH3+; Figures 1 (left) and 3, respectively). For fac-[Re(CO)3L]n complexes in which the nitrogen anchoring the dangling group is a terminal secondary amine (i.e., coordinated secondary amine not linking two chelate rings), the NH proton could be endo (projecting away from the face defined by the tridentate ligand, as in 9, Figure 3) or exo (projecting into this face, as in 12, Figure 4).
Figure 1.

Perspective drawings of the complex in [Re(CO)3(DTGH)-NNO]PF6·H2O (5 PF6·H2O) (left) and [Re(CO)3(DTG)-NNN]·H2O (6·H2O) (right) with 50% probability for the thermal ellipsoids. The PF6− anion and H2O molecule are omitted for clarity.
Figure 4.

Perspective drawing of the complex in [Re(CO)3(DTA)-NNN]·CH3OH (12·CH3OH) with 50% probability for the thermal ellipsoids. The methanol molecule is omitted for clarity.
Figure 2.

Perspective drawing of [Re(CO)3(UEDDAH)-NNO] (8) with 50% probability for the thermal ellipsoids.
Figure 3.
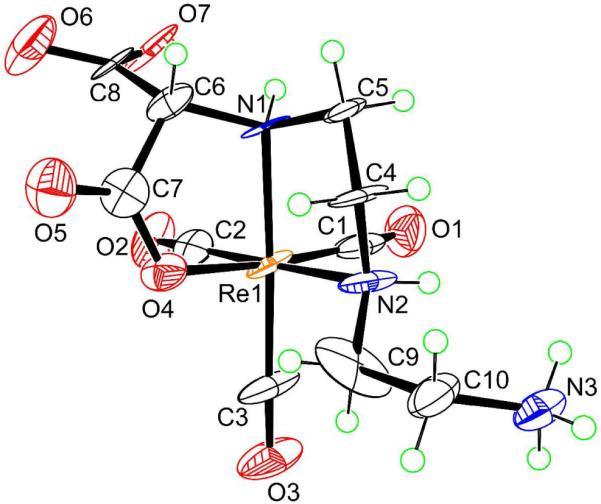
Perspective drawing of the complex in [Re(CO)3(DTMH)-NNO]·2H2O (9·2H2O) with 50% probability for the thermal ellipsoids. The H2O molecules are omitted for clarity.
Except for 6, which in the solid has essentially a mirror plane containing the O2–C2–Re–N2 bonds (cf. Figures 1-4 for numbering), the complexes are asymmetric and the unit cells contain a racemic mixture. The structure of only one enantiomer is illustrated. All complexes exhibit significant intermolecular hydrogen bonding in the solid.
Spectroscopic Properties
The use of 1H NMR spectra to study ReI analogues of TcI radiopharmaceuticals is less advanced than in the study of ReV analogues of TcV radiopharmaceuticals.15,21,23,71-74 Here, we employed 1H NMR spectra to provide needed evidence for assessing the overall geometry of the new fac-[Re(CO)3L]n complexes, especially when crystals could not be obtained. Fundamental studies are ongoing in our program to employ simple ligands lacking dangling groups to interpret the causes of unusual NMR chemical shifts in Re analogues of 99mTc agents with dangling groups.75 Although we note briefly a few such shifts that appear to be characteristic of some geometric features, the elucidation of the underlying factors influencing these shifts is not the focus of the current study.
Identification of coordination mode relies on comparison of the NMR spectra of the free and coordinated ligand. For free ligands, the signals for the methylene group adjacent to the carboxyl group are singlets, and the signals of the protons of each ethylene bridge are two triplets. After ligand coordination, the methylene group protons give rise to two strongly coupled doublets having coupling constants (J = ~17 Hz) consistent with geminal coupling because the protons become non-equivalent in an asymmetric environment. However, when the ligand coordinates in such a way that the resultant Re complex has mirror symmetry (mirror plane through one of the three carbonyl groups, as in 6), the signal for the methylene group protons of a dangling acetate side chain is a singlet. The methylene signals do not have very different shifts or coupling constants whether the group is in a coordinated or dangling acetate group. The key observations providing information on structure are the presence or absence of coupling and the number of signals. However, from past work,3 the doublets of a coordinated acetate group have a larger coupling constant than those for a dangling group and we use this criterion for attributing a particular set of doublets to coordinated or dangling acetate groups. In addition, if the coordinated acetate group is attached to a secondary amine, one acetate doublet exhibits weak NH/CH coupling (J ~7 Hz) because the H-C-N-H torsion angle is ca. ~20-30° for this CH proton. The signal for the second CH proton (H-C-N-H torsion angle ca. 90°) remains a doublet. On the other hand, both doublets of the dangling acetate group are further split by the coupling to an NH proton if the dangling group is attached to a secondary amine terminating the chelate ring.3
Also after coordination, the two triplets of each ethylene bridge are further split into four multiplets; these sometimes overlap with each other or with other signals. These features are consistent with the facial tridentate coordination mode of these polyaminocarboxylate ligands. In general, because the CH signals can be observed under all pH conditions, the CH signals were more useful than NH signals in assessing the coordination mode of ligands in products that did not form X-ray quality crystals.
In some cases, NH signals were also studied (see below). In our previous studies of fac-[Re(CO)3L]n analogues of 99mTc renal agents with dangling carboxyl groups, we found unusual shift dispersion of NH 1H NMR signals in several solvents, including D2O and DMSO-d6 (where L = S-methyl-L-cysteine,4 methionine,4 lanthionine,6 or ethylenediamine-N,N’-diacetic acid3). Although much more study is needed, some guidelines are developing. NH protons projecting into the face defined by the tridentate ligand (designated as exo-NH protons) sometimes have unusually upfield shifts compared to NH protons projecting away from this face (designated as endo-NH protons), even for the same primary amine. An endo-NH proton in a terminal secondary amine groups has a shift that is typically less downfield than that of the NH of a secondary amine anchoring the two chelate rings. The latter protons also project away from the tridentate face of the pseudo octahedron. However, complexes of ligands with dangling groups and asymmetric centers have complicated spectra with signals having shifts not readily understood, as mentioned above.6 In studies with prototypical simpler ligands directed at investigating the possible cause of the NH dispersion,75 we concluded that the more downfield shift of the endo-NH signals vs. the exo-NH signals is related to greater solvent exposure of the endo-NH proton. Hydrogen bonding in solution causes downfield shifts.75
N versus O Binding
To evaluate factors affecting the relative binding ability of N versus O donors, we selected the DTGH ligand because it is symmetrical and has both one carboxyl group and one tertiary amine, but it also has two primary amines.
Compounds Derived from the DTGH Ligand
Treatment of fac-[Re(CO)3(H2O3)+ with DTGH (1, Chart 1) at reflux gave two products, 5 (RT = 3.8 min) and 6 (RT = 7.6 min) (Scheme 1). Both 5 and 6 were isolated and fully characterized, including by X-ray crystallography (see above). The product favored at low pH (5) is [Re(CO)3(DTGH)-NNO]+; the NNO coordination mode consists of two amine (one primary and one tertiary) and one carboxyl group donors, leaving the –CH2CH2NH3+ group dangling (Figure 1, left). The product favored at high pH (6) is [Re(CO)3(DTG)-NNN]; the NNN coordination mode consists of three amine groups of the ligand, and the CH2CO2− moiety is the pendant arm (Figure 1, right). As expected for complexes with different coordination modes, the 1H NMR spectra of 5 and 6 differ greatly. For 5 (cf. Figure S1 in Supporting Information, see Figure 1 (left) for atom labeling), two signals (4.94 and 4.32 ppm) are attributable to the coordinated primary amine N1H2 group. The N1H signal at 4.94 ppm had cross-peaks to two of the signals of the four resolved ethylene bridge multiplets (3.19 and 2.75 ppm); these CH signals, which had cross-peaks to each other, were assigned to the C7H2 group (next to N1H2). Two almost overlapping signals at 3.02 and 3.05 ppm (these signals were better resolved in DMSO-d6) had cross-peaks with both H7 signals and were assigned to the C6H2 group. The signals (3.86 and 3.69 ppm) of the C9H2 group next to the protonated primary amine group (N3H3+) of the dangling ethylammonium arm are about 0.3 ppm downfield from the signal for the C8H2 protons (multiplet at 3.48 ppm integrating for two protons) next to the coordinated tertiary amine (N2). The signals of the methylene group of the coordinated acetate group of 5 appeared as a set of doublets with a coupling constant of 16.8 Hz at 3.99 and 3.75 ppm. In contrast to this complicated spectrum, 6 has a very simple spectrum (in D2O at pH 8.7 there are two multiplets at 3.26 and 3.17 ppm for eight ethylene protons and one singlet at 4.12 ppm for two methylene protons of the dangling acetate group); these values are consistent with the symmetry of 6.
The ratio of products 5 and 6 obtained from reaction mixtures at reflux temperatures depended on the pH of the reaction mixture (Scheme 1). While 5 and 6 were formed in nearly equal abundance (45 : 55 ratio of 5 : 6) at pH ~7, 5 (in a protonated form) was the major product (~90%) at pH ~3-5, whereas 6 was the only product (100%) at pH ~9-12 (Table 3). To determine whether products from the synthetic reactions were thermodynamically or kinetically controlled, we assessed both whether the products interconvert and whether interconversion occurs on a relevant time scale. Experiments conducted at both elevated and room temperatures are described in the next paragraph.
Table 3.
Relative Abundance of Members of Product Pairs Under Acidic, Neutral, and Basic Conditions (from Preparative Reaction Mixtures, Except As Noted)
| form of complex at pH 7 | composition (%) |
||
|---|---|---|---|
| pH 3-5 | pH ~ 7 | pH 9-12 | |
| [Re(CO)3(DTGH)-NNO]+ (5) | 90 | 45 | 0 |
| [Re(CO)3(DTG)-NNN] (6) | 10 | 55 | 100 |
| [Re(CO)3(UEDDAH)-ONO] (7) | 96 | 2 | 0 |
| [Re(CO)3(UEDDA)-NNO]− (8) | 4 | 98 | 100 |
| [Re(CO)3(DTMH)-NNO] (9) | 100 | 45 | 0 |
| [Re(CO)3(DTM)-NNN]- (10) | 0 | 55 | 100 |
| [Re(CO)3(DTAH)-NNO]+ (11) | 98a | 42 | 20b |
| [Re(CO)3(DTA)-NNN] (12) | 2a | 58 | 80b |
Equilibrated mixture (pH ~ 2) at 100 °C, starting from isolated 12.
Equilibrated mixtures (pH ~ 8) at both 25 °C and 100 °C, starting from isolated 12.
HPLC and 1H NMR techniques were used to measure the relative abundance of the [Re(CO)3(DTGH)-NNO]+(5) and [Re(CO)3(DTG)-NNN] (6) products present in solutions prepared with each pure isomer and maintained at various pH values at room temperature for 3 or more days. No changes were observed from pH 3 to pH 5 for 5 and from pH 9 to pH 12 for 6. Mixtures of 5 and 6 were observed between pH 5 and pH 9. The [Re(CO)3(DTGH)-NNO]+ (5) product undergoes complete conversion to the [Re(CO)3(DTG)-NNN] (6) product at high pH (> 9) almost immediately. However, interconversion of 5 and 6 was much slower at neutral pH and room temperature. At pH 7.0, the same final mixture (30 : 70) of isomers 5 : 6 was observed after about one week whether we started with [Re(CO)3(DTGH)-NNO]+ (5) or [Re(CO)3(DTG)-NNN] (6). Note that for the preparative reaction solution, which was heated at reflux, a 45 : 55 ratio of 5 : 6 was achieved at pH 7.5 in a short time (~1 h). Because these results suggest that the formation reactions at reflux starting with L and the fac-[Re(CO)3(H2O3)+ cation were under thermodynamic, not kinetic control, we also heated a solution of 6 at pH ~3 and found that it converted to 5 in 1-1.5 h, a time comparable to that of the typical preparative reaction. These results show clearly that the typical labeling conditions will give the thermodynamic distribution at elevated temperature, regardless of pH, but that at room temperature the change in coordination mode occurs readily only at high pH, where the dangling amine exists to a high extent in the unprotonated state (pKa of the CH2CH2NH3+ group = ~9-11). This observation, in turn, implies that the change in coordination mode may involve either an SN2 mechanism with the free amine attacking the ReI center or an SN1CB reaction with a deprotonated coordinated amine labilizing the Re-O bond, possibly generating an intermediate with reduced coordination number. Extensive studies would need to be performed in order to establish the likely mechanism, but some results described below suggest that the SN1CB mechanism is unlikely.
Compounds Derived from the Other Ligands
Because other polyaminocarboxylate ligands (2, 3, and 4) afforded isomers showing a pH dependence of the coordination mode reflective of that observed for isomers formed by 1 (Table 3), the products formed by each polyaminocarboxylate ligand were studied in less depth than those from 1. We shall discuss mainly the noteworthy results in solution for each set of compounds derived from these ligands.
Compounds Derived from the UEDDAH2 Ligand
UEDDAH2 (2, Chart 1) has two amine nitrogen and two carboxyl oxygen donors available for coordination. As observed for other ligands studied here, the UEDDAH2 ligand also formed a pair of complexes differing in coordination mode. The coordination modes were ONO or NNO, depending on the pH of the reaction mixture. Syntheses performed at low pH and moderate temperature (30 to 40 °C) led to the formation of one major complex, in the neutral zwitterionic form, [Re(CO)3(UEDDAH)-ONO] (7, RT = 8.8 min; ~96% yield by HPLC). The 1H NMR spectrum of 7 in D2O (pH 4.0) is very simple and confirmed the formation of only one isomer, having a dangling –CH2CH2NH3+ moiety. The two acetates give only one pair of methylene doublets (at 3.72 and 3.93 ppm, J = 16.8 Hz, each integrating for two protons) indicating a symmetrical coordination of the UEDDAH− ligand via the two carboxylates and the tertiary amine group. A very similar spectrum with J = 16.8 Hz for the acetate methylene doublets was obtained for the Re analogue of the promising [99mTc(CO)3(nitrilotriacetate)]2− renal imaging agent.3,8 As found for 5 (the other product made here with the –CH2CH2NH3+ moiety dangling from a tertiary N), the multiplet (3.72 ppm) for the protons of the CH2 group of 7 next to the protonated amine group is 0.31 ppm downfield from the multiplet (3.41 ppm) for the protons of the CH2 group next to the coordinated tertiary amine.
However, syntheses performed at pH ~7 and moderate temperature (30 to 40 °C) produced a different new complex, [Re(CO)3(UEDDA)-NNO]−, which formed almost exclusively (8, RT = 14.5 min, ~98%). Depending on the protonation state of the dangling uncoordinated carboxyl group, the complex has a negative charge (deprotonated carboxyl at and above physiological pH) or neutral (protonated carboxylate group at low pH). The latter form of 8 ([Re(CO)3(UEDDAH)-NNO]) was obtained as crystals (Figure 2).
The 1H NMR spectrum of 8 revealed the presence of four multiplets (each integrating for one proton) and two distinct sets of two sharp doublets, confirming the unsymmetrical NNO coordination of the UEDDA2− ligand and the asymmetry of 8. On the basis of previous studies in which the complexes contained ligands allowing unambiguous assignments of coordinated and dangling acetate, the CH2 signals,3,8 the doublets with the lower J (16.4 Hz, 4.21 and 3.96 ppm), are attributed to the C8H2 protons of the uncoordinated acetate group and the doublets with higher J (17.6 Hz, 4.18 and 4.02 ppm) are attributed to the C5H2 protons of the coordinated acetate group. Complex 8 persisted under all pH conditions investigated at room temperature, including at low pH, where X-ray quality crystals were grown, allowing a structural determination establishing the NNO coordination mode (Figure 2). We then investigated whether 8 would convert to 7 (with the ONO coordination mode) at low pH and elevated temperature. Heating at reflux a pH 2 aqueous solution of 8 converted ~25% of 8 to 7, giving the equilibrium ratio of 25 : 75 (7 : 8). As in other cases, immediately after the pH of this solution was adjusted to 9 at room temperature, the only product detected by HPLC was 8. Thus, 7 (with the ONO coordination mode) is a kinetic product formed at low pH and moderate temperature (25 - 40 °C). In contrast to the behavior of other pairs of complexes, in which both exist in comparable amounts under neutral conditions, 7 rearranges almost completely to 8 (NNO coordination mode), the thermodynamic product, under neutral conditions. Indeed, even at low pH, 8 is much more favored than 7 when samples of 8 are heated under conditions allowing a change in coordination mode.
Compounds Derived from the DTMH2 Ligand
Treatment of [Re(CO)3(H2O3)+ with DTMH2 (3, Chart 1) at pH ~7 gave a mixture of two products, [Re(CO)3(DTMH)-NNO] (9; RT = 3.5 min) and [Re(CO)3(DTM)-NNN]− (10; RT = 6.8 min), in an almost 45 : 55 ratio of 9 : 10. The molecular structure of the pseudo-octahedral neutral complex 9 reveals that the DTMH− ligand is coordinated via an NNO donor set (the two neutral secondary amines and one terminal carboxylate group) (Figure 3); two groups, –CO2− and –CH2CH2NH3+, are dangling. This neutral zwitterion form of 9, [Re(CO)3(DTMH)-NNO], crystallized from low pH (~3.5) aqueous solution. However, at pH ~3 only the cationic form of 9 ([Re(CO)3(DTMH2)-NNO]+) with a protonated carboxylic group) was observed. Likewise, only [Re(CO)3(DTM)-NNN]− (10) was observed for preparative reactions at pH ~10. Although it could be separated and characterized, all attempts to obtain crystals suitable for X-ray crystallography of this NNN product, 10, have been unsuccessful.
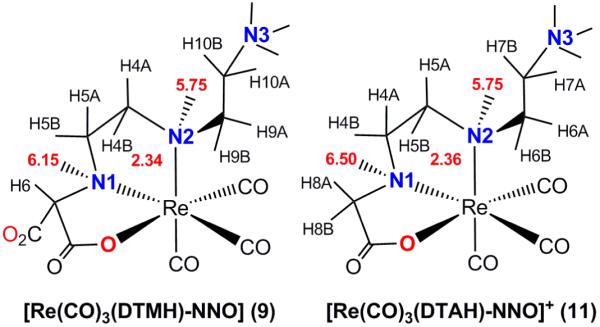
The 1H NMR spectrum of 9 in aqueous solution at pH 6 (Figure S2 in Supporting Information), in accord with its X-ray structure, consists of the two NH signals, (6.15 (s, 1H, N1H) and 5.75 ppm (m, 1H, N2H)), a singlet for C6H-CO2− (4.01 ppm; the H6 signal is a singlet because it has a ca. 90° torsion angle relationship to the N1H amine proton), and several multiplets with very similar chemical shifts, indicating that the signals of one ethylene bridge belong to a dangling arm. The NMR spectrum of 9 revealed a very characteristic upfield signal at 2.34 ppm assigned by COSY to H4B (Chart 2) on the basis of the H4B-C4-N2-H2 torsion angle value of −161.36° from the X-ray crystal structure (Figure 3). Such a ~2.3 ppm signal for a proton of the methylene group attached to the coordinated secondary amine (N2H) terminating the chelate ring has been observed in related Re(CO)3 complexes.3
Chart 2.
On the other hand, the eight clearly separated multiplets (each integrating for one proton) for the two ethylene bridges of 10 in H2O at pH 9 (cf. Figure S3 in Supporting Information) are consistent with facial coordination of all three amine groups. While no NH signal was observed for the dangling –CH2CH2NH3+ group in 9, as expected because the protons of an uncoordinated protonated primary amine group are undoubtedly in fast exchange with water, two signals (5.21 and 3.98 ppm) were present in H2O for the coordinated primary amine protons in 10, identified by the presence of a COSY cross-peak between these signals.
No changes were observed by NMR and HPLC for up to 10 days for solutions at room temperature of 9 at pH 3 to 5 or 10 at pH 9 to 12, results similar to those observed for solutions of the Re(CO)3(DTG) complexes, 5 and 6. In addition, the Re(CO)3(DTM) complexes slowly converted into each other at room temperature when the pH was changed from acidic to basic (9 converted to 10) and from basic to acidic (10 converted to 9), indicating that both 9 and 10 are the thermodynamically favored products at the pH conditions under which they were isolated (9 at pH 3.5 and 10 at pH 10).
Compounds Derived from the DTAH Ligand
After the high pH synthesis of 10, it was purified on a Sephadex G-15 column. The purification revealed a small amount of another new fac-[Re(CO)3L]n complex having an HPLC RT of 7.8 min (12). Analytical, spectroscopic, and X-ray crystallographic methods revealed that 12, neutral [Re(CO)3(DTA)-NNN], contains the fully deprotonated DTA ligand, facially coordinated through three amine donor groups (Figure 4). Under alkaline conditions, heating is well known to lead to decarboxylation of carboxylic acids with a β-carbonyl group, such as the malonic acid moiety of DTMH2. Because 10 was stable at high pH and no decarboxylation was observed, some decarboxylation of the DTMH2 ligand must have occurred during the synthesis of 10. The newly formed acetate group (–CH2CO2−) of the resulting DTAH ligand is dangling in 12 (Figure 4).
Complex 12 was kept in solution at pH ~2 at room temperature. Analysis by HPLC of the low pH solution revealed the presence of a second peak (RT = 3.6 min, [Re(CO)3(DTAH)-NNO]+ (11)) in addition to the peak of 12 (7.8 min); the peak for 11 grew very slowly and after 4 days the peaks were present in a 30 : 70 ratio (11 : 12). However, when a solution of 12 at pH ~ 2 was heated for 30 min, 12 converted almost completely (~98%) to 11. At this low pH, the dangling group of 12 is a carboxylic acid, and apparently the [Re(CO)3(DTAH)-NNN]+ cation does not easily convert to the NNO product with a dangling protonated ethylamine group at room temperature; however, the conversion is achieved at elevated temperature. About 1 h after adjusting the pH of the low pH solution to ~8 and at room temperature, the ratio changed to 85 : 15 (11 : 12), but after additional heating at ~ 100 °C for 30 min the ratio changed to 20 : 80 (11 : 12), and it did not change with time, even at elevated temperature; thus, 20 : 80 is the thermodynamic distribution of products at pH ~8. To verify these results, we synthesized the DTAH ligand in order to investigate whether this ligand could in fact form [Re(CO)3(DTAH)-NNO]+ (11) by reaction with fac-[Re(CO)3(H2O)3]+. Indeed, HPLC analysis of the reaction mixture of DTAH with the precursor at pH 6 to 7 and 70 °C for 2 h showed two peaks (RT = 3.6 min for 11 and RT = 7.8 min for 12) in a 42 : 58 ratio.
The 1H NMR spectrum of the more abundant product is identical with that of [Re(CO)3(DTA)-NNN] (12). The 1H NMR spectrum (at pH 3) of the less abundant product ([Re(CO)3(DTAH)-NNO]+, 11) has relatively downfield acetate signals (3.53ppm, d, J = 17.6 Hz; 3.91ppm, dd, J = 17.6, 7.6 Hz; see Experimental Section). This pattern is characteristic of the NNO coordination mode of facially coordinated five membered chelate rings such as we previously observed in Re(CO)3(ENDAC) and Re(CO)3(ENAC).3 We can assign the doublet of doublets at 3.91 ppm of 11 H8B (Chart 2) because the H1-N1-C8-H8B torsion angle is expected to be ~20-30° (a weak H1/H8B coupling).3 We assign the doublet at 3.53 ppm to H8A (Chart 2) for which a ca. 90° torsion angle relationship to the N1H proton is expected.
The pH 3 spectrum of 11 also exhibits an NH signal with a shift of 5.75 ppm (as we observed for 9) and the unusually upfield multiplet at 2.36 ppm, also as we observed for 9. The multiplet is assigned to H5B (Chart 2) on the chelate ring carbon attached to the coordinated secondary amine terminating the ring on the basis of the assignment for 9. This similarity indicates that the both 11 and 9 have the same configuration at the coordinated secondary amine group. Note that the X-ray structure of 9 (Figure 3) establishes that this is the configuration at N2 having an endo-NH proton. The somewhat downfield 5.75 ppm shift is consistent with our past studies of related complexes with an endo-NH proton in the coordinated secondary amine terminating a chelate ring.3,75 The other NH signal of 11 has a more downfield chemical shift, 6.50 ppm, consistent with an NH signal for a proton on the central N1.
In addition to the other similarities, the signals for the dangling CH2CH2NH3+ group have similar shifts in 11 and 9. Thus the proposed similarity in the structures for 11 and 9 (as shown in Scheme 1) with an NNO coordination mode and a dangling –CH2CH2NH3+ attached to a secondary amine N terminating one chelate ring is well supported by the NMR data. It is interesting that the exo dangling group attached to a secondary N of 11 and 9 lacks the somewhat downfield multiplet characteristic of this dangling group in 5 and 7, complexes for which the dangling group is attached to a tertiary N and is therefore necessarily endo. Further research on other compounds is needed before one can determine if the differences in shift are related to the type of nitrogen to which the dangling group is attached or to the endo vs exo disposition of the dangling group.
The more downfield NH signal for 12 at 6.55 ppm can be assigned to the N2H (the central coordinated amine) and the more upfield NH shift at 5.17 ppm can be attributed to N1H (an exo-NH proton, Figure 4); this assignment is consistent with our past studies of related complexes with amines terminating a chelate ring.3,75 The C8H2 signals for the dangling acetate have similar shifts (3.79 and 3.74 ppm) giving an AB spin system. This has an octuplet pattern arising from geminal coupling 3J = 16.8 Hz combined with the vicinal coupling (~7 Hz) to the NH. Similar vicinal coupling of ~ 7 Hz has been observed previously and is attributable to the expected rapid free rotation of the uncoordinated acetate group around the N-C bond.3
Effect of Base on the Ease of Interconversion of Coordination Mode
When D2O solutions of the various products were made basic (above pH 8), the CH signals sharpened and exhibited simpler coupling patterns. The NH signals diminished in size and eventually disappeared. However, the shifts of the CH signals did not change with this increase in basicity, even at very high pH > 10.The relatively rapid NH/ND exchange indicates some NH deprotonation, because this process requires coordinated amine deprotonation.21,23,76 However, the absence of the shifting of the CH signals indicates deprotonation is not favorable enough to produce detectable amounts of NH-deprotonated complexes. This behavior is in contrast to our findings for ReVO analogues of 99mTcVO radiopharmaceuticals, for which detectable amounts of NH-deprotonated forms are present even under physiological conditions.15,21,23,71-74
As mentioned, the rapid NH/ND exchange under basic conditions means that a minor amount of a species with a deprotonated coordinated N must exist. However, we can rule out this species as an intermediate during a change in coordination mode. 7 does not have a coordinated amine bearing an NH group. However, both the 7 to 8 and the 5 to 6 change in coordination mode occur readily in basic solution; the likely reason for the facile process is that the dangling amine becomes deprotonated and undergoes intramolecular exchange with a coordinated carboxyl.
Conclusions
On the basis of the observations in this study, where chelate ligands forming five-membered chelate rings were compared, we are able to provide some guiding principles for radiopharmaceutical design. At elevated temperature, the rate of formation and equilibration of the ReI complexes is sufficiently rapid that equilibrium is reached in an hour or less. Because TcI complexes are expected to equilibrate more rapidly than ReI complexes, most high-temperature preparations of TcI tracers should be at equilibrium. Experience suggests that the TcI products persist at room temperature, and thus it is desirable to form the tracer under conditions in which one or primarily one product is present. In low-pH solutions, the product with one carboxylate oxygen bound to the fac-{ReI(CO)3}+ core, leaving one potential amine donor as a dangling protonated group, is often both kinetically and thermodynamically favored. A greater affinity of the fac-{ReI(CO)3}+ core for the amine group relative to the carboxylate donor in high pH aqueous solution is established by the isolation of Re tricarbonyl complexes in which the polyaminocarboxylate ligand binds through the amine nitrogen donors in preference to carboxylate oxygen donors. Thus, for renal tracers, labeling can be done at elevated T at low pH, and the sample can then be converted to the N coordination mode at room temperature by raising the pH. Basic conditions facilitate the change in coordination mode to the species with the dangling carboxyl group whether or not the coordinated amines in the tracer formed at low pH have a dissociable proton. Just before injection, the pH of all 99mTc tracer doses is always adjusted to 7.4.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R37 DK038842). The authors thank Dr. Kenneth Hardcastle of Emory University for useful discussions and for determining the structures by using instruments supported by NIH Grant No. S10-RR13673 and NSF Grant No. CHE 9974864. We also thank Theshini Perera (Louisiana State University) for help in preparing the figures.
References
- 1.Richards P. In: Technetium in Chemistry and Nuclear Medicine 3. Nicolini M, Bandoli G, Mazzi U, editors. Cortina International; Verona, Italy: 1990. pp. 5–9. [Google Scholar]
- 2.Bandoli G, Dolmella A, Porchia M, Refosco F, Tisato F. Coord. Chem. Rev. 2001;214:43–90. [Google Scholar]
- 3.Lipowska M, Cini R, Tamasi G, Xu X, Taylor AT, Marzilli LG. Inorg. Chem. 2004;43:7774–7783. doi: 10.1021/ic049544i. [DOI] [PubMed] [Google Scholar]
- 4.He H, Lipowska M, Xu X, Taylor AT, Carlone M, Marzilli LG. Inorg. Chem. 2005;44:5437–5446. doi: 10.1021/ic0501869. [DOI] [PubMed] [Google Scholar]
- 5.Lipowska M, He H, Malveaux E, Xu X, Marzilli LG, Taylor A. J. Nucl. Med. 2006;47:1032–1040. [PMC free article] [PubMed] [Google Scholar]
- 6.He H, Lipowska M, Xu X, Taylor AT, Marzilli LG. Inorg. Chem. 2007;46:3385–3394. doi: 10.1021/ic0619299. [DOI] [PubMed] [Google Scholar]
- 7.He H, Lipowska M, Christoforou AM, Marzilli LG, Taylor AT. Nucl. Med. Biol. 2007;34:709–716. doi: 10.1016/j.nucmedbio.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lipowska M, Marzilli LG, Taylor AT. J. Nucl. Med. 2009;50:454–460. doi: 10.2967/jnumed.108.058768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nosco DL, Beaty-Nosco JA. Coord. Chem. Rev. 1999;184:91–123. [Google Scholar]
- 10.Trejtnar F, Laznicek M. Q. J. Nucl. Med. 2002;46:181–194. [PubMed] [Google Scholar]
- 11.Shikano N, Kanai Y, Kawai K, Ishikawa N, Endou H. J. Nucl. Med. 2004;45:80–85. [PubMed] [Google Scholar]
- 12.Hansen L, Marzilli LG, Eshima D, Malveaux E, Folks R, Taylor A. J. Nucl. Med. 1994;35:1198–1205. [PubMed] [Google Scholar]
- 13.Taylor A, Hansen L, Eshima D, Malveaux E, Folks R, Shattuck L, Lipowska M, Marzilli LG. J. Nucl. Med. 1997;38:821–826. [PubMed] [Google Scholar]
- 14.Taylor A, Lipowska M, Hansen L, Malveaux E, Marzilli LG. J. Nucl. Med. 2004;45:885–891. [PubMed] [Google Scholar]
- 15.Marzilli LG, Banaszczyk MG, Hansen L, Kuklenyik Z, Cini R, Taylor A., Jr. Inorg. Chem. 1994;33:4850–4860. [Google Scholar]
- 16.Edwards DS, Cheesman EH, Watson MW, Maheu LJ, Nguyen SA, Dimitre L, Nason T, Watson AD, Walovitch R. In: Technetium and Rhenium in Chemistry and Nuclear Medicine 3. Nicolini M, Bandoli G, Mazzi U, editors. Cortina International; Verona. Italy: 1990. pp. 433–444. [Google Scholar]
- 17.Grummon G, Rajagopalan R, Palenik GJ, Koziol AE, Nosco DL. Inorg. Chem. 1995;34:1764–1772. [Google Scholar]
- 18.Johannsen B, Spies H. Top. Curr. Chem. 1996;176:77–121. [Google Scholar]
- 19.Rajagopalan R, Grummon GD, Bugaj J, Hallemann LS, Webb EG, Marmion ME, Vanderheyden J-L, Srinivasan A. Bioconjugate Chem. 1997;8:407–415. doi: 10.1021/bc9700401. [DOI] [PubMed] [Google Scholar]
- 20.Chatterjee M, Achari B, Das S, Banerjee R, Chakrabarti C, Dattagupta JK, Banerjee S. Inorg. Chem. 1998;37:5424–5430. doi: 10.1021/ic970577q. [DOI] [PubMed] [Google Scholar]
- 21.Hansen L, Lipowska M, Melendez E, Xu X, Hirota S, Taylor A, Marzilli LG. Inorg. Chem. 1999;38:5351–5358. [Google Scholar]
- 22.Hansen L, Xu X, Lipowska M, Taylor A, Marzilli LG. Inorg. Chem. 1999;38:2890–2897. doi: 10.1021/ic981151u. [DOI] [PubMed] [Google Scholar]
- 23.Hansen L, Hirota S, Xu X, Taylor A, Marzilli LG. Inorg. Chem. 2000;39:5731–5740. doi: 10.1021/ic000183q. [DOI] [PubMed] [Google Scholar]
- 24.Lipowska M, He H, Malveaux E, Taylor AT, Marzilli LG. In: Technetium, Rhenium and Other Metals in Chemistry and Nuclear Medicine 7. Mazzi U, editor. Servizi Grafici Editoriali snc; Padova, Italy: 2006. pp. 115–116. [Google Scholar]
- 25.Eshima D, Taylor A. Semin. Nucl. Med. 1992;22:61–73. doi: 10.1016/s0001-2998(05)80082-0. [DOI] [PubMed] [Google Scholar]
- 26.Van Nerom CG, Bormans GM, De Roo MJ, Verbruggen A. Eur. J. Inorg. Chem. 1993;20:738–746. doi: 10.1007/BF00180902. [DOI] [PubMed] [Google Scholar]
- 27.Bormans GM, Cleynhens BJ, De Roo MJK, Verbruggen AM. Eur. J. Inorg. Chem. 1992;19:271–277. doi: 10.1007/BF00175141. [DOI] [PubMed] [Google Scholar]
- 28.Gianolli L, Dosio F, Matarrese M, Colombo F, Cutler C, Stepnial-Biniakiewicz D, Deutsch E, Savi A, Lucignani G, Fazio F. Nucl. Med. Biol. 1996;23:927–933. doi: 10.1016/s0969-8051(96)00135-7. [DOI] [PubMed] [Google Scholar]
- 29.Vanbilloen HP, Dezutter NA, Cleynhens BJ, Verbruggen AM. Eur. J. Inorg. Chem. 1997;24:1374–1379. doi: 10.1007/s002590050163. [DOI] [PubMed] [Google Scholar]
- 30.Schibli R, Schubiger AP. Eur. J. Nucl. Med. 2002;29:1529–1542. doi: 10.1007/s00259-002-0900-8. [DOI] [PubMed] [Google Scholar]
- 31.Alberto R. Top. Curr. Chem. 2005;252:1–44. [Google Scholar]
- 32.Alberto R. J. Organomet. Chem. 2007;692:1179–1186. [Google Scholar]
- 33.Egli A, Alberto R, Tannahill L, Schibli R, Abram U, Schaffland A, Waibel R, Tourwe D, Jeannin L, Iterbeke K, Schubiger PA. J. Nucl. Med. 1999;40:1913–1917. [PubMed] [Google Scholar]
- 34.Waibel R, Alberto R, Willuda J, Finnern R, Schibli R, Stichelberger A, Egli A, Abram U, Mach J-P, Plückthun A, Schubiger AP. Nat. Biotechnol. 1999;17:897–901. doi: 10.1038/12890. [DOI] [PubMed] [Google Scholar]
- 35.von Guggenberg E, Behe M, Behr TM, Saurer M, Seppi T, Decristoforo C. Bioconjugate Chem. 2004;15:864–871. doi: 10.1021/bc0300807. [DOI] [PubMed] [Google Scholar]
- 36.Ramesh C, Bryant B, Nayak T, Revankar CM, Anderson T, Carlson KE, Katzenellenbogen JA, Sklar LA, Norenberg JP, Prossnitz ER, Arterburn JB. J. Am. Chem. Soc. 2006;128:14476–14477. doi: 10.1021/ja066360p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alves S, Correia JDG, Gano L, Rold TL, Prasanphanich A, Haubner R, Rupprich M, Alberto R, Decristoforo C, Santos I, Smith CJ. Bioconjugate Chem. 2007;18:530–537. doi: 10.1021/bc060234t. [DOI] [PubMed] [Google Scholar]
- 38.Causey PW, Besanger TR, Schaffer P, Valliant JF. Inorg. Chem. 2008;47:8213. doi: 10.1021/ic800775w. [DOI] [PubMed] [Google Scholar]
- 39.Alberto R, Schibli R, Egli A, Schubiger AP, Abram U, Kaden TA. J. Am. Chem. Soc. 1998;120:7987–7988. doi: 10.1021/ic980112f. [DOI] [PubMed] [Google Scholar]
- 40.Alberto R, Ortner K, Wheatley N, Schibli R, Schubiger AP. J. Am. Chem. Soc. 2001;123:3135–3136. doi: 10.1021/ja003932b. [DOI] [PubMed] [Google Scholar]
- 41.Alberto R, Schibli R, Waibel R, Abram U, Schubiger AP. Coord. Chem. Rev. 1999;190-192:901–919. [Google Scholar]
- 42.Schibli R, La Bella R, Alberto R, Garcia-Garayoa E, Ortner K, Abram U, Schubiger PA. Bioconjugate Chem. 2000;11:345–351. doi: 10.1021/bc990127h. [DOI] [PubMed] [Google Scholar]
- 43.Banerjee SR, Levadala MK, Lazarova N, Wei L, Valliant JF, Stephenson KA, Babich JW, Maresca KP, Zubieta J. Inorg. Chem. 2002;41:6417–6425. doi: 10.1021/ic020476e. [DOI] [PubMed] [Google Scholar]
- 44.Pietzsch H-J, Gupta A, Reisgys M, Drews A, Seifert S, Syhre R, Spies H, Alberto R, Abram U, Schubiger AP, Johannsen B. Bioconjugate Chem. 2000;11:414–424. doi: 10.1021/bc990162o. [DOI] [PubMed] [Google Scholar]
- 45.Kramer DJ, Davison A, Davis WM, Jones AG. Inorg. Chem. 2002;41:6181–6183. doi: 10.1021/ic025887+. [DOI] [PubMed] [Google Scholar]
- 46.Riondato M, Camporese D, Martín D, Suades J, Alvarez-Larena A, Mazzi U. Eur. J. Inorg. Chem. 2005;20:4048–4055. [Google Scholar]
- 47.Lazarova N, Babich JW, Valliant J, Schaffer P, James S, Zubieta J. Inorg. Chem. 2005;44:6763–6770. doi: 10.1021/ic050735a. [DOI] [PubMed] [Google Scholar]
- 48.Schibli R, Katti KV, Volkert WA, Barnes CL. Inorg. Chem. 2001;40:2358–2362. doi: 10.1021/ic001284r. [DOI] [PubMed] [Google Scholar]
- 49.Mundwiler S, Waibel R, Spingler B, Kunze S, Alberto R. Nucl. Med. Biol. 2005;32:473–484. doi: 10.1016/j.nucmedbio.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 50.Adams KM, Marzilli LG. Inorg. Chem. 2007;46:4926–4936. doi: 10.1021/ic062410f. [DOI] [PubMed] [Google Scholar]
- 51.Adams KM, Marzilli PA, Marzilli LG. Inorg. Chem. 2007;46:9172–9181. doi: 10.1021/ic701038f. [DOI] [PubMed] [Google Scholar]
- 52.Alberto R, Pak JK, van Staveren D, Mundwiler S, Benny P. Biopolymers. 2004;76:324–333. doi: 10.1002/bip.20129. [DOI] [PubMed] [Google Scholar]
- 53.Taylor A, Lipowska M, Marzilli LG. J. Nucl. Med. 2010 doi: 10.2967/jnumed.109.070813. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stephenson KA, Zubieta J, Banerjee SR, Lavadala MK, Taggart L, Ryan L, McFarlane N, Boreham DR, Maresca KP, Babich JW, Valliant JF. Bioconjugate Chem. 2004;15:128–136. doi: 10.1021/bc034128s. [DOI] [PubMed] [Google Scholar]
- 55.Liu Y, Pak JK, Schmutz P, Bauwens M, Mertens J, Knight H, Alberto R. J. Am. Chem. Soc. 2006;128:15996–15997. doi: 10.1021/ja066002m. [DOI] [PubMed] [Google Scholar]
- 56.Rattat D, Eraets K, Cleynhens B, Knight H, Fonge H, Verbruggen A. Tetrahedron Lett. 2004;45:2531–2534. [Google Scholar]
- 57.Wei L, Babich JW, Zubieta J. Inorg. Chim. Acta. 2005;358:3691–3700. [Google Scholar]
- 58.Lipowska M, Hansen L, Marzilli LG, Taylor A. J. Nucl. Med. 2001;42:259P. [PubMed] [Google Scholar]
- 59.Lipowska M, He H, Xu X, Marzilli LG, Taylor A. J. Label. Compd. Radiopharm. 2005;48:S258. [Google Scholar]
- 60.McLendon G, MacMillan DT, Hariharan M, Martell AE. Inorg. Chem. 1975;14:2322–2326. [Google Scholar]
- 61.McLendon G, Motekaitis RJ, Martell AE. Inorg. Chem. 1975;14:1993–1996. [Google Scholar]
- 62.Forsterling H-D, Stuk L, Barr A, McCormick WD. J. Phys. Chem. 1993;97:2623–2621. [Google Scholar]
- 63.Bruker APEX2. Bruker AXS, Inc.; Madison Wisconsin, USA: 2007. [Google Scholar]
- 64.Bruker SAINT. Bruker AXS, Inc.; Madison Wisconsin, USA: 2007. [Google Scholar]
- 65.Bruker SADABS. Bruker AXS, Inc.; Madison Wisconsin, USA: 2008. [Google Scholar]
- 66.Sheldrick GM. Acta Crystallogr. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 67.Wilson AJC. International Tables for X-ray Crystallography. C. Kynoch, Academic Publishers; Dordrecht: 1992. pp. 500–502.pp. 219–222. Tables 6.1.1.4. and 4.2.6.8. [Google Scholar]
- 68.Mundwiler S, Candreia L, Hafliger P, Ortner K, Alberto R. Bioconjugate Chem. 2004;15:195–202. doi: 10.1021/bc034171f. [DOI] [PubMed] [Google Scholar]
- 69.Allali M, Cousinie S, Gressier M, Tessier C, Beauchamp AL, Coulais Y, Dartiguenave M, Benoist E. Inorg. Chim. Acta. 2006;359:2128–2134. [Google Scholar]
- 70.Wang W, Hor TSA, Yan YK. Inorg. Chim. Acta. 2006;359:3754–3762. [Google Scholar]
- 71.Hansen L, Xu X, Yue KT, Taylor A, Marzilli LG. Inorg. Chem. 1996;35:2785–2791. [Google Scholar]
- 72.Hansen L, Yue KT, Xu X, Lipowska M, Taylor A, Jr., Marzilli LG. J. Am. Chem. Soc. 1997;38:8965–8972. [Google Scholar]
- 73.Lipowska M, Hansen L, Cini R, Xu X, Choi H, Taylor AT, Marzilli LG. Inorg. Chim. Acta. 2002;339:327–340. [Google Scholar]
- 74.Lipowska M, Hansen L, Xu X, Marzilli PA, Taylor A, Marzilli LG. Inorg. Chem. 2002;41:3032–3041. doi: 10.1021/ic011325z. [DOI] [PubMed] [Google Scholar]
- 75.Christoforou AM, Marzilli PA, Fronczek FR, Marzilli LG. Inorg. Chem. 2007;46:11173–11182. doi: 10.1021/ic701576u. [DOI] [PubMed] [Google Scholar]
- 76.Hansen L, Xu X, Yue KT, Kuklenyik Z, Taylor A, Marzilli LG. Inorg. Chem. 1996;35:1958–1966. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.