

Abstract
Anthrax lethal toxin (LT) contributes to the immune evasion strategy of B. anthracis by impairing the function of cells of the immune system, such as macrophages and dendritic cells (DCs). Macrophages from certain inbred mice strains undergo rapid death upon LT treatment mediated by caspase-1 activation dependent on Nalp1b, an inflammasome component. Rapid LT-induced death is however not observed in macrophages from human and many mouse strains. Here, we focused on the responses of various murine DCs to LT. Using a variety of knock-out mice, we found that depending on the mouse strain, death of bone marrow derived DCs and macrophages was mediated either by a fast Nalp1b and caspase-1 dependent, or by a slow caspase-1 independent pathway that was triggered by the impairment of MEK1/2 pathways. Caspase-1 independent death was observed in cells of different genetic backgrounds and interestingly occurred only in immature DCs. Maturation, triggered by different types of stimuli, led to full protection of DCs. These studies illustrate that the cellular damage inflicted by LT depends not only on the innate responses but also on the maturation stage of the cell, which modulates the more general caspase-1 independent responses.
INTRODUCTION
Many pathogenic bacteria have developed mechanisms to overcome host immune defenses. In the case of Bacillus anthracis, the causative agent of anthrax, immune evasion is mediated by various virulence factors, particularly a bipartite protein toxin called Lethal Toxin (LT) (Baldari et al., 2006). LT is composed of two subunits: the Protective Antigen (PA; 83kDa) and the Lethal Factor (LF; 90kDa). PA mediates binding to target cells via ubiquitous receptors (Bradley et al., 2001; Scobie et al., 2003), and is also responsible for the delivery of LF to the cytoplasm (Abrami et al., 2005). LF is a metalloprotease that targets all Mitogen-Activated Protein Kinase Kinases (MAPKKs) with the exception of MEK5 (Duesbery et al., 1998; Vitale et al., 1998; Vitale et al., 2000), thereby impairing MAPK signalling in intoxicated cells. MAP kinases (ERK, p38 and JNK families) are involved in diverse functions including cell survival, differentiation, proliferation and stress responses (Roux and Blenis, 2004); therefore, the consequences of their cleavage by LF will depend on the cell type.
The general view is that the role of Lethal toxin (LT, combination of PA+LF) during anthrax infection is to cripple, or even fully dismantle, the immune system of the host, thus permitting an unrestrained spread of bacteria and the progression of the disease (Moayeri and Leppla, 2004; Baldari et al., 2006). This view is based on the observations that LT impairs the function of neutrophiles and monocytes (During et al., 2005; Kassam et al., 2005), T cells (Comer et al., 2005; Fang et al., 2005; Paccani et al., 2005), B cells (Fang et al., 2006), dendritic cells (Agrawal et al., 2003; Alileche et al., 2005; Brittingham et al., 2005; Tournier et al., 2005; Cleret et al., 2006) and macrophages (Pellizzari et al., 1999; Erwin et al., 2001).
The most dramatic effect is observed on macrophages from certain strains of inbred mice, which rapidly (< 4 h) lyse upon exposure to LT (Friedlander, 1986; Friedlander et al., 1993). The underlying mechanism, which is not fully understood, was recently found to involve Nalp1b dependent activation of caspase-1 (Boyden and Dietrich, 2006). The gene encoding for Nalp1b is highly polymorphic, with 5 alleles identified among 18 mice strains (Boyden and Dietrich, 2006). Thus, LT-induced Nalp1b dependent activation of caspase-1 can be observed only in macrophages of certain strains, correlating with a higher sensitivity to the toxin (Boyden and Dietrich, 2006). Macrophages from many mice strains, as well as from humans, however show a far lower sensitivity. How death is triggered in these cells is unknown.
Nalp1b is a member of the NOD-like receptor family (NLR), a family of cytoplasmic proteins involved in the recognition of microbial products or danger signals (Creagh and O’Neill, 2006; Meylan et al., 2006). Upon binding of specific ligands, certain NLRs, such as Nalp1 and Nalp3, were shown to assemble into multimeric protein complexes called inflammasomes (Martinon and Tschopp, 2004), which recruit procaspase-1 allowing its autoproteolytic activation. Active caspase-1 subsequently mediates the processing of the important inflammatory mediators interleukin (IL) 1β, IL-18 and IL-33 (Martinon and Tschopp, 2004; Schmitz et al., 2005). The existence of other substrates for caspase-1 has been proposed based in particular on the observation that caspase-1 mediates macrophage cell death upon infection with Salmonella typhimurium (Mariathasan et al., 2006) or Shigella flexneri (Hilbi et al., 1998) and promotes cell survival upon membrane damage by pore forming toxins (Gurcel et al., 2006).
The observation that rapid macrophage death upon LT treatment is dependent on the Nalp1b/caspase-1 axis suggests that cell death is a consequence of the innate immune response of the cells rather than the direct action of the toxin. Our interest was to further evaluate the role of cellular innate responses to LT focusing on dendritic cells (DCs), the most efficient antigen-presenting cells of the immune system. DCs play a crucial role both in initiating and regulating the adaptive immune response (Gatti and Pierre, 2003; Steinman et al., 2003). In their immature form, DCs patrol the peripheral tissues where they efficiently take up antigens, which they will present to naïve T cells in lymphoid organs upon maturation (Trombetta and Mellman, 2005). Treatment of DCs with LT was found to prevent the secretion of cytokines in response to microbial products (Agrawal et al., 2003; Brittingham et al., 2005; Tournier et al., 2005; Cleret et al., 2006). With the exception of one study that reports strain dependent LT sensitivity as for macrophages (Alileche et al., 2005), LT is not thought to affect DC viability.
Our interest was to get a better understanding of the cellular mechanisms modulating the LT sensitivity of DCs and in particular investigate the role of Nod-like receptors and interacting proteins. For this we have made use of different strains of mice carrying different alleles of Nalp1b, as well as single knock out mice deficient in caspase-1 and other inflammasome components. The studies were performed on bone marrow derived DCs in which the maturation state was tightly controlled and revealed that DCs sensitivity is not only dependent on the genetic background, but also on the maturation stage of the cells.
RESULTS
Lethal Toxin triggers death of immature DCs
Cell death in response to anthrax lethal toxin (LT) has been extensively studied on murine macrophages and its dependence on genetic factors is well established (Friedlander et al., 1993; Boyden and Dietrich, 2006). The susceptibility of DCs, however, still remains controversial. Besides one study reporting strain dependent susceptibility of murine DCs (Alileche et al., 2005), other studies reported functional impairment without loss of viability in response to LT treatment of human and murine DCs (Agrawal et al., 2003; Brittingham et al., 2005; Tournier et al., 2005). We wanted to clarify this issue by using DCs obtained from C57BL/6J (B6) mice, whos macrophages and DCs are thought to have low sensitivity to LT induced death, similarly to human cells (Alileche et al., 2005; Brittingham et al., 2005). DCs were prepared from bone marrow and kept in their immature state, monitored by the absence of the co-stimulatory molecule and maturation marker CD86 at the cell surface. Treatment for 24 h with lethal toxin (LT, composed of PA+LF) led to an increase in the population of 7AAD and Annexin V stained cells, markers of dead and apoptotic cells respectively (Fig. 1A). This increase in cell death required the delivery of enzymatically active LF, since the catalytically inactive LF mutant, E687C, had no effect, nor did a furin resistant PA mutant, PASNKE, which is unable to escort LF into cells (Gordon et al., 1995; Abrami et al., 2006)(Fig. 1B, upper row). The preparations of toxins were controlled by adding PA and LF alone, which, as expected, had not effect on cell viability.
Figure 1. Lethal toxin kills immature DCs.
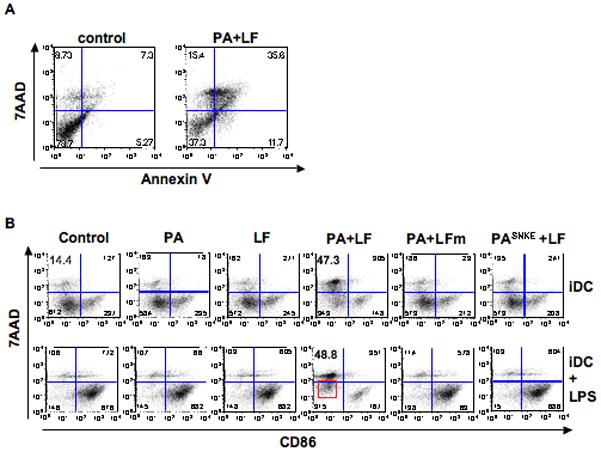
A) B6 immature DCs were treated for 24 h with 500ng/ml PA and 20ng/ml LF, or left untreated (control). Dead and apoptotic cells were detected by FACS after staining with Annexin V-FITC and 7AAD and represented for the gated CD11c+ population. Numbers in the corners of the gates represent the % of cells within the CD11c+ population. Dead cells are 7AAD+/Annexin V+. B) DCs obtained from B6 mice were incubated with different combinations of anthrax toxins and their mutant forms during 24 h. PA (wild type or mutant) was added at 500ng/ml, and LF (wild type or mutant) was added at 20ng/ml. LPS (100ng/ml) was added 8 h before the end of the incubation to the indicated cells (iDC+LPS). Cells were collected and analyzed by flow cytometry after staining with anti CD86-PE and 7AAD. Here is represented the double staining on gated CD11C+ cells treated with medium alone (control), PA alone, LF alone, PA+LF wild type, PA+LFm (inactive mutant E687C) and PASNKE+LF (a furin resistant mutant form of PA). Numbers in each gate correspond to the % of cells within the CD11c+ population. Note the higher 7AAD staining on PA+LF treated cells subsequently treated with LPS (red square) (See Suppl. Fig. 3B for statistical significance). Data from one representative experiment out of 3 is shown.
Although significant cell death was observed when immature DCs were treated with the fully active toxin, some 50% of the cells remained viable (7AAD low) after 24 h. Interestingly these cells did not express maturation markers at their surface (Suppl. Fig. 1A), indicating that PA and LF did not induce maturation by themselves. We subsequently monitored whether LT-treated immature DCs would undergo maturation upon LPS treatment, with the aim of testing the functionality of this population of surviving cells. When LPS was added to immature DCs previously treated for 16 h with LT, it led only to a minor increase in surface expression of the co-stimulatory molecule CD86 when compared to control cells (Fig. 1B lower panels and Suppl. Fig. 1B). Also LPS-triggered cytokine secretion (IL-1α, IL-1β, IL-6, IL-12, MIP-1b, TNF-α) was very low for LT-treated DCs (Suppl. Fig. 2), in agreement with previous observations (Agrawal et al., 2003; Brittingham et al., 2005; Tournier et al., 2005; Cleret et al., 2006). The limited increase in surface expression of maturation markers and cytokine secretion could be interpreted as an inhibitory effect of LT on DC maturation. However, although 50% of LT-treated cells were not maximally 7AAD-positive, they did exhibit a significantly increased 7AAD mean fluorescence staining after LPS treatment (Fig. 1B boxed cells and Suppl. Fig. 3B), indicative of a pre or early apoptotic stage (Philpott et al., 1996) as confirmed by an also significantly higher fluorescence for Annexin V staining (Suppl. Fig. 3A boxed cells and Suppl. Fig. 3B). Thus, LPS stimulation on LT-treated cells appears to act synergistically with the toxin in inducing DC death, as observed for macrophages (Park et al., 2002).
Together, these observations show that anthrax lethal toxin did not trigger DC maturation in DCs from B6 mice, but induced cell death of some 50% of immature DCs and caused the entire DC population to lose its ability to respond to LPS likely due to the initiation of cell death pathways.
Maturation protects DCs from anthrax lethal toxin killing
The synergistic effect of LT and LPS on immature DCs suggested an interplay between LT induced death and DC maturation pathways. This prompted us to study the effects of LT on B6 DCs that had undergone maturation by a prior 8 h LPS treatment. Remarkably, mature DCs, expressing CD86 at their surface, remained 7AAD (Fig. 2A) and Annexin V (not shown) negative after exposure to the fully active LT. This observation suggested that maturation induced a protective state. To confirm this, 7AAD staining was monitored as a function of LT treatment time on both immature and mature DCs. As shown in Fig. 2B, a slow, time dependent, increase in 7AAD positive cells was observed for immature DCs, an effect that was increased by the subsequent addition of LPS (Fig. 2C). In contrast, LT had no effect on mature DCs (Fig. 2D). Death of mature DCs was observed at long times (48 h) even under control conditions, provably due to the absence of survival signals provided by T cells (Hou and Van Parijs, 2004). The resistance of mature DCs to the toxin was not due to the low doses of LF used in these experiments since, even at concentrations of LF above 1 μg/ml (10 nM), the protective effect was maintained (Fig. 2E).
Figure 2. Mature DCs are protected from LT-killing.
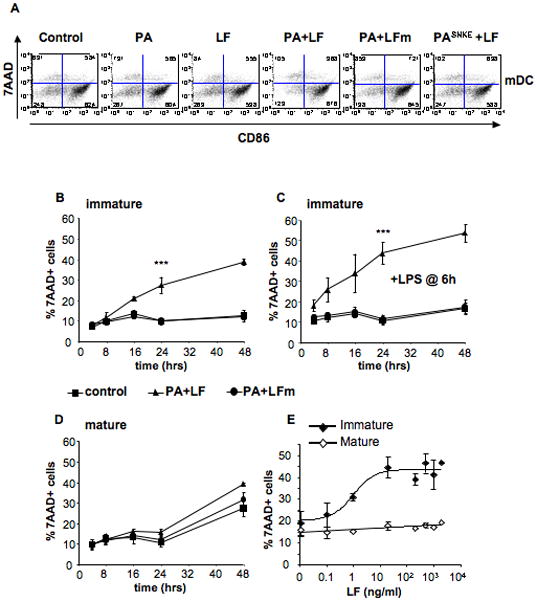
A) B6 derived DCs were treated during 8 h with 100ng/ml LPS to induce maturation, and subsequently treated with combinations of anthrax toxin and their mutant forms as in Fig. 1B). 7AAD and CD86 staining is represented after 24 h of toxin treatment on the CD11c+ population. Data from one representative experiment (n=3). B,C) DCs obtained from B6 mice were incubated with media, PA+LF or PA+LFmutant (500ng/ml of PA and 20ng/ml of LF or LF E687C mutant) during 4, 8, 16, 24 and 48 h, in the absence (B) or presence (C) of 100 ng/ml LPS during the last 6 h of incubation (or added 30min after the toxins for the 4 h time point). The percentage of dead cells (7AAD positive) on the CD11c+ population at each time point is represented. ***: Treatment with PA+LF during 24 h leads to a significant increase in the % of 7AAD+ cells compared with control cells (p<0.0001 obtained with data from 9 independent experiments done in duplicates or triplicates). D) DCs obtained from B6 mice were incubated with 100ng/ml LPS to induce maturation of the cells. After 8 h, toxins were added as in B–C) and the percentage of 7AAD positive cells on the CD11c+ population after the indicated times of incubation with toxin is represented. No significant difference in the % of 7AAD+ cells is observed between PA+LF and control cells at 24 h (p=0.9 obtained with data from 9 independent experiments done in duplicates or triplicates). E) DCs from B6 mice were matured by 8 h LPS treatment (mature) or left immature and subsequently treated for 24 h with different concentrations of LF (0.1, 1, 20, 200, 500, 1000 and 2000 ng/ml) and PA (500ng/ml or 2000ng/ml for the two higher concentrations of LF). The percentage of 7AAD+ cells on the CD11c+ gated population is represented as mean±SD. Data from 3 independent experiments done in triplicates.
Existence of Nalp1b/caspase-1 dependent and independent LT-induced death pathways
The above-described experiments were all performed on DCs obtained from B6 mice, which carry allele 2 of Nalp1b (Boyden and Dietrich, 2006). Allele 2 (and allele 4) encodes a protein that is not able to activate caspase-1 in response to LT (here termed type 2 Nalp1b), in contrast to the protein encoded by allele 1 (type 1 Nalp1b) (Boyden and Dietrich, 2006). To investigate the importance of this genetic difference, we extended our studies to bone marrow derived DCs from 129S1/svImJ (129S) mice that carry type 1 Nalp1b (Boyden and Dietrich, 2006). Immature DCs obtained from 129S mice were significantly more susceptible than B6 derived DCs and died with a fast kinetics in response to LT treatment (Fig. 3A). Moreover, maturation did not confer sustained protection from LT-induced death to these cells (Fig. 3B, C) in contrast to what we observed on B6 derived DCs (Fig. 2) and suggesting a different mechanism of death.
Figure 3. DC susceptibility to LT depends on genetic factors.
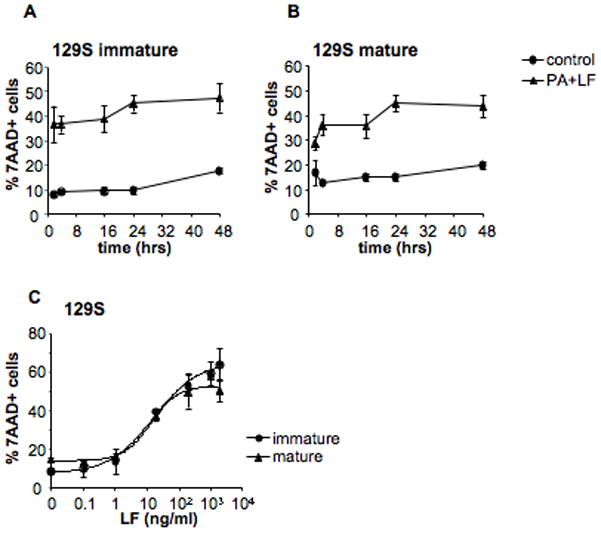
A) DCs were obtained from 129S mice (type 1 Nalp1b) and were incubated with media or PA+LF (500ng/ml of PA and 20ng/ml of LF) during 2, 4, 16, 24 and 48 h. B) DCs obtained from 129S mice were incubated with 100ng/ml LPS to induce maturation of the cells. After 8 h, toxins were added as in A). The percentage of dead cells (7AAD positive) on the CD11c+ population at each time point is represented. C) DCs from 129S mice were matured by 8 h LPS treatment (mature) or left immature and subsequently treated for 24 h with different concentrations of LF (0.1, 1, 20, 200, 500, 1000 and 2000 ng/ml) and PA (500ng/ml or 2000ng/ml for the two higher concentrations of LF). The percentage of 7AAD+ cells on the CD11c+ gated population is represented as mean±SD. Data from 3 independent experiments done in triplicates.
These observations support the proposed role of type 1 Nalp1b in LT-induced death of DCs, at least at early time points (Boyden and Dietrich, 2006), reflecting the activation of a rapid death pathway against which maturation did not provide protection.
We next investigated whether LT-induced type 1 Nalp1b dependent death of DCs required caspase-1. Murine Nalp1b indeed has a caspase recruitment domain (CARD) that potentially allows it to interact with caspase-1 (See Fig. 10B). We used caspase-1 knock out mice with either the B6 or the 129S genetic background (Nalp1b type 2 and 1, respectively). At short times of toxin exposure (4 h), caspase-1−/− DCs remained viable, irrespective of their genetic background (Fig. 4A, C). These findings agree well with what was observed on macrophages after 4 h (Boyden and Dietrich, 2006). However, the absence of caspase-1 did not prevent cell death. It only delayed it, since the number of 7AAD positive cells was similar in caspase-1−/− and caspase-1+/+ DCs after 24 h, again for both Nalp1b genetic backgrounds (Fig. 4A, C). The same cell death profiles were obtained using Annexin V staining, Trypan Blue exclusion and Neutral Red retention (not shown). Interestingly, even in the 129S (type 1 Nalp1b) background, maturation had a strong protective effect on LT induced death of caspase- 1−/− DCs (Fig. 4B, D–F). Protection was observed even at late time points (Fig. 4B, D) and at high concentrations of toxin (Fig. 4E, F), an effect that was not observed for the control 129S (Fig. 4F). Thus, the response of caspase-1−/− DCs to LT is strongly modulated by maturation in different genetic backgrounds (Fig. 4 vs. Fig 2D).
Figure 10. DC susceptibility to LT is modulated by Nalp1b and maturation.
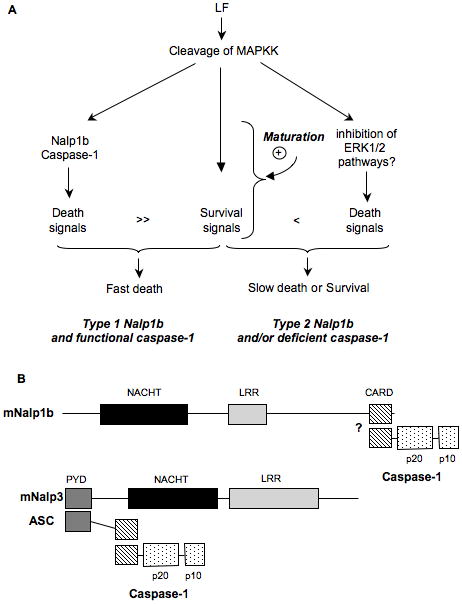
A) Model to explain DC responses to LT based on the activation of two death pathways (Nalp1b/caspase-1 dependent or independent) by LT. Survival pathways upregulated upon DC maturation are able to counteract only the Nalp1b/caspase-1 independent pathway. B) Schematic representation of the domain composition of murine Nalp1b and Nalp3 and their interacting proteins. “?”: a direct interaction between mNalp1b and caspase-1 is hypothesized based on data obtained with human Nalp1 (Faustin et al., 2007).
Figure 4. Caspase-1 is involved in rapid but not slow DC death induced by LT.
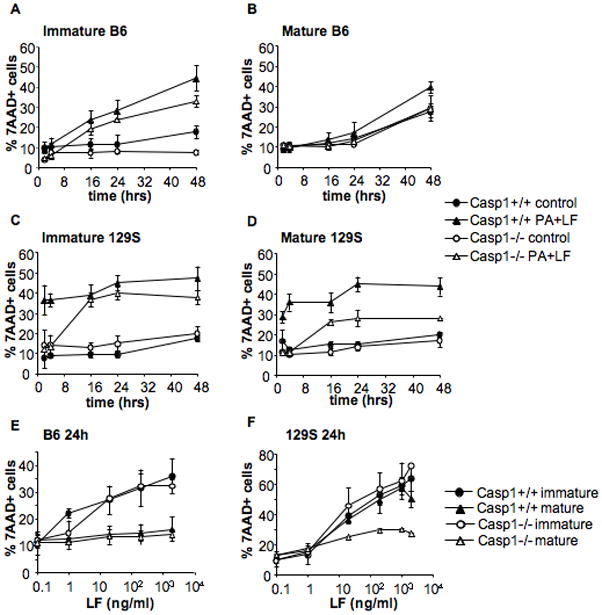
DCs were obtained from Caspase-1−/− mice in B6 (A,B,E) or 129S (C,D,F) background and their parental Caspase-1+/+ B6 and 129S mice. A,C) Immature DCs were incubated with media (control) or 500ng/ml PA and 20ng/ml LF for 2, 4, 16, 24 or 48 hours. B,D) DCs were treated with 100ng/ml LPS for 8 h to induce maturation, and incubated with toxins as in A,C). E,F) DCs were matured by 8 h LPS treatment (mature) or left immature and subsequently treated for 24 h with different concentrations of LF (0.1, 1, 20, 200 and 2000 ng/ml) and PA (500ng/ml or 2000ng/ml for the higher concentration of LF). The percentage of 7AAD+ cells on the CD11c+ gated population is represented as mean±SD. Data from at least 3 independent experiments done in triplicates.
Taken together, these observations suggested that LT can induce at least two death pathways in DCs: 1) a pathway dependent on type 1 Nalp1b and caspase-1 that leads to rapid death whether DCs are immature or mature, and 2) a pathway independent of caspase-1 that is only effective in immature, but not mature, DCs and is observed at late times (> 4 h).
Since LT-induced caspase-1-independent death is only detected at late times points (i.e. 24 h), we thought it might have been overlooked in previous studies on macrophages (Boyden and Dietrich, 2006). Macrophages were obtained from the bone marrow of caspase-1−/− and WT mice, treated with different concentrations of PA+LF and stained with 7AAD to monitor cell death. After 4 h of toxin treatment, macrophages from caspase-1−/− mice in the129S background were less sensitive than those from WT 129S mice (Fig. 5A), a difference that disappeared at later times (24 h, Fig. 5B). We also tested macrophages from WT and caspase-1−/− mice in a B6 background. As expected, no death was observed after 4 h and only slightly after 24 h (Fig 5A, B). After 48 h however, 40% of dead cells were observed for both WT and caspase-1−/− (Fig. 5C). The behavior of caspase-1−/− macrophages, in both genetic backgrounds, indeed demonstrates that both DCs and macrophages posses a caspase-1 independent cell death pathway induced by anthrax toxin, in agreement with recently published observations using caspase-1 inhibitors (Muehlbauer et al., 2007).
Figure 5. Caspase-1 is not essential for LT induced death on macrophages.
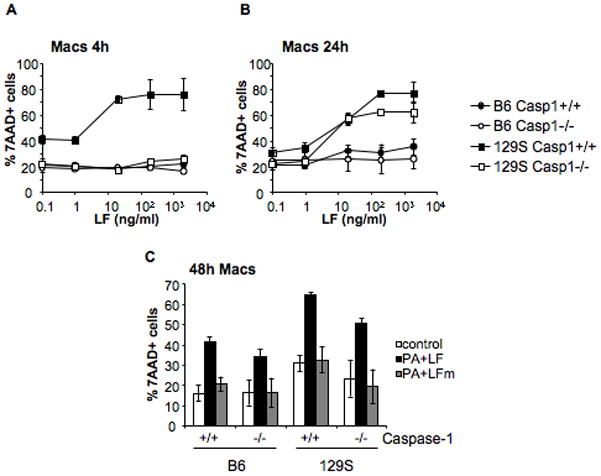
Bone marrow derived macrophages were obtained from Caspase-1−/− mice on 129S background and Caspase-1−/− mice on B6 background. Macrophages obtained from bone marrow of the respective Caspase-1+/+ parental strains were used as controls. Cells were treated with different concentrations of LF (0.1, 1, 20, 200 and 2000 ng/ml) and PA (500ng/ml or 2000ng/ml for the higher concentration of LF) during 4 h (A) or 24 h (B). C) Cells were treated for 48 h with media, PA+LF or PA+LFmutant (500ng/ml of PA and 20ng/ml of LF or LF E687C mutant). The percentage of 7AAD+ cells is represented as mean±SEM. Data from 3 independent experiments done in triplicates.
Protection by maturation acts downstream MAPKK cleavage by LT
In DCs with different genetic backgrounds, the caspase-1 independent death pathway induced by LT is strongly modulated by the maturation state of the cells. More specifically, we found that DC maturation decreases the susceptibility of cells to anthrax toxin. Since maturation is known to induce a down regulation of certain forms of endocytosis and to enhance lysosomal acidification and proteolysis (Trombetta and Mellman, 2005), we asked if toxin resistance reflected reduced binding, uptake, or entry of PA or LF. Binding of PA was similar in immature and mature DCs suggesting similar levels of surface expressed receptors (PA63 on Fig. 6A). Endocytosis was followed by measuring the appearance of the SDS-resistant PA heptamer, a form that is generated by the low endosomal pH upon arrival to early endosomes (Milne et al., 1994; Abrami et al., 2004). In both immature and mature DCs, the SDS-resistant heptamer could readily be detected after 20 min of toxin uptake (Fig. 6A) indicating that endocytosis of the toxin occurred at similar rate. This is consistent with the fact that clathrin-mediated endocytosis, which is the entry route of anthrax toxin (Abrami et al., 2003), is not affected by maturation (Garrett et al., 2000) in contrast to macropinocytosis and phagocytosis (Garrett et al., 2000; West et al., 2004).
Figure 6. Protection is not linked to differential entry of the toxin.
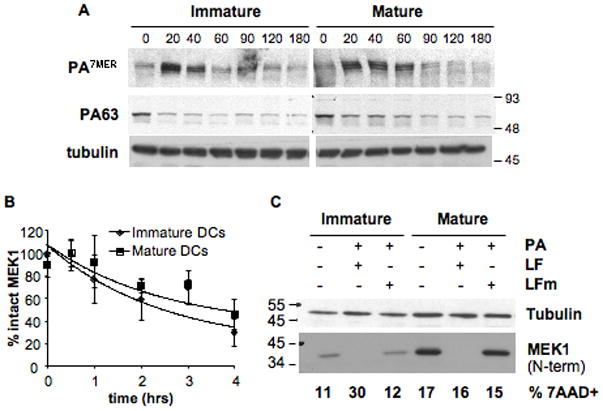
B6 derived DCs were matured over-night with LPS or left immature and a binding assay was performed (A,B): cells were incubated with 500ng/ml of trypsin-nicked PA (Abrami et al., 2003) and 200ng/ml LF, for 1 h on ice. Then washed with toxin free media and transferred at 37°C for different periods of time (in min.). 40 μg of total cell extracts were analyzed by western blotting to detect PA (A), LF processed MEK1 (anti Nterm antibody) and total MEK1 (anti Cterm antibody) (B, see also Suppl. Fig. 4). Equal loading is assessed by an antibody anti-tubulin. B) The amount of intact MEK1 (MEK1 Nterm on Suppl. Fig. 4) was quantified by densitometry, normalized to the amount of total MEK1 (detected with the antibody anti Cterm) as loading control, and the resulting values were normalized to the amount of intact MEK1 (Nterm) at time t=0. The kinetics of decrease of intact MEK1 were compared for immature and mature DCs. Data is expressed as mean±SD from 4 independent experiments. C) DCs from B6 mice were matured by 8 h LPS treatment or left immature, and treated for 24 h with 500ng/ml PA and 20ng/ml LF or LFm (E687C inactive mutant). 40μg of each cell extract were analyzed by western blot with an antibody against the Nterm part of MEK1 to detect only intact MEK1. Equal loading was assessed with an anti-tubulin antibody. An aliquot of the same cells was stained with 7AAD to determine the amount of dead cells by flow cytometry, indicated as % within the CD11c+ population. Blots from one representative experiment (n≥3).
The kinetics of LF cytoplasmic delivery, monitored by the rate of LF-induced N-terminal cleavage of MEK1, were also similar in immature vs. mature cells (Fig. 6B, Suppl. Fig. 4). Similar results were obtained for LF-induced cleavage of MKK3, the MAPKK upstream of p38 (not shown). To rule out that the protective effect of maturation was due to de novo MAPKK synthesis that could overcome LF inactivation of the kinases, we compared the levels of intact MEK1 on immature and mature DCs at long times of toxin exposure. Full length MEK1 remained undetectable in both mature and immature DCs even 24 h after toxin addition, despite the fact that mature DCs expressed higher amounts of MEK1. Importantly, there was no correlation between levels of intact MEK1 and cell death in mature DCs (Fig. 6C). Altogether these observations indicate that the protective effect of maturation operates down-stream of MAPKK cleavage by LT.
The effect of LT on B6 derived DCs could be mimicked by the specific inhibitor of MEK1/2 kinases (U0126) that prevents activation of the ERK1/2 pathway (Fig. 7A). In 129S derived DCs, however, inhibition of MEK1/2 pathways does not mimic LT effects on mature DCs, which are resistant to the inhibitor but not to the toxin (Fig. 7B). This is not surprising, since the type 1 Nalp1b/caspase-1 dependent death pathway is not overcome by maturation. In the absence of caspase-1, however, inhibition of MEK1/2 pathways in 129S DCs totally mimics LT treatment (Fig. 7C), raising the possibility that the caspase-1 independent death of DCs is triggered by the specific inhibition of MEK1/2 by LT.
Figure 7. Inhibition of MEK1/2 mimics the effects of LT in DCs.
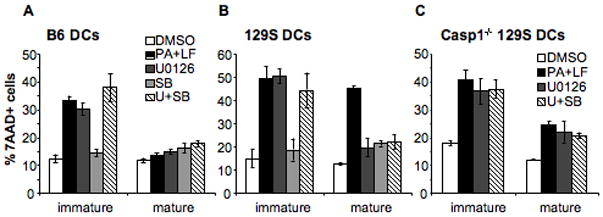
DCs were obtained from B6 mice (A), 129S mice (B) and Caspase-1−/− mice in 129S background (C). DCs were matured by 8 h LPS treatment or left immature and treated for 24 h with 2 μl DMSO (DMSO), 500ng/ml PA and 20ng/ml LF in the presence of DMSO (PA+LF), 5 μM U0126 (MEK1/2 specific inhibitor) (U0126), 20 μM SB203580 (p38 specific inhibitor) (SB) or 5 μM U0126 + 20 μM SB203580 (U+SB). The percentage of 7AAD+ cells on the CD11c+ gated population is represented as mean±SEM. Data from at least 3 independent experiments done in triplicates.
Taken together, these experiments show that LT binding, endocytosis, cytoplasmic delivery of LF and MAPKK cleavage occur similarly in immature and mature DCs indicating that the reduced sensitivity of mature DCs is not due to a prevention of LF action or the preservation of the LF targets but rather to the generation of a phenotype that rendered cells insensitive to the inactivation of MAPK pathways, in particular that of ERK1/2.
Protection to LT is independent of the maturation stimulus and is reached early during the maturation process
As described above, LT-induced caspase-1 independent DC death can be inhibited or counteracted by LPS triggered maturation. A similar protection was obtained when triggering maturation with other bacterial products such as peptidoglycan or Bacillus anthracis extracts, or an inhibitor of GSK3β (SB216763) (Fig. 8A) that induces a non-inflammatory form of DC maturation (Jiang et al., 2007). These results indicate that the precise pathway leading to maturation matters less than the end point at which protection is reached. Surprisingly, this point was reached early during the maturation process, since even when the maturation stimuli were added to the cells simultaneously with the toxin, protection was observed (Fig. 8B, C). Whereas DCs treated with both LPS and toxin expressed CD86 at their surface (Fig. 8C), their cytokine secretion was very low (Fig. 8D). Altogether our results suggest that early signaling events induced by maturation stimuli are able to protect cells from LT induced death even though LT subsequently inhibits cytokine secretion.
Figure 8. Protection by maturation is independent on maturation stimuli and is achieved at an early maturation stage.
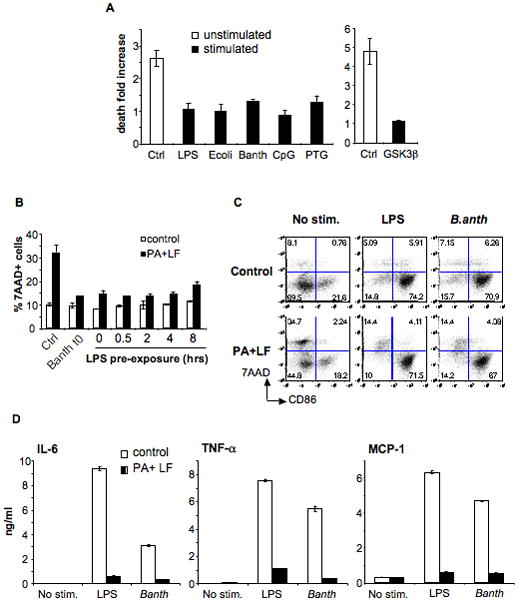
A) DCs were obtained from B6 mice and treated (stimulated) or not (unstimulated) with different maturation stimuli for 8 h: LPS (lipopolysaccharide), E. coli (heat inactivated Escherichia coli), B. anth (sonicated extract of Bacillus anthracis), CpG (CpG DNA), PTG (peptidoglycan); or 16 h: GSK3β (inhibitor of GSK3β, SB216763). Toxins were added during 24 h (PA, 500ng/ml and LF, 20ng/ml) and death was measured by flow cytometry of 7AAD staining on the CD11c+ population. Due to the high autoflorescence of the GSK3β inhibitor, death on that condition was measured by Trypan Blue staining on light microscopy on the total cell population. The fold increase in the number of dead cells is represented for each condition with respect to each control (% of dead cells on toxin treated condition divided by % of dead cells on control condition). Data from a representative experiment done in triplicates (n=3) B) DCs were obtained from B6 mice and treated or not with LPS or a sonicated extract of B.anthracis for the indicated times before addition of PA (500ng/ml) and LF (20ng/ml). t0 indicates that the maturation stimuli and the toxin were added simultaneously. The percentage of 7AAD+ cells within the CD11c+ population after 24 h of toxin treatment is indicated as mean±SD. C–D) DCs were obtained from B6 mice and treated (for 24 h) or not with PA (500ng/ml) and LF (20ng/ml) together with LPS or B.anthracis extract or in the absence of maturation stimuli (No stim.) C) Cells were stained for FACS analysis. The double staining with 7AAD and anti-CD86 on the CD11c+ population is represented. Note that toxin treatment only induced death to CD86− (immature) cells. D) Cytokines secreted on the DCs media were measured by FACS using the Cytometric Bead Array system. Data from one representative experiment done in triplicates (n=3).
The above observations combined with those presented in Figure 2 show that modulation of the responses of DCs to LT by LPS depends on when this maturation stimulus is added: when added after the toxin, LPS promoted DC death (Fig. 1B, 2C) in a manner reminiscent to what was observed in macrophages (Park et al., 2002), whereas when added prior or concomitantly with the toxin, LPS protected cells from death (Fig. 2A, D and Fig. 8B, C).
The inflammasome components Nalp3 and ASC are not involved in the response to LT
The observation that maturation stimuli lead to the establishment of a protective phenotype in a B6, type 2 Nalp1b, background, raised the possibility that a survival component/pathway is rapidly upregulated. It has been previously reported, and confirmed here (Suppl. Fig. 5) that LPS-induced maturation of DCs leads to the rapid upregulation of the mRNA of Nalp3 (Sutterwala et al., 2006), another member of the NLR family (Sutterwala et al., 2006, Gurcel, 2006 #3332; Mariathasan and Monack, 2007). We therefore analyzed the effect of LT on DCs from Nalp3−/− mice (Mariathasan et al., 2006; Sutterwala et al., 2006). To study the role of Nalp3 in LT-induced responses both in the presence and in the absence of the caspase-1 dependent death pathway, we made use of two different strains of Nalp3−/− mice, which had the same genetic background (B6) but carried different alleles of Nalp1b (type 1 and type 2) because they were generated from ES cells obtained from 129SvEvBrd (Sutterwala et al., 2006) and B6 mice (Mariathasan et al., 2006), respectively. Due to the proximity between the Nalp1b and the Nalp3 loci, the Nalp3−/− mice generated from 129SvEvBrd ES cells still carried the 129S allele of Nalp1b after 8 generations of backcrossing with B6 mice (not shown). Wild type DCs from 129S and B6 mice were used as controls for the Nalp3−/− derived DCs carrying type 1 and type 2 Nalp1b, respectively. We observed that in the presence of a type 1 Nalp1b, Nalp3 deficiency rendered immature DCs significantly more sensitive to LT than the 129S controls (Fig. 9A). Maturation had no effect on the sensitivity of type1 Nalp1b expressing Nalp3 −/− DCs (Fig. 9B, E), not surprising given the weak modulation by maturation of the caspase-1 dependent death pathway. However, in the absence of the caspase-1 dependent death pathway triggered by LT (i.e. in a type 2 Nalp1b background), Nalp3 deficiency had no effect on the susceptibility of DCs to LT, that died with a slow kinetics and were protected by maturation similarly as wild type cells (Fig. 9C, D, F). Similar results were obtained using DCs derived from mice defective in the adaptor protein ASC (Apoptosis-associated Speck-like protein containing a CARD) (Masumoto et al., 1999; Conway et al., 2000), which contains a Pyrin and a CARD domain and is required for the assembly of the Nalp3 inflammasome (Suppl. Fig. 6).
Figure 9. Nalp3 deficiency differentially affects DC susceptibility to LT.
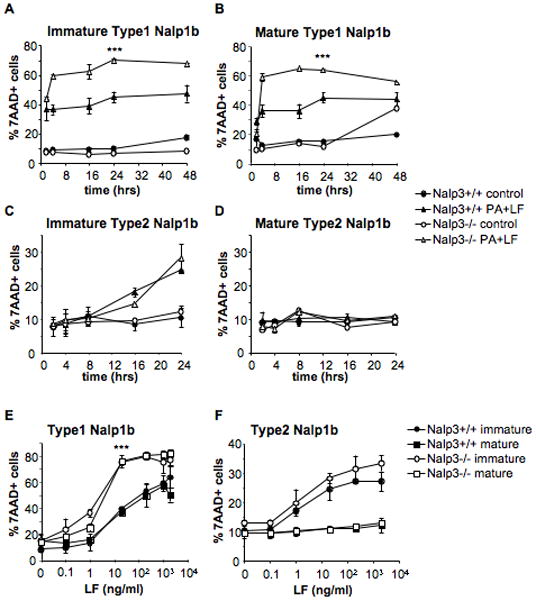
DCs were obtained from Nalp3−/− mice in B6 background, carrying type 1 Nalp1b (Sutterwala et al., 2006)(A,B,E) or type 2 Nalp1b (Mariathasan et al., 2006) (C,D,F) and their controls Nalp3+/+ 129S and B6 mice, respectively. A,C) Immature DCs were incubated with media (control) or 500ng/ml PA and 20ng/ml LF for 2, 4, 16, 24 or 48 hours. B,D) DCs were treated with 100ng/ml LPS for 8 h to induce maturation, and incubated with toxins as in A,C). E,F) DCs were matured by 8 h LPS treatment (mature) or left immature and subsequently treated for 24 h with different concentrations of LF (0.1, 1, 20, 200, 1000 and 2000 ng/ml) and PA (500ng/ml or 2000ng/ml for the two higher concentrations of LF). The percentage of 7AAD+ cells on the CD11c+ gated population is represented as mean±SD. ***: Type 1-Nalp3−/− DCs are significantly more susceptible than Nalp3+/+ control cells, in both conditions of maturation (p≤0.0001 comparing the %7AAD+ cells on Nalp3−/− vs Nalp3+/+ DCs after 24h treatment with PA+LF (20ng/ml)). Data from 3 independent experiments done in triplicates.
These results suggest that Nalp3 might have an inhibitory role on the activation of the Nalp1b inflammasome triggered by LT in DCs expressing type 1 Nalp1b. However, neither Nalp3 nor ASC seem to play any role in modulating DCs susceptibility to the LT-induced caspase-1 independent death pathway observed in type 2 Nalp1b expressing cells.
DISCUSSION
Recent studies have illustrated that anthrax lethal toxin contributes to the immune evasion of B. anthracis by impairing the function of cells from the immune system (reviewed in (Baldari et al., 2006)). The mechanisms of functional impairment, and of cell death however are not fully understood. Our goal was to better understand the cellular response to anthrax lethal toxin and whether sensitivity was dependent on the maturation state in addition to the genetic background. We here find that the consequence of cleavage of MAP kinase kinases by LT –assuming there are no other targets for the LF metalloprotease– is sensed by multiple mechanisms, leading to a diversity of cellular responses, the relative strength of which depends on the genetic background and the maturation stage of the cells.
Activation of cell death pathways in response to LT
When expressing type 1 Nalp1b, DCs respond to LT treatment by switching on a rapid death pathway that depends on the activation of caspase-1, as do macrophages (Boyden and Dietrich, 2006). Murine Nalp1b most likely interacts directly with caspase-1 via its C-terminal CARD domain (Caspase recruitment domain) (Fig. 10B) as recently shown for human Nalp1 (Faustin et al., 2007). Previous studies, however, reported that human Nalp1 interacts with caspase-1 via its N-terminal pyrin domain– absent in murine Nalp1b– and the adaptor protein ASC (Martinon et al., 2002). Our preliminary results obtained with DCs from ASC−/− mice in a Balb/cJ background (carrying type 1 Nalp1b), show no impairment in caspase-1 dependent death of ASC−/− DCs treated with LT (unpublished observations), arguing against a role of ASC in the assembly of the murine Nalp1b inflammasome. We cannot however exclude, the existence of interaction or competition between Nalp1b and Nalp3, since the absence of Nalp3 led to higher susceptibility to LT of DCs carrying type 1 Nalp1b, suggesting a role of Nalp3 as a negative regulator of Nalp1b activation. Nevertheless, this effect could also be explained by genetic differences in loci other than Nalp1b, since the genetic backgrounds of type 1 Nalp1b-Nalp3−/− mice and their controls were different (B6 and 129S, respectively). Fully addressing this issue will require the generation of Nalp3−/− mice in the 129S background.
It is not known how LT triggers the activation of Nalp1b and caspase-1. Recent reports proposed that this process requires K+ efflux (Wickliffe et al., 2007) and proteasomal activity (Squires et al., 2007; Wickliffe et al., 2007), but the mechanism by which LT triggers ionic fluxes and the proteasome substrates involved in Nalp1b regulation are not yet defined. The molecular mechanism by which caspase-1 induces rapid cell death also remains unknown. Caspase-1 is mostly considered as an inflammatory caspase, mediating the processing and secretion of interleukins such as IL-1β and IL-18, however increasing evidence in the literature points towards the existence of additional targets. For example, activation of caspase-1 is critical for macrophage death triggered by infection with Salmonella (Hersh et al., 1999; Brennan and Cookson, 2000) and Shigella (Hilbi et al., 1998), by a form of programmed cell death termed pyroptosis that is linked to inflammation (reviewed in (Fink and Cookson, 2007)).
Rapid cell death in response to LT, however, does not occur in human macrophages and DCs (Alileche et al., 2005; Brittingham et al., 2005; Kassam et al., 2005). Similarly, DCs obtained from mice carrying type 2 Nalp1b, such as B6 mice, or type 4 Nalp1b such as DBA/2J (our unpublished observations) do not rapidly lyse upon treatment with LT. LT does not induce caspase-1 activation in these DCs (not shown), in agreement with previous findings on macrophages (Boyden and Dietrich, 2006). In contrast to this latter study, however, we found that in the absence of caspase-1 activation– whether in type 2 Nalp1b expressing or caspase-1−/− cells–, DCs and macrophages were still susceptible to the toxin, except that kinetics of cell death were delayed. This shows the existence of a second cell death pathway triggered by LT that is independent of caspase-1, in agreement with recent observations using chemical inhibition of caspase-1 on macrophages (Muehlbauer et al., 2007). The discrepancies between studies might be due to the short time frames studied by Boyden et al. (4–6 h) or to the single method used (neutral red) to assess cell viability (Boyden and Dietrich, 2006).
DC maturation modulates cellular responses to LT
Given the similarities between death induced by LT in DCs from B6 mice and in human DCs (Alileche et al., 2005), caspase-1 independent cell death appears to be the more relevant cellular response of human cells to LT. We report here that the caspase-1 independent death pathway is strongly modulated by maturation of DCs. Indeed, maturation, induced by a variety of stimuli, rendered type 2 Nalp1b expressing DCs fully resistant to LT. Interestingly, mature DCs were also more resistant to death caused by chemical inhibition of MEK1/2 pathways, a treatment that phenocopied the effects of LT on DCs where caspase-1 was not activated. Inhibition of p38 pathways, however, did not mimic LT effects, in contrast to what was observed in macrophages co-stimulated with LPS and LT (Park et al., 2002). Thus, the caspase-1 independent death pathway induced by LT in DCs could be due to the inactivation of the MEK1/2-ERK1/2 pathways upon cleavage by LF.
The fact that maturation stimuli led to LT-protection of DCs from B6 mice could explain some of the many discrepancies in the literature. When DCs were exposed to B. anthracis spores, cells remained viable, maturation markers were upregulated but cytokine secretion was impaired (Brittingham et al., 2005; Tournier et al., 2005). This cellular response is very similar to the one we have observed when simultaneously adding the toxin and the maturation stimulus (Fig. 8C, D). One possibility therefore is that upon spore addition to cells, the onset of maturation and the initiation of intoxication occurred within a very short period of each other. Agrawal et al. (Agrawal et al., 2003) performed their study on splenic DCs. This method of DC preparation requires physical disruption of cell-cell interactions and replating, operations that trigger DC maturation by a process that is mimicked by the GSK3s inhibitor (Jiang et al., 2007). Since toxin addition presumably occurred shortly after DC preparation, cells were probably again in a situation where stimulation and toxin addition occurred within a short time frame. The possibility that these splenic DCs (Agrawal et al., 2003) received a maturation stimulus is supported by the high death rate of the untreated control DC, i.e. 80% cell death after 48 h. In the absence of survival signals provided by T cells, viability of mature DCs is indeed impaired in contrast to that of immature DCs (Wong et al., 1997).
The molecular mechanism by which mature DCs become resistant to LT induced caspase-1 independent death remains to be defined. Cell survival is known to be stimulated upon activation of the NF-KB pathway, which is triggered upon DC maturation (Karin and Lin, 2002; Ouaaz et al., 2002). Some of the NF-KB target genes encode for members of the Bcl-2 protein family, which are known to be important players in both pro-survival and pro-apoptotic pathways (Cory and Adams, 2002). In particular the death gene Bnip3 was found to be down regulated through a mechanism involving NF-KB (Baetz et al., 2005), a gene we found to be down regulated 20 fold in DCs following maturation (unpublished). Therefore, the increased survival of mature DCs could be due to the down regulation of death genes and upregulation of anti-apoptotic proteins such as Bcl-XL (our unpublished observations and (Lundqvist et al., 2002; Tureci et al., 2003; Hou and Van Parijs, 2004)), which is crucial for survival of DCs in vitro and in vivo (Hon et al., 2004; Hou and Van Parijs, 2004).
Concluding remarks
Based on the here-described observations, we propose the following model to explain DC responses to LT (Fig. 10A). After LT entry and MAPKK cleavage, at least two death pathways are activated. One death pathway depends on the type 1 Nalp1b/caspase-1 axis and leads to rapid death. Although this pathway can be slightly modulated by maturation dependent survival signals, it cannot be overcome. This pathway is found in DCs from certain mouse strains but not in human DCs. The second death pathway, possibly triggered by the impairment of ERK1/2 pathways, is caspase-1 independent and is strongly modulated, or counteracted, by maturation-dependent survival signals. The upregulation of pro-survival signals induced by maturation may account for complete survival of mature type 2 Nalp1b expressing DCs to LT.
DC killing by LT might have a role in the pathogenesis of an anthrax infection, particularly at early phases of pulmonary anthrax. Upon phagocytosis of B. anthracis spores, lung resident DCs traffic to lymph nodes while allowing germination of the bacteria within (Brittingham et al., 2005; Cleret et al., 2006). The expression of toxin proteins by vegetative bacilli will impair the function of these DCs at the level of cytokine secretion and mounting an immune response, permitting further multiplication of the bacteria and production of toxins. These toxins will have effects even on immature DCs, which will succumb, perturbing the host immune system even further and allowing the progression of the disease.
The present study illustrates that the effects of a given toxin are sensed by multiple mechanisms leading to the onset of different signaling cascades, the efficiencies of which are modulated by the genetic background, the type and developmental stage of cells and the amplitude of the toxic insult. Some of the future challenges are clearly to determine what events are sensed by the cells, i.e. which is the true ligand of the Nalp1b leucine rich repeat domain, which are the targets of caspase-1 involved in cell death triggered by LT, and what are the molecular requirements of the caspase-1 independent death pathway and of their modulation by the maturation stage of the cells.
MATERIALS AND METHODS
Mouse strains, Toxins and reagents
C57BL/6J and 129S1/svImJ mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Nalp3−/− mice carrying type 1 Nalp1b and ASC−/− mice were described (Sutterwala et al., 2006). These mice were backcrossed for 8 generations with C57BL/6J, but given the proximity between the Nalp1b and the Nalp3 loci, Nalp3−/− mice still carried the type 1 Nalp1b form from 129SvEvBrd ES cells. Femurs from Nalp3−/− mice carrying type 2 Nalp1b were obtained from (Mariathasan et al., 2006). Caspase 1−/− mice were developed in R. Flavell’s laboratory (Kuida et al., 1995) on 129SvEvBrd background and obtained from J. Galán laboratory (Yale University, New Haven) (Lara-Tejero et al., 2006). Caspase 1−/− mice on C57BL/6J background were obtained from R. Flavell’s laboratory (Yale University, New Haven) after backcrossing for 5 generations with C57BL/6J strain. The Nalp1b genotype of all mice used was confirmed to be either the same as 129S1/svImJ strain (type 1) or as C57BL/6J strain (type 2) by PCR on genomic DNA using the following primers: for type 1 Nalp1b: 5′-AGGGTAGAGTGCTCTCACTGAAGTCA-3 ′ and 5 ′-AGTGCATAT/CCTTGCTGTTTTTCCATA-3′ (forward and reverse primer, respectively); for type 2 Nalp 1b : 5 ′-GCAGTCATGTCTTACATCTTGGAGGA-3 ′ and 5 ′-CATTTGTCTGTGACACTGTGGCTTCC-3′ (forward and reverse primer, respectively). Toxins were a gift from S. Leppla, prepared as described (Leppla, 1988), including wild type PA, PASNKE (Gordon et al., 1995), LF and LF E687C (Klimpel et al., 1994). FITC-Annexin V and Fluorochrome-conjugated antibodies for FACS analysis were all purchased from Pharmingen, BD. 7AAD (7-aminoactinomycin D), Peptidoglycan from S. aureus and LPS from Salmonella typhimurium were obtained from Sigma-Aldrich. Polyclonal antibodies anti PA and LF were a gift from S. Leppla (Liu and Leppla, 2003). Anti C-term MEK1 antibody was purchased from Santa Cruz Biotechnology. Anti N-term MEK1 antibody was obtained from Upstate Biotechnology. Anti α-tubulin antibody was obtained from Sigma. Secondary anti rabbit and mouse IgG HRP-antibodies were from Pierce.
Mouse DC culture
Bone marrow-derived DCs were prepared and cultured as described previously ((Pierre et al., 1997), (Inaba et al., 1992)) from males 7–9 week-old. At day 5 the cultures contained many aggregates of immature DCs loosely attached to the plate. When indicated, cells were matured by addition of 100ng/ml LPS for at least 6 h. In some experiments maturation was induced by addition of heat inactivated bacteria (1.3μl/ml MAX Efficiency DH5α competent cells, Invitrogen), 5μl of sonicated extract of Bacillus anthracis (gift of C. Montecucco), 10μg/ml of Peptidoglycan, or 1nM CpG DNA (5′-TCCATGACGTTCCTGACGTT-3′) for 8 h. GSK3β inhibitor (SB216763) was used over-night at 10μM to induce maturation to day 4 DCs. When mature DCs were used, their phenotype was checked by flow cytometry (see below). Mature DCs present high staining of CD86, CD80 and MHCII, whereas immature DCs were high CD11c, low CD86 and intermediate MHCII staining.
Experiments with bone marrow derived macrophages
Murine bone marrow-derived macrophages were prepared and cultured as described previously (Racoosin and Swanson, 1989). Cells were plated at 4×105 cells/ml in 6well plates in media containing 5ng/ml of recombinant mouse M-CSF (Sigma). Fresh media was added after 3 days and cells were used for experiments after 6 days in culture. Toxin treatment was performed in identical conditions as for DCs experiments. Cell viability was assayed by flow cytometry of 7AAD staining after harvesting cells with cold PBS+5mM EDTA.
Toxin treatment
Immature or mature DCs (treated for 8 h with LPS) at 106 cells/ml were incubated with 500ng/ml of PA and 20ng/ml of LF during 24 h (different forms of the toxins, concentrations and times of incubation are indicated otherwise). To study the response of toxin-treated DCs to maturation stimuli, LPS was added to toxin-treated or untreated cells during the last 6 or 8 h of incubation, as indicated in figure legends. When the toxin treatment was shorter than 6 h, LPS was added together with the toxin for the indicated times.
Inhibitor studies
Immature or mature DCs were treated with 5μM MEK1/2 inhibitor (U0126, Signaling Technology Inc.), 20μM p38 inhibitor (SB203580, LC Laboratory) or both, for 24 h. Control cells were treated with the same volume of DMSO.
Flow cytometry analysis
Loosely attached cells were harvested by pipeting and stained for 30 min on ice with several fluorochrome conjugated antibodies to detect DC and maturation markers, washed in PBS+1%FCS and then evaluated on a FACSCalibur™ (Becton Dickinson). Anti-mouse antibodies used were: APC anti-CD11c, PE anti-CD86 and FITC anti-MHCII I-Ab. Dead or apoptotic cells were detected by staining for 30 min on ice with FITC-Annexin V and 1min with 7AAD. FACS data were analyzed using FlowJo software (FlowJo, LLC). Cells on the gated CD11c+ population are shown in the figures.
Western blot analysis
DCs were harvested, washed with PBS+0.5%BSA and lysed in HB-1% NP40 (HB: 2.9 mM imidazole and 250mM sucrose, pH 7.4) containing Roche mini tablet protease inhibitors cocktail following manufacturer’s instructions. Protein quantification was done with Pierce BCA kit. Proteins were loaded at 40μg prot/lane and separated on a 4–20% acrylamide precast novex gel (Invitrogen) or on a 12% acrylamide gel using the miniprotean system of BioRad, under reducing conditions and transferred to nitrocellulose membranes (Schleicher and Schuell) by semi-dry transfer or wet transfer.
Binding assays
Immature or mature DCs at 106 cells/ml (treated over-night with LPS) were washed 5min with cold IM (Gibco RPMI 1640, 10mM Hepes, 1% FCS) and incubated 1 h on ice with IM + toxin (trypsin-nicked PA83 500ng/ml + LF 200ng/ml) (Abrami et al., 2003). Cells were washed with cold IM and incubated at 37°C for the indicated times. After the desired internalization time, cells were collected, washed with cold PBS+0.5% BSA and lysed in HB-1% NP40.
Measurements of cytokine secretion
Multiplex analysis of 20 mouse cytokines was performed on culture supernatants of toxin-treated or control DCs using Luminex system by John Conolly (Baylor Institute for Immunology Research, Dallas). BD Cytometric Bead Array mouse inflammation kit was used following manufacturer instructions.
Real time PCR
CD11c-positive DCs were purified with magnetic micro-beads conjugated to anti–mouse CD11c mAb (clone N418; Miltenyi Biotech) for 15 min at 4°C. CD11c-positive DCs were then separated by passing cells over a MACS MS+ column held in a VarioMACS magnetic separator (Miltenyi Biotech). RNA was extracted from 6×106 CD11+ cells using RNA easy miniextraction kit (Qiagen) and 1μg was used for reverse transcription with Superscript II (Invitrogen) using random hexamers. A 1/20 dilution of the resulting cDNA was used to perform real-time PCR using the CYBR-Green reagent (Applied Biosystems) in an ABI Prism SDS 7900 HT system (Applied Biosystems) with the following primers: Nalp3 (forward 5′-CGAGACCTCTGGGAAAAAGCT-3 ′, reverse 5 ′-GCATACCATAGAGGAATGTGATGTACA-3 ′ ), A S C ( forward 5 ′-GCTGCAAACGACTAAAGAAGAGTCT-3 ′, reverse 5 ′-GCTGTACTCTGAGCAGGGACACT-3′). The 4 murine housekeeping genes used to normalize the mRNA values of the genes of interest were: Eukaryotic Translation Elongation Factor 1 A1 (EEF1A1), TBP (TATA binding protein), Gapdh (Glyceraldehide-3-phosphate dehydrogenase) and Rps9 (ribosomal protein S9).
Statistical analysis
Data presented are obtained from at least 3 independent experiments. In experiments ran in triplicates, data represents mean ± standard deviation (SD) or standard error (SEM), as indicated in figure legends. T-student p values were obtained by considering a 2-tail distribution with equal variance using Microsoft Excel and GraphPad Prism softwares.
Supplementary Material
A) DCs from B6 mice were treated for 24 h with 500ng/ml of PA+ 20ng/ml LF or LF E687C mutant (LFm). Control cells were left untreated. Surface expression of CD86, MHCII and CD80 was monitored by FACS and represented on the population of CD11c+ cells. B) B6 derived DCs were treated as in A) and LPS (100ng/ml) was added for the last 8 h of toxin incubation to induce maturation. Surface expression of CD86, MHCII and CD80 was analyzed by FACS and represented on CD11c+ gated cells.
B6 immature DCs were treated for 24 h with 500ng/ml of PA + 1 or 20 ng/ml of LF or LF E687C mutant (LFm). Control cells were left untreated. LPS 100ng/ml was added during the last 8 h of incubation when indicated (+LPS). The concentration of several cytokines measured by Luminex system in the culture supernatants is represented as mean±SD.
A) DCs obtained from B6 mice were incubated with different combinations of anthrax toxins and their mutant forms during 24 h. PA (wild type or mutant) was added at 500ng/ml, and LF (wild type or mutant) was added at 20ng/ml. LPS (100ng/ml) was added 8 h before the end of the incubation to the indicated cells (iDC+LPS). Cells were collected and analyzed by flow cytometry after staining with anti Annexin V-FITC and 7AAD. Here is represented the double staining on gated CD11C+ cells treated with medium alone (control), PA alone, LF alone, PA+LF wild type, PA+LFm (inactive mutant E687C) and PASNKE+LF (a furin resistant mutant form of PA). Numbers in each gate correspond to the % of cells within the CD11c+ population. Note the higher 7AAD and Annexin V staining on PA+LF treated cells subsequently treated with LPS (red square). Data from one representative experiment (n=3). B) DCs obtained from B6 mice were incubated during 24 h with PA+LF (500ng/ml and 20ng/ml, respectively) or left untreated. LPS (100ng/ml) was added 8 h before the end of the incubation to the indicated cells (iDC+LPS). Represented the increase of mean fluorescence intensity of 7AAD and Annexin V stainings (fluorescence on PA+LF treated cells – fluorescence in control cells) on the population of CD11c+ surviving cells (7AAD negative) after treatment. LPS treatment causes a significant increase in fluorescence of both markers (p= 0.004 for 7AAD; p>0.0001 for Annexin V). Represented as mean±SEM from data from 6 independent experiments done in triplicate.
B6 derived DCs were matured over-night with LPS or left immature. Cells were incubated with 500ng/ml of trypsin-nicked PA (Abrami et al., 2003) and 200ng/ml LF, for 1 h on ice. Then washed with toxin free media and transferred at 37°C for different periods of time (in min.). 40 μg of total cell extracts were analyzed by western blotting to detect LF processed MEK1 (anti Nterm antibody), total MEK1 (anti Cterm antibody) or LF.
RNA was extracted from B6 derived DCs treated with 100ng/ml LPS for 3 or 8 h or left untreated (No LPS). The levels of Nalp3 and ASC mRNA were quantified by Taqman real-time RT-PCR and normalized to the values obtained for 4 different housekeeping genes (see materials and methods). Values were normalized to the levels of mRNA on control cells (No LPS) and represented as fold change as mean±SD. Data from one representative experiment run in triplicates (n=3).
DCs were obtained from ASC−/− mice in B6 background (Sutterwala et al., 2006) and their parental ASC+/+ B6 mice. A) Immature DCs were incubated with media (Ctrl) or 500ng/ml PA and 20ng/ml LF for 2, 4, 8, 16, 24 or 48 hours. B) DCs were treated with 100ng/ml LPS for 8 h to induce maturation, and incubated with toxins as in A). C) DCs were matured by 8 h LPS treatment (mDC) or left immature (iDC) and subsequently treated for 24 h with different concentrations of LF (0.1, 1, 20, 200 and 2000 ng/ml) and PA (500ng/ml or 2000ng/ml for the higher concentration of LF). The percentage of 7AAD+ cells on the CD11c+ gated population is represented as mean±SEM. Data from 4 independent experiments done in triplicates.
Acknowledgments
This work was supported by the Swiss National Science Foundation (G.v.d.G.) and the NIH (IM). N.R. is recipient of a long term FEBS post-doctoral fellowship. G. v.d.G. is an international scholar of the Howard Hughes Medical Institute. I. M. is an affiliate member of the Ludwig Institute for Cancer Research. We thank Stephen H. Leppla for generously providing toxins and antibodies, C. Montecucco for the sonicated extract of B.anthracis, M. Docquier (Plateforme Génomique du NCCR «Frontiers in Genetics») for her assistance on the quantitative RT-PCR experiments, W. Reith lab for providing with cDNAs, M-L. Tejero from J. Galan’s lab for providing with Caspase-1−/− mice on 129S1/svImJ background and S. Mariathasan for providing with femurs from Nalp3−/− mice carrying type 2 Nalp1b. We thank all the members of van der Goot, Mellman and Warren labs for helpful discussions.
References
- Abrami L, Leppla SH, van der Goot FG. Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J Cell Biol. 2006;172(2):309–20. doi: 10.1083/jcb.200507067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Lindsay M, Parton RG, Leppla SH, van der Goot FG. Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J Cell Biol. 2004;166:645–651. doi: 10.1083/jcb.200312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol. 2003;160(3):321–8. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Reig N, van der Goot FG. Anthrax toxin: the long and winding road that leads to the kill. Trends Microbiol. 2005;13(2):72–8. doi: 10.1016/j.tim.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Agrawal A, Lingappa J, Leppla SH, Agrawal S, Jabbar A, Quinn C, Pulendran B. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature. 2003;424(6946):329–34. doi: 10.1038/nature01794. [DOI] [PubMed] [Google Scholar]
- Alileche A, Serfass ER, Muehlbauer SM, Porcelli SA, Brojatsch J. Anthrax lethal toxin-mediated killing of human and murine dendritic cells impairs the adaptive immune response. PLoS Pathog. 2005;1(2):e19. doi: 10.1371/journal.ppat.0010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetz D, Regula KM, Ens K, Shaw J, Kothari S, Yurkova N, Kirshenbaum LA. Nuclear factor-kappaB-mediated cell survival involves transcriptional silencing of the mitochondrial death gene BNIP3 in ventricular myocytes. Circulation. 2005;112(24):3777–85. doi: 10.1161/CIRCULATIONAHA.105.573899. [DOI] [PubMed] [Google Scholar]
- Baldari CT, Tonello F, Paccani SR, Montecucco C. Anthrax toxins: A paradigm of bacterial immune suppression. Trends Immunol. 2006;27(9):434–40. doi: 10.1016/j.it.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin.[see comment] Nature Genetics. 2006;38(2):240–4. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414(6860):225–9. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. 2000;38(1):31–40. doi: 10.1046/j.1365-2958.2000.02103.x. [DOI] [PubMed] [Google Scholar]
- Brittingham KC, Ruthel G, Panchal RG, Fuller CL, Ribot WJ, Hoover TA, et al. Dendritic cells endocytose Bacillus anthracis spores: implications for anthrax pathogenesis. J Immunol. 2005;174(9):5545–52. doi: 10.4049/jimmunol.174.9.5545. [DOI] [PubMed] [Google Scholar]
- Cleret A, Quesnel-Hellmann A, Mathieu J, Vidal D, Tournier JN. Resident CD11c+ lung cells are impaired by anthrax toxins after spore infection. J Infect Dis. 2006;194(1):86–94. doi: 10.1086/504686. [DOI] [PubMed] [Google Scholar]
- Comer JE, Chopra AK, Peterson JW, Konig R. Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infect Immun. 2005;73(12):8275–81. doi: 10.1128/IAI.73.12.8275-8281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KE, McConnell BB, Bowring CE, Donald CD, Warren ST, Vertino PM. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res. 2000;60(22):6236–42. [PubMed] [Google Scholar]
- Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2(9):647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- Creagh EM, O’Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27(8):352–7. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280(5364):734–7. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- During RL, Li W, Hao B, Koenig JM, Stephens DS, Quinn CP, Southwick FS. Anthrax lethal toxin paralyzes neutrophil actin-based motility. J Infect Dis. 2005;192(5):837–45. doi: 10.1086/432516. [DOI] [PubMed] [Google Scholar]
- Erwin JL, DaSilva LM, Bavari S, Little SF, Friedlander AM, Chanh TC. Macrophage-derived cell lines do not express proinflammatory cytokines after exposure to Bacillus anthracis lethal toxin. Infect Immun. 2001;69(2):1175–7. doi: 10.1128/IAI.69.2.1175-1177.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Cordoba-Rodriguez R, Lankford CS, Frucht DM. Anthrax lethal toxin blocks MAPK kinase-dependent IL-2 production in CD4+ T cells. J Immunol. 2005;174(8):4966–71. doi: 10.4049/jimmunol.174.8.4966. [DOI] [PubMed] [Google Scholar]
- Fang H, Xu L, Chen TY, Cyr JM, Frucht DM. Anthrax lethal toxin has direct and potent inhibitory effects on B cell proliferation and immunoglobulin production. J Immunol. 2006;176(10):6155–61. doi: 10.4049/jimmunol.176.10.6155. [DOI] [PubMed] [Google Scholar]
- Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25(5):713–24. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. 2007 doi: 10.1111/j.1462-5822.2007.01036.x. [DOI] [PubMed] [Google Scholar]
- Friedlander AM. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem. 1986;261(16):7123–6. [PubMed] [Google Scholar]
- Friedlander AM, Bhatnagar R, Leppla SH, Johnson L, Singh Y. Characterization of macrophage sensitivity and resistance to anthrax lethal toxin. Infect Immun. 1993;61(1):245–52. doi: 10.1128/iai.61.1.245-252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Chen LM, Kroschewski R, Ebersold M, Turley S, Trombetta S, et al. Developmental control of endocytosis in dendritic cells by Cdc42. Cell. 2000;102(3):325–34. doi: 10.1016/s0092-8674(00)00038-6. [DOI] [PubMed] [Google Scholar]
- Gatti E, Pierre P. Understanding the cell biology of antigen presentation: the dendritic cell contribution. Curr Opin Cell Biol. 2003;15(4):468–73. doi: 10.1016/s0955-0674(03)00069-3. [DOI] [PubMed] [Google Scholar]
- Gordon VM, Klimpel KR, Arora N, Henderson MA, Leppla SH. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infection and Immunity. 1995;63(1):82–87. doi: 10.1128/iai.63.1.82-87.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell. 2006;126(6):1135–45. doi: 10.1016/j.cell.2006.07.033. [DOI] [PubMed] [Google Scholar]
- Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci U S A. 1999;96(5):2396–401. doi: 10.1073/pnas.96.5.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, Banerjee S, et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J Biol Chem. 1998;273(49):32895–900. doi: 10.1074/jbc.273.49.32895. [DOI] [PubMed] [Google Scholar]
- Hon H, Rucker EB, 3rd, Hennighausen L, Jacob J. bcl-xL is critical for dendritic cell survival in vivo. J Immunol. 2004;173(7):4425–32. doi: 10.4049/jimmunol.173.7.4425. [DOI] [PubMed] [Google Scholar]
- Hou WS, Van Parijs L. A Bcl-2-dependent molecular timer regulates the lifespan and immunogenicity of dendritic cells. Nat Immunol. 2004;5(6):583–9. doi: 10.1038/ni1071. [DOI] [PubMed] [Google Scholar]
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, Jiang S, et al. Disruption of E-Cadherin-Mediated Adhesion Induces a Functionally Distinct Pathway of Dendritic Cell Maturation. Immunity. 2007 doi: 10.1016/j.immuni.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Kassam A, Der SD, Mogridge J. Differentiation of human monocytic cell lines confers susceptibility to Bacillus anthracis lethal toxin. Cell Microbiol. 2005;7(2):281–92. doi: 10.1111/j.1462-5822.2004.00458.x. [DOI] [PubMed] [Google Scholar]
- Klimpel KR, Arora N, Leppla SH. Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Molecular Microbiology. 1994;13(6):1093–1100. doi: 10.1111/j.1365-2958.1994.tb00500.x. [DOI] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267(5206):2000–3. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Lara-Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, et al. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J Exp Med. 2006;203(6):1407–12. doi: 10.1084/jem.20060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppla SH. Production and purification of anthrax toxin. Methods Enzymol. 1988;165:103–16. doi: 10.1016/s0076-6879(88)65019-1. [DOI] [PubMed] [Google Scholar]
- Liu S, Leppla SH. Cell surface tumor endothelium marker 8 cytoplasmic tail-independent anthrax toxin binding, proteolytic processing, oligomer formation and internalization. J Biol Chem. 2003;278:5227–34. doi: 10.1074/jbc.M210321200. [DOI] [PubMed] [Google Scholar]
- Lundqvist A, Nagata T, Kiessling R, Pisa P. Mature dendritic cells are protected from Fas/CD95-mediated apoptosis by upregulation of Bcl-X (L) Cancer Immunol Immunother. 2002;51(3):139–44. doi: 10.1007/s00262-002-0265-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7(1):31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117(5):561–74. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274(48):33835–8. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442(7098):39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J Biol Chem. 1994;269:20607–20612. [PubMed] [Google Scholar]
- Moayeri M, Leppla SH. The roles of anthrax toxin in pathogenesis. Curr Opin Microbiol. 2004;7(1):19–24. doi: 10.1016/j.mib.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Muehlbauer SM, Evering TH, Bonuccelli G, Squires RC, Ashton AW, Porcelli SA, et al. Anthrax lethal toxin kills macrophages in a strain-specific manner by apoptosis or caspase-1-mediated necrosis. Cell Cycle. 2007;6(6):758–66. doi: 10.4161/cc.6.6.3991. [DOI] [PubMed] [Google Scholar]
- Ouaaz F, Arron J, Zheng Y, Choi Y, Beg AA. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity. 2002;16(2):257–70. doi: 10.1016/s1074-7613(02)00272-8. [DOI] [PubMed] [Google Scholar]
- Paccani SR, Tonello F, Ghittoni R, Natale M, Muraro L, D’Elios MM, et al. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J Exp Med. 2005;201(3):325–31. doi: 10.1084/jem.20041557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297(5589):2048–51. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- Pellizzari R, Guidi-Rontani C, Vitale G, Mock M, Montecucco C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett. 1999;462(1–2):199–204. doi: 10.1016/s0014-5793(99)01502-1. [DOI] [PubMed] [Google Scholar]
- Philpott NJ, Turner AJ, Scopes J, Westby M, Marsh JC, Gordon-Smith EC, et al. The use of 7-amino actinomycin D in identifying apoptosis: simplicity of use and broad spectrum of application compared with other techniques. Blood. 1996;87(6):2244–51. [PubMed] [Google Scholar]
- Pierre P, Turley SJ, Gatti E, Hull M, Meltzer J, Mirza A, et al. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature. 1997;388(6644):787–92. doi: 10.1038/42039. [DOI] [PubMed] [Google Scholar]
- Racoosin EL, Swanson JA. Macrophage colony-stimulating factor (rM-CSF) stimulates pinocytosis in bone marrow-derived macrophages. J Exp Med. 1989;170(5):1635–48. doi: 10.1084/jem.170.5.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68(2):320–44. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23(5):479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci U S A. 2003;100(9):5170–4. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires RC, Muehlbauer SM, Brojatsch J. Proteasomes control caspase-1 activation in anthrax lethal toxin-mediated cell killing. J Biol Chem. 2007;282(47):34260–7. doi: 10.1074/jbc.M705687200. [DOI] [PubMed] [Google Scholar]
- Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, et al. Critical Role for NALP3/CIAS1/Cryopyrin in Innate and Adaptive Immunity through Its Regulation of Caspase-1. Immunity. 2006;24(3):317–27. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Tournier JN, Quesnel-Hellmann A, Mathieu J, Montecucco C, Tang WJ, Mock M, et al. Anthrax edema toxin cooperates with lethal toxin to impair cytokine secretion during infection of dendritic cells. J Immunol. 2005;174(8):4934–41. doi: 10.4049/jimmunol.174.8.4934. [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- Tureci O, Bian H, Nestle FO, Raddrizzani L, Rosinski JA, Tassis A, et al. Cascades of transcriptional induction during dendritic cell maturation revealed by genome-wide expression analysis. Faseb J. 2003;17(8):836–47. doi: 10.1096/fj.02-0724com. [DOI] [PubMed] [Google Scholar]
- Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J. 2000;352(Pt 3):739–45. [PMC free article] [PubMed] [Google Scholar]
- Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem Biophys Res Commun. 1998;248(3):706–11. doi: 10.1006/bbrc.1998.9040. [DOI] [PubMed] [Google Scholar]
- West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, et al. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science. 2004;305(5687):1153–7. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- Wickliffe KE, Leppla SH, Moayeri M. Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell Microbiol. 2007 doi: 10.1111/j.1462-5822.2007.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong BR, Josien R, Lee SY, Sauter B, Li HL, Steinman RM, Choi Y. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med. 1997;186(12):2075–80. doi: 10.1084/jem.186.12.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A) DCs from B6 mice were treated for 24 h with 500ng/ml of PA+ 20ng/ml LF or LF E687C mutant (LFm). Control cells were left untreated. Surface expression of CD86, MHCII and CD80 was monitored by FACS and represented on the population of CD11c+ cells. B) B6 derived DCs were treated as in A) and LPS (100ng/ml) was added for the last 8 h of toxin incubation to induce maturation. Surface expression of CD86, MHCII and CD80 was analyzed by FACS and represented on CD11c+ gated cells.
B6 immature DCs were treated for 24 h with 500ng/ml of PA + 1 or 20 ng/ml of LF or LF E687C mutant (LFm). Control cells were left untreated. LPS 100ng/ml was added during the last 8 h of incubation when indicated (+LPS). The concentration of several cytokines measured by Luminex system in the culture supernatants is represented as mean±SD.
A) DCs obtained from B6 mice were incubated with different combinations of anthrax toxins and their mutant forms during 24 h. PA (wild type or mutant) was added at 500ng/ml, and LF (wild type or mutant) was added at 20ng/ml. LPS (100ng/ml) was added 8 h before the end of the incubation to the indicated cells (iDC+LPS). Cells were collected and analyzed by flow cytometry after staining with anti Annexin V-FITC and 7AAD. Here is represented the double staining on gated CD11C+ cells treated with medium alone (control), PA alone, LF alone, PA+LF wild type, PA+LFm (inactive mutant E687C) and PASNKE+LF (a furin resistant mutant form of PA). Numbers in each gate correspond to the % of cells within the CD11c+ population. Note the higher 7AAD and Annexin V staining on PA+LF treated cells subsequently treated with LPS (red square). Data from one representative experiment (n=3). B) DCs obtained from B6 mice were incubated during 24 h with PA+LF (500ng/ml and 20ng/ml, respectively) or left untreated. LPS (100ng/ml) was added 8 h before the end of the incubation to the indicated cells (iDC+LPS). Represented the increase of mean fluorescence intensity of 7AAD and Annexin V stainings (fluorescence on PA+LF treated cells – fluorescence in control cells) on the population of CD11c+ surviving cells (7AAD negative) after treatment. LPS treatment causes a significant increase in fluorescence of both markers (p= 0.004 for 7AAD; p>0.0001 for Annexin V). Represented as mean±SEM from data from 6 independent experiments done in triplicate.
B6 derived DCs were matured over-night with LPS or left immature. Cells were incubated with 500ng/ml of trypsin-nicked PA (Abrami et al., 2003) and 200ng/ml LF, for 1 h on ice. Then washed with toxin free media and transferred at 37°C for different periods of time (in min.). 40 μg of total cell extracts were analyzed by western blotting to detect LF processed MEK1 (anti Nterm antibody), total MEK1 (anti Cterm antibody) or LF.
RNA was extracted from B6 derived DCs treated with 100ng/ml LPS for 3 or 8 h or left untreated (No LPS). The levels of Nalp3 and ASC mRNA were quantified by Taqman real-time RT-PCR and normalized to the values obtained for 4 different housekeeping genes (see materials and methods). Values were normalized to the levels of mRNA on control cells (No LPS) and represented as fold change as mean±SD. Data from one representative experiment run in triplicates (n=3).
DCs were obtained from ASC−/− mice in B6 background (Sutterwala et al., 2006) and their parental ASC+/+ B6 mice. A) Immature DCs were incubated with media (Ctrl) or 500ng/ml PA and 20ng/ml LF for 2, 4, 8, 16, 24 or 48 hours. B) DCs were treated with 100ng/ml LPS for 8 h to induce maturation, and incubated with toxins as in A). C) DCs were matured by 8 h LPS treatment (mDC) or left immature (iDC) and subsequently treated for 24 h with different concentrations of LF (0.1, 1, 20, 200 and 2000 ng/ml) and PA (500ng/ml or 2000ng/ml for the higher concentration of LF). The percentage of 7AAD+ cells on the CD11c+ gated population is represented as mean±SEM. Data from 4 independent experiments done in triplicates.