

Abstract
Angiotensin II (Ang II) induces reactive oxygen species (ROS) production by human vascular smooth muscle cells (hVSMCs). ROS have been implicated in the development of both acute stress-induced premature senescence (SIPS) and chronic replicative senescence. Global oxidative DNA damage triggers SIPS and telomere DNA damage accelerates replicative senescence, both mediated via p53. This study tests the hypothesis that DNA is an important target for Ang II–induced ROS leading to senescence via telomere-dependent and independent pathways. DNA damage was quantified using the Comet assay, telomere DNA length by Southern blotting and hVSMC senescence by senescence-associated β-galactosidase staining. Exposure to Ang II increased DNA damage in hVSMCs within 4 hours. Inhibition by an AT1 receptor antagonist (losartan metabolite: E3174) or catalase, confirmed that Ang II–induced DNA damage was AT1 receptor-mediated, via the induction of ROS. Acute exposure to Ang II resulted in SIPS within 24 hours that was prevented by coincubation with E3174 or catalase. SIPS was associated with increased p53 expression but was not dependent on telomere attrition because overexpression of human telomerase did not prevent Ang II–induced SIPS. Exposure to Ang II over several population doublings accelerated the rate of telomere attrition (by >2-fold) and induced premature replicative senescence of hVSMCs—an effect that was also attenuated by E3174 or catalase. These data demonstrate that Ang II–induced ROS-mediated DNA damage results in accelerated biological aging of hVSMCs via 2 mechanisms: (1) Acute SIPS, which is telomere independent, and (2) accelerated replicative senescence which is associated with accelerated telomere attrition.
Keywords: vascular smooth muscle cell, senescence, angiotensin II, reactive oxygen species, DNA damage
Angiotensin II (Ang II) has been widely implicated in the pathogenesis of cardiovascular disease and the vascular smooth muscle cell (VSMCs) is an important target for its actions. Numerous cell signaling pathways have been implicated, several of which involve reactive oxygen species (ROS) generation via NAD(P)H oxidase activity.1-4 Many previous studies have shown that Ang II stimulates the intracellular accumulation of ROS, particularly H2O2, in VSMCs which play a critical role in cell signaling.2,5,6 In the vascular system, the cell signaling functions of H2O2 account for the pathological effects on growth, angiogenesis, hypertrophy and inflammation of Ang II.3
DNA is particularly susceptible to damage by ROS but surprisingly this has received little attention in the context of cardiovascular disease. In cultured cells, critical DNA damage induces rapid “stress induced premature senescence” (SIPS).7 In addition to SIPS, vascular cells may also undergo premature replicative senescence via accelerated attrition of telomeres.8,9 The telomeres are chromosomal protective cap structures, characterized by long stretches of DNA repeat sequences (TTAGGG) that shorten with replication and cell aging. The high GGG content renders telomeres particularly susceptible to oxidative damage by ROS and this is an important cause of accelerated telomere attrition.10 Cells with critically shortened or damaged telomeres enter a state of “replicative senescence”.11,12 Unlike SIPS which is rapid (within days), primary cultures of human cells undergo replicative senescence over several weeks or months in culture, depending on their original telomere length, the magnitude of stress, and their rate of proliferation. The states of SIPS and replicative senescence share common molecular signals which usually involve stabilization of p53, increased expression of p21, and subsequent hypophosphorylation of Rb protein which mediates growth arrest.7 The cyclin-dependent kinase inhibitor, p16, may also play a role in mediating growth arrest by some stressors.7 It is likely that both SIPS and replicative senescence occur simultaneously in populations of vascular cells and their progenitors and that this contributes to manifestations of disease in aging vascular tissues.13-16
The senescent cell phenotype and shortened telomeres have both been strongly implicated in the pathogenesis of vascular disease.13,16 Senescent cells are commonly observed in the vascular wall at sites prone to atheroma in both humans and animal models of disease.8,17-20 Moreover, VSMCs within atherosclerotic plaques show morphological and biochemical features of senescence,8,17 oxidative DNA damage, shorter telomeres, and activation of the p53-p21 axis9 suggesting that human cells undergo accelerated replicative senescence mediated by ROS. In addition, recent studies have emphasized a strong link between shortened telomeres and an increased risk of premature cardiovascular morbidity and mortality.21,22 Consequently, understanding the key mediators and mechanisms for vascular cell senescence has become an area of great interest.
In this study, we examined the hypothesis that Ang II induces human VSMC senescence via ROS-mediated DNA damage. We also hypothesized that senescence would occur via 2 DNA damage-dependent pathways, ie, SIPS—a telomere independent pathway, and accelerated replicative senescence, associated with accelerated telomere attrition.
Materials and Methods
hVSMCs were obtained from explants of human saphenous vein23 and hTERT-SMC generated from normal hVSMCs as described.24,25 The effect of Ang II on ROS production, DNA damage, telomere attrition, and senescence was evaluated by supplementing culture medium with Ang II. For chronic exposure experiments, ie, up to 30 days, the medium overlying cells was supplemented with Ang II on alternate days.
Superoxide production was determined by lucigenin chemiluminescence1 and nuclear DNA damage was measured using Comet assay.26 Telomere DNA length was estimated by measuring the Terminal Restriction Fragment (TRF) length via Southern blotting.27 Senescence-associated β-galactosidase (SA β-gal) activity was measured as originally described.28 An expanded Materials and Methods section is available in the supplemental materials (available online at http://circres.ahajournals.org).
Results
ROS Production in Human Saphenous Vein Smooth Muscle Cells
Ang II–induced ROS generation via NADPH oxidase activity has been reported in cultured VSMCs,1,2,29 although not for cells derived from human saphenous veins. In lysates of these cells, NADPH induced a 3- to 4-fold increase in lucigenin chemiluminescence (see supplemental Figure). In contrast, NADH was relatively ineffective. Similarly, cyclooxygenase and xanthine oxidoreductase were not significant sources of ROS under these conditions, because neither antimycin A/succinate nor xanthine increased chemiluminescence (see supplemental Figure). However, ROS production was significantly inhibited by prior incubation with diphenylene iodonium (DPI) (flavoprotein inhibitor; 83% inhibition) and apocynin (specific inhibitor of NADPH oxidase sub-unit assembly; 25%) confirming involvement of NADPH oxidase. Incubation with tiron (superoxide scavenger; 73% inhibition) (see supplemental Table) confirmed the detection of superoxide. Mechanisms involving mitochondrial electron transport and cyclooxygenase did not contribute to ROS generation by NADPH as neither rotenone (2μmol/L) nor indomethacin (20μmol/L) significantly decreased chemiluminescence (see supplemental Table). Together these data demonstrate for the first time, that NADPH oxidase–mediated superoxide production is detectable in saphenous vein hVSMCs.
Effect of Ang II on Superoxide Production by hVSMCs
Serum-deprived cells were incubated with Ang II for up to 24 hours before measurement of superoxide production. Ang II (10−7mol/L) greatly potentiated NADPH-stimulated superoxide production in VSMCs (Figure 1A). Quantitation of the area under the curve showed that the Ang II effect was rapid (within 1 hour), consistent with previous reports using human arterial VSMCs,29 and persisted for at least 24 hours (Figure 1B).
Figure 1.

Ang II induces NADPH-mediated superoxide generation in hVSMCs. A, NADPH-mediated superoxide production by Ang II (10−7 mol/L) treated cells (24 hours) relative to control cells. Superoxide generation determined at 40 second intervals was plotted over a total period of 40 minutes. Data are representative of 4 separate experiments. B, Time-dependent increase in superoxide production in Ang II–treated cells. Bars represent mean+SEM; n=5 (P<0.01 for all time points compared with control). AUC indicates total area under curve.
Real-time determination of lucigenin chemiluminescence demonstrated that Ang II–induced ROS production by hVSMCs was mediated via the AT1 receptor, as effects were blocked by preincubation with the Losartan metabolite, E3174 (Merck; 10−5 mol/L), a potent, selective, and specific AT1 receptor antagonist (Figure 2A). In cell lysates derived from Ang II–treated cells, addition of tiron (10mmol/L), a relatively selective and nontoxic superoxide scavenger, inhibited chemiluminescence confirming that superoxide was being detected (Figure 2B). Both apocynin and DPI completely blocked the Ang II–dependent chemiluminescence, strongly suggesting superoxide was derived from NADPH-oxidase activity (Figure 2C and 2D). Although DPI is an effective, yet nonspecific flavoprotein inhibitor, apocynin specifically inhibits NADPH oxidase subunit assembly in cells. Rotenone did not block superoxide production in lysates derived from Ang II–treated VSMCs, confirming the lack of mitochondrial involvement (Figure 2E).
Figure 2.

Ang II induces superoxide production in hVSMCs via the AT1 receptor and NADPH oxidase. VSMCs were preincubated with Ang II for 1 hour then lysates used for measurement of superoxide by real-time lucigenin chemiluminescence. This was measured at 40-second intervals and plotted over 5 minutes. In A, cells were preincubated with E3174 before Ang II. In B through E, inhibitors were added to VSMC lysates. For all panels: black indicates control; red, Ang II; green, inhibitor alone; purple, Ang II+inhibitor. E3174 (A, 10−5 mol/L), tiron (B, 10×10−3 mol/L), apocynin (C, 100×10−6 mol/L) and DPI (D, 50×10−6 mol/L) inhibited superoxide production in cells treated with Ang II (10−7 mol/L) for 60 minutes. Rotenone (E, 2×10−6 mol/L) did not affect superoxide production. Data are representative of 3 to 5 experiments.
Ang II Induces DNA Damage in hVSMCs via the AT1 Receptor
The “Comet assay”26 has been used extensively as a very sensitive measure of DNA damage in cells and was used to investigate nuclear DNA damage induced by Ang II (Figure 3A through 3C). The culture medium overlaying hVSMCs was supplemented with Ang II for 4 or 24 hours before measurement of damage. Ang II induced DNA damage in hVSMCs which was significant by 24 hours (Figure 3D). Allowing cells to take up catalase by preincubation with the enzyme (300 U/mL for 3 hours at 37°C) inhibited Ang II–induced DNA damage suggesting that damage was ROS-mediated and more specifically required hydrogen peroxide generation. This result does not imply necessarily that hydrogen peroxide persists for 24 hours, because downstream mediators may actually mediate damage. Furthermore, endogenous peroxidases and DNA repair may also explain the delayed nature of DNA damage detection. Nevertheless, however, the catalase data show that the production of hydrogen peroxide was necessary for the DNA damage observed at 24 hours after Ang II treatment of hVSMCs. Moreover, the DNA damage was AT1 receptor-mediated as it was completely prevented by coincubation with E3174 (Figure 3E). Together these data confirm that Ang II, via the AT1 receptor and hydrogen peroxide production results in DNA damage in hVSMCs. Because ROS production and DNA damage have the capacity to profoundly influence cell viability, we next investigated whether Ang II could induce either SIPS and/or accelerated replicative senescence via telomere attrition.
Figure 3.

Ang II–induced DNA damage in hVSMCs. A, Image of a cell nucleoid using the Comet assay. Limited migration of DNA toward the anode indicates the cell has low level DNA damage. B, Nucleoid showing more significant migration of DNA into the tail of the Comet indicates greater DNA damage. C, DNA breaks within long loops of DNA allow them to migrate toward the anode, giving rise to a Comet-like appearance in heavily damaged nucleoids. D, DNA damage was measured using the Comet assay in hVSMCs treated with Ang II (10−6 mol/L)±catalase preincubation (300 Units/mL; n=3). Bars represent mean+SEM. (*P<0.05 vs control). E, Ang II induces DNA damage in hVSMCs via the AT1 receptor. Cells were treated with Ang II (10−6 mol/L)±E3174 (10−5 mol/L) for 20 hours. Bars represent mean+SEM. (*P<0.01 and **P<0.001 vs control; ***P<0.001 vs Ang II–treated cells; n=3).
Ang II Induces SIPS in Cultured hVSMCs
SIPS has been demonstrated in several cell types, including hVSMCs.30 SA-β-gal activity is widely used as a marker of senescence of cells in culture and in tissues.28 In this study, VSMCs were treated with Ang II for 24 hours and stained for SA-β-gal activity 24 hours after return to normal media. Ang II caused a dose-dependent increase in senescence, maximal at 10−8 mol/L (Figure 4A). Coincubation with either E3174 or catalase prevented Ang II–induced SIPS, suggesting this was AT1 receptor-mediated and dependent on hydrogen peroxide (Figure 4B). This rapid induction of senescence is characteristic of SIPS and a ≈1.5-fold increase in p53 and in p21 was consistently observed (Figure 4C) suggesting that cell cycle arrest and senescence occurred in response to DNA damage. Using Western blotting we were unable to detect significant changes in expression of the CDK inhibitor, p16INK4a (data not shown).
Figure 4.

Ang II accelerates SIPS in human VSMCs. A, hVSMCs were treated with Ang II for 24 hours, then replated at 1×105 cells per well for 24 hours, before staining for SA-β-gal. Data are shown as mean+SEM; n=5 (*P<0.05 and #P<0.01 compared with control). B, Ang II–induced hVSMC SIPS is dependent on AT1 receptor and ROS generation. hVSMCs were pretreated with E3174 (10−5 mol/L for 1 hour) or catalase (300Units/mL for 3 hours) before induction of senescence with 10−8 mol/L Ang II for 24 hours. Bars represent mean+SEM; n=5. (#P<0.05 vs control; *P<0.05 and **P<0.01 vs Ang II alone). C, Ang II induces expression of p53 and p21 within 24 hours. VSMCs were treated with 10−8 mol/L Ang II for 24 hours and p53 and p21 detected by Western blotting, compared with β-actin and α-tubulin loading controls. Bars represent mean+SEM of 2 experiments. D, Confirmation of TERT expression in hTERT-SMCs. Whole cell lysates from hTERT-SMCs and hVSMCs were blotted for hTERT using α-tubulin loading control. HeLa cell extract was used as a positive control. E, Ang II dose-dependently increases hTERT-SMC senescence. Quiescent cells were treated with Ang II for 24 hours and reseeded at 5×104 cells per well (12 well) for 24 hours, before staining for SA-β-gal. Bars represent mean+SEM; n=3 (*P<0.05 compared with control).
Ang II–Induced SIPS Occurs via Telomere-Independent Mechanism
We repeated the acute exposure to Ang II using hVSMCs transfected with TERT to preserve telomeres.24 Overexpression of TERT in these cells was confirmed by Western blotting (Figure 4D). Importantly, Ang II–induced SIPS was not prevented by TERT-overexpression, suggesting that SIPS was not mediated by telomere damage and was thus telomere-independent (Figure 4E).
Ang II Accelerates Telomere Loss in hVSMCs
Telomere attrition has been described as a consequence of oxidative DNA damage10 and promotes premature replicative senescence. Not all cells exposed to Ang II undergo SIPS. As the telomere is a vulnerable target for ROS10 we considered whether continuing exposure to Ang II would result in accelerated telomere attrition and premature replicative senescence. Using Southern blotting,27 we showed that Ang II markedly accelerated the rate of telomere loss (>2-fold versus control) in a dose-dependent manner (Figure 5A). Coincubation with E3174 markedly attenuated Ang II–induced telomere attrition, confirming this effect was mediated via the AT1 receptor (Figure 5B).
Figure 5.

Ang II causes telomere attrition in hVSMCs. A, Dose-response of Ang II on telomere length in hVSMCs, for 24 hours. Mean percentage telomere changes per PD with SEM shown; n=5. (P<0.001 for all doses compared with control). B, Ang II treatment accelerates hVSMC telomere attrition via the AT1 receptor. Telomere length was measured in hVSMCs grown continuously in media supplemented daily for 7 days with Ang II (10−6 mol/L)±E3174 (10−5 mol/L). *P<0.05 vs all other treatments; n=9; n=3 for E3174 alone.
Telomerase activity preserves telomere DNA length in some cells, and inhibition of telomerase has been proposed as a mechanism for telomere loss in response to Ang II–derived ROS.31 Using a sensitive PCR ELISA we were unable to detect telomerase activity in cultured hVSMCs, either before or after exposure to Ang II, thereby discounting modulation of telomerase activity as a primary mechanism for Ang II–induced telomere loss in adult hVSMC (data not shown). These data suggest that Ang II promotes accelerated attrition of telomere DNA in adult hVSMCs via ROS-mediated DNA damage, rather than by inhibition of telomerase activity.
Ang II Induces Replicative Senescence in hVSMCs
Critical loss of telomere DNA has been associated with reduced cellular proliferation and premature cell senescence.27 SA-β-gal activity was used to enumerate senescent hVSMCs after exposure to Ang II over a period of 30 days in culture. Ang II induced a dose-dependent increase in cell senescence with a maximal effect at 10−8 mol/L (Figure 6). Taken together with the data on telomere length, this finding suggests that over time in culture, Ang II accelerated the loss of telomere DNA and this was associated with premature replicative senescence.
Figure 6.
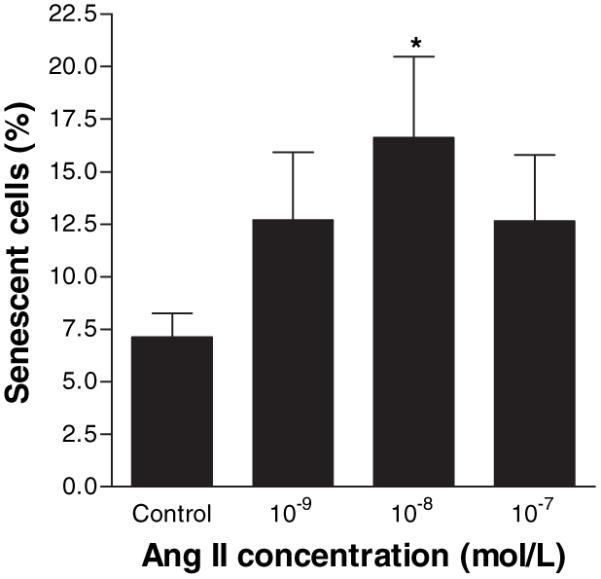
Ang II accelerates replicative senescence in human VSMCs. VSMCs were treated on alternate days for 30 days with Ang II, then replated at 1×105 cells per well for 24 hours. Cells were then stained for SA-β-gal activity. Bars represent mean+SEM, n=6. *P<0.05 compared with control.
Discussion
The present study demonstrates that Ang II–induced ROS production leads to DNA damage and the accelerated biological aging of hVSMCs via 2 mechanisms: (1) SIPS, which is telomere independent, and (2) accelerated replicative senescence which is associated with telomere attrition.
A common paradigm for the induction of cell senescence is loss of telomere integrity or critical shortening of telomeres, resulting in a “DNA damage response” and cell cycle arrest via activation of CDKIs.32 Telomere shortening occurs progressively with every cell division due the “end-replication problem”, eventually leading to critical shortening of telomeres which triggers replicative senescence. This process is accelerated by exposure of cells to oxidant stress and DNA damage.10,33 The telomere region of DNA is especially vulnerable to oxidative damage.34,35 Moreover, the repair of DNA within the telomere is inefficient relative to the rest of the genome.36 We show that the average loss of TRF length in hVSMCs in culture is ≈1.0% per population doubling, consistent with our previous data on human vascular endothelial cells27 and studies using cultured VSMCs.24,37 We also show that treatment with Ang II markedly accelerated this loss by 2.5-fold, thereby predisposing hVSMCs to accelerated replicative senescence.
Cell senescence may also arise more rapidly, in a manner independent of the slower, replication-dependent attrition of telomere DNA. This mechanism, termed “premature senescence” or SIPS, involves activation of identical CDKIs to those activated in replicative senescence.7 Induction of SIPS typically arises from treatment of cells with subcytotoxic levels of agents that generate ROS, eg, UV-irradiation, peroxides.38,39 To date, 2 possible mechanisms have been proposed to account for the induction of SIPS by ROS: (1) oxidant-induced p38 phosphorylation,40 and (2) critical but sublethal DNA damage with transient activation of p53 and subsequent expression of p2141; although the dependence of p21 expression on p53 has recently been disputed.42 We show that Ang II rapidly induces oxidative DNA damage in hVSMCs, associated with an increase in p53 and rapid onset of senescence. These observations demonstrate the capacity of Ang II to induce SIPS via a mechanism dependent on the associated DNA damage. Importantly, our data using hTERT-expressing VSMCs confirm that SIPS is independent of telomere attrition—as similarly observed in other cell types.43,44
Our data are consistent with the hypothesis that the fate of the hVSMCs exposed to Ang II is dependent on the magnitude of the resulting DNA damage. We propose that severe and lethal DNA damage would result in apoptosis, consistent with reports of Ang II–induced apoptosis in some cell types.45-48 Sublethal but critical levels of DNA damage would rapidly induce p53-dependent SIPS. Lower levels of DNA damage sustained over several population doublings would target and accelerate the gradual erosion of telomeres, promoting premature replicative senescence. It is likely that all 3 responses would occur in the same cell population, the individual cells’ response to Ang II depending on the biological age of the cell, their individual sensitivity to Ang II, and the efficiency of their oxidant defense and DNA repair mechanisms. Further work is required to evaluate this hypothesis.
The development of vascular cell senescence is increasingly recognized as an important mechanism in the pathogenesis of vascular disease.14,49 Senescent vascular cells are not benign and may undergo a phenotypic shift to a more inflammatory and atherogenic phenotype.8,50 Cells with a senescence phenotype have been identified in human and experimental animal arteries at sites prone to atherosclerosis.8,17-20 In rabbits, denuded arteries show evidence of extensive SA-β-gal staining.20 Similar SA-β-gal staining has been detected in diseased coronary arteries but not in healthier internal mammary arteries in humans.8 It has also been shown that the senescent cells persist in the vascular wall14 and so may inhibit natural repair by inhibiting recruitment of progenitor cells. The importance of vascular cell senescence in determining the function of an aging vascular system has stimulated interest in identifying the molecular mediators and pathways for its induction. Our data strongly support the hypothesis that Ang II may play a key role in the development of human vascular cell senescence.
Telomere length is also used as a marker of vascular cell aging in vivo. When senescence is observed in the vascular wall, those cells often show evidence of telomere attrition. For example, VSMCs with shortened telomeres have been identified in established atherosclerotic plaques51 and at sites of enhanced vascular wall stress in humans, ie, those sites predisposed to atheroma.52 Evidence for the clinical significance of shortened telomeres is now emerging albeit fuelled by literature using the leukocyte telomere as a surrogate marker for vascular wall aging.49 Cawthon and colleagues showed strong associations between reduced telomere DNA length in adults and premature mortality from cardiovascular disease.21 Furthermore, people with shorter telomeres have a 3-fold higher risk of premature MI22 and patients with coronary artery disease have shorter leukocyte telomeres compared with disease-free controls.22,53 Moreover, a recent report links shorter telomere DNA with CVD risk factors and subsequent risk of MI or stroke.54 The link between oxidative damage, telomeres, and vascular senescence has been further strengthened by studies detailing telomere-dependent senescence of VSMCs within human atherosclerotic plaques via a mechanism dependent on oxidative DNA damage and cell cycle arrest.9 Our data support and extend this concept by suggesting a pathway from oxidative DNA damage through to senescence, with the hypothesis that the magnitude of DNA damage determines whether cells enter senescence through a telomere-independent SIPS pathway, or a replication and telomere-dependent pathway.
Our data demonstrate Ang II–dependent induction of senescence in hVSMCs via the induction of ROS from NADPH oxidase and subsequent DNA damage. These findings are supported by a recent report suggesting that Ang II induced premature senescence in human aortic smooth muscle cells over a period of 3 days.30 These authors suggested that telomere attrition was not involved because telomere length did not change over 3 days in culture. Our current observations, using hTERT-expressing cells provide much more conclusive evidence that Ang II induces SIPS via a telomere-independent pathway which is ROS and DNA damage dependent. Both studies support the importance of the p53-p21 pathway in the development of Ang II–induced SIPS. Consistent with our findings, Ang II was recently reported to accelerate endothelial progenitor cell senescence via ROS generation and downregulation of telomerase.31 In contrast, our data suggest that Ang II–induced DNA damage, rather than its impaired repair, is the primary mechanism for accelerated telomere loss in adult human cells. This is not surprising mindful of the much greater telomerase activity in progenitor cells. Nevertheless, the senescence of both resident mature vascular cells and progenitor cells may play a role in the biological aging and associated pathology of vascular tissues.
Our hypothesis that Ang II may play a direct role in accelerating vascular aging processes is also supported by reports showing that treatment of experimental animals with drugs that inhibit the renin-angiotensin system, markedly increases survival and prevents many of the pathological changes associated with cardiovascular aging.55 Our studies extend these observations and provide the first direct evidence that Ang II acting via its AT1 receptor can induce DNA damage and senescence, either with or without accelerated telomere attrition, in adult hVSMCs.
Our study has some limitations. Firstly, our model uses human saphenous vein SMCs. Although ROS production by Ang II has been studied in both human and murine VSMCs taken from different vascular sites,1,2,56 ours is the first data on induction of DNA damage and senescence by Ang II using saphenous vein cells. However, it is likely that these data are applicable to other sources of VSMCs that respond to Ang II. Secondly, this study uses cultured cells in vitro to investigate mechanisms of senescence rather than a direct examination of senescence in vivo. However, the evidence cited above suggests that the mechanisms of senescence are likely to be similar in both settings. Finally, Dikalov et al57 recently critically reviewed the sensitivity and applicability of lucigenin chemiluminescence in the detection of ROS in vascular cells. Potential artifacts arising from lucigenin auto-oxidation were thought, on balance of the evidence in the literature, to be insignificant. Although this remains a possibility, in many cases lucigenin detection of ROS in vascular cells has been corroborated by alternate assays and our data are consistent with these findings.
In conclusion, our data show that Ang II causes DNA damage in hVSMCs via NADPH oxidase-derived ROS and that this results in SIPS or replicative senescence. These findings suggest novel mechanisms to directly implicate Ang II in the pathogenesis of human vascular cell aging and vascular disease.
Supplementary Material
Acknowledgments
The authors wish to thank Hash Patel for his help.
Sources of Funding
This project was supported by a grant from the British Heart Foundation PG/2001110. The authors are grateful to Merck & Co Inc for the gift of EXP3174.
Footnotes
Disclosures
Bryan Williams has received independent investigator-led grant support and honoraria from Merck for lectures and consultancy.
References
- 1.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 2.Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- 3.Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- 4.Mehta PK, Griendling KK. Angiotensin II Cell Signaling: Physiological and Pathological Effects in the Cardiovascular System. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 5.Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022–15029. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- 6.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 7.Toussaint O, Medrano EE, von Zglinicki T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp Gerontol. 2000;35:927–945. doi: 10.1016/s0531-5565(00)00180-7. [DOI] [PubMed] [Google Scholar]
- 8.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–1544. doi: 10.1161/01.cir.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- 9.Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, Bennett M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006;99:156–164. doi: 10.1161/01.RES.0000233315.38086.bc. [DOI] [PubMed] [Google Scholar]
- 10.von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- 11.Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, Greider CW, Harley CB. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A. 1992;89:10114–10118. doi: 10.1073/pnas.89.21.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 13.Minamino T, Miyauchi H, Yoshida T, Tateno K, Kunieda T, Komuro I. Vascular cell senescence and vascular aging. J Mol Cell Cardiol. 2004;36:175–183. doi: 10.1016/j.yjmcc.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- 15.Gorenne I, Kavurma M, Scott S, Bennett M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc Res. 2006;72:9–17. doi: 10.1016/j.cardiores.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Ballard VL, Edelberg JM. Stem cells and the regeneration of the aging cardiovascular system. Circ Res. 2007;100:1116–1127. doi: 10.1161/01.RES.0000261964.19115.e3. [DOI] [PubMed] [Google Scholar]
- 17.Ross R, Wight TN, Strandness E, Thiele B. Human atherosclerosis. I. Cell constitution and characteristics of advanced lesions of the superficial femoral artery. Am J Pathol. 1984;114:79–93. [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett MR, Macdonald K, Chan SW, Boyle JJ, Weissberg PL. Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ Res. 1998;82:704–712. doi: 10.1161/01.res.82.6.704. [DOI] [PubMed] [Google Scholar]
- 19.Liao S, Curci JA, Kelley BJ, Sicard GA, Thompson RW. Accelerated replicative senescence of medial smooth muscle cells derived from abdominal aortic aneurysms compared to the adjacent inferior mesenteric artery. J Surg Res. 2000;92:85–95. doi: 10.1006/jsre.2000.5878. [DOI] [PubMed] [Google Scholar]
- 20.Fenton M, Barker S, Kurz DJ, Erusalimsky JD. Cellular senescence after single and repeated balloon catheter denudations of rabbit carotid arteries. Arterioscler Thromb Vasc Biol. 2001;21:220–226. doi: 10.1161/01.atv.21.2.220. [DOI] [PubMed] [Google Scholar]
- 21.Cawthon RM, Smith KR, O’Brien E, Sivatchenko A, Kerber RA. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet. 2003;361:393–395. doi: 10.1016/S0140-6736(03)12384-7. [DOI] [PubMed] [Google Scholar]
- 22.Brouilette S, Singh RK, Thompson JR, Goodall AH, Samani NJ. White cell telomere length and risk of premature myocardial infarction. Arterioscler Thromb Vasc Biol. 2003;23:842–846. doi: 10.1161/01.ATV.0000067426.96344.32. [DOI] [PubMed] [Google Scholar]
- 23.O’Callaghan CJ, Williams B. Mechanical strain-induced extracellular matrix production by human vascular smooth muscle cells: role of TGF-beta(1) Hypertension. 2000;36:319–324. doi: 10.1161/01.hyp.36.3.319. [DOI] [PubMed] [Google Scholar]
- 24.McKee JA, Banik SS, Boyer MJ, Hamad NM, Lawson JH, Niklason LE, Counter CM. Human arteries engineered in vitro. EMBO Rep. 2003;4:633–638. doi: 10.1038/sj.embor.embor847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klinger RY, Blum JL, Hearn B, Lebow B, Niklason LE. Relevance and safety of telomerase for human tissue engineering. Proc Natl Acad Sci U S A. 2006;103:2500–2505. doi: 10.1073/pnas.0508184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins AR, Dusinska M, Gedik CM, Stetina R. Oxidative damage to DNA: do we have a reliable biomarker? Environ Health Perspect. 1996;104(Suppl 3):465–469. doi: 10.1289/ehp.96104s3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hastings R, Qureshi M, Verma R, Lacy PS, Williams B. Telomere attrition and accumulation of senescent cells in cultured human endothelial cells. Cell Prolif. 2004;37:317–324. doi: 10.1111/j.1365-2184.2004.00315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res. 2002;90:1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 30.Kunieda T, Minamino T, Nishi J, Tateno K, Oyama T, Katsuno T, Miyauchi H, Orimo M, Okada S, Takamura M, Nagai T, Kaneko S, Komuro I. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation. 2006;114:953–960. doi: 10.1161/CIRCULATIONAHA.106.626606. [DOI] [PubMed] [Google Scholar]
- 31.Imanishi T, Hano T, Nishio I. Angiotensin II accelerates endothelial progenitor cell senescence through induction of oxidative stress. J Hypertens. 2005;23:97–104. doi: 10.1097/00004872-200501000-00018. [DOI] [PubMed] [Google Scholar]
- 32.Sharpless NE, DePinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest. 2004;113:160–168. doi: 10.1172/JCI20761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurz DJ, Decary S, Hong Y, Trivier E, Akhmedov A, Erusalimsky JD. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J Cell Sci. 2004;117:2417–2426. doi: 10.1242/jcs.01097. [DOI] [PubMed] [Google Scholar]
- 34.Oikawa S, Kawanishi S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 1999;453:365–368. doi: 10.1016/s0014-5793(99)00748-6. [DOI] [PubMed] [Google Scholar]
- 35.Henle ES, Han Z, Tang N, Rai P, Luo Y, Linn S. Sequence-specific DNA cleavage by Fe2+-mediated fenton reactions has possible biological implications. J Biol Chem. 1999;274:962–971. doi: 10.1074/jbc.274.2.962. [DOI] [PubMed] [Google Scholar]
- 36.von Zglinicki T, Pilger R, Sitte N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic Biol Med. 2000;28:64–74. doi: 10.1016/s0891-5849(99)00207-5. [DOI] [PubMed] [Google Scholar]
- 37.Minamino T, Mitsialis SA, Kourembanas S. Hypoxia extends the life span of vascular smooth muscle cells through telomerase activation. Mol Cell Biol. 2001;21:3336–3342. doi: 10.1128/MCB.21.10.3336-3342.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Q, Ames BN. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci U S A. 1994;91:4130–4134. doi: 10.1073/pnas.91.10.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dumont P, Burton M, Chen QM, Gonos ES, Frippiat C, Mazarati JB, Eliaers F, Remacle J, Toussaint O. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic Biol Med. 2000;28:361–373. doi: 10.1016/s0891-5849(99)00249-x. [DOI] [PubMed] [Google Scholar]
- 40.Frippiat C, Dewelle J, Remacle J, Toussaint O. Signal transduction in H2O2-induced senescence-like phenotype in human diploid fibroblasts. Free Radic Biol Med. 2002;33:1334–1346. doi: 10.1016/s0891-5849(02)01044-4. [DOI] [PubMed] [Google Scholar]
- 41.Chen JH, Ozanne SE, Hales CN. Heterogeneity in premature senescence by oxidative stress correlates with differential DNA damage during the cell cycle. DNA Repair (Amst) 2005;4:1140–1148. doi: 10.1016/j.dnarep.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 42.Zdanov S, Debacq-Chainiaux F, Toussaint O. Knocking down p53 with siRNA does not affect the overexpression of p21WAF-1 after exposure of IMR-90 hTERT fibroblasts to a sublethal concentration of H2O2 leading to premature senescence. Ann N Y Acad Sci. 2007;1100:316–322. doi: 10.1196/annals.1395.034. [DOI] [PubMed] [Google Scholar]
- 43.Naka K, Tachibana A, Ikeda K, Motoyama N. Stress-induced premature senescence in hTERT-expressing ataxia telangiectasia fibroblasts. J Biol Chem. 2004;279:2030–2037. doi: 10.1074/jbc.M309457200. [DOI] [PubMed] [Google Scholar]
- 44.Gorbunova V, Seluanov A, Pereira-Smith OM. Expression of human telomerase (hTERT) does not prevent stress-induced senescence in normal human fibroblasts but protects the cells from stress-induced apoptosis and necrosis. J Biol Chem. 2002;277:38540–38549. doi: 10.1074/jbc.M202671200. [DOI] [PubMed] [Google Scholar]
- 45.Yamada T, Horiuchi M, Dzau VJ. Angiotensin II type 2 receptor mediates programmed cell death. Proc Natl Acad Sci U S A. 1996;93:156–160. doi: 10.1073/pnas.93.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamada T, Akishita M, Pollman MJ, Gibbons GH, Dzau VJ, Horiuchi M. Angiotensin II type 2 receptor mediates vascular smooth muscle cell apoptosis and antagonizes angiotensin II type 1 receptor action: an in vitro gene transfer study. Life Sci. 1998;63:PL289–PL295. doi: 10.1016/s0024-3205(98)00448-2. [DOI] [PubMed] [Google Scholar]
- 47.Akishita M, Nagai K, Xi H, Yu W, Sudoh N, Watanabe T, Ohara-Imaizumi M, Nagamatsu S, Kozaki K, Horiuchi M, Toba K. Renin-angiotensin system modulates oxidative stress-induced endothelial cell apoptosis in rats. Hypertension. 2005;45:1188–1193. doi: 10.1161/01.HYP.0000165308.04703.f2. [DOI] [PubMed] [Google Scholar]
- 48.Li Y, Song YH, Mohler J, Delafontaine P. ANG II induces apoptosis of human vascular smooth muscle via extrinsic pathway involving inhibition of Akt phosphorylation and increased FasL expression. Am J Physiol Heart Circ Physiol. 2006;290:H2116–H2123. doi: 10.1152/ajpheart.00551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuster JJ, Andres V. Telomere biology and cardiovascular disease. Circ Res. 2006;99:1167–1180. doi: 10.1161/01.RES.0000251281.00845.18. [DOI] [PubMed] [Google Scholar]
- 50.Minamino T, Yoshida T, Tateno K, Miyauchi H, Zou Y, Toko H, Komuro I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation. 2003;108:2264–2269. doi: 10.1161/01.CIR.0000093274.82929.22. [DOI] [PubMed] [Google Scholar]
- 51.Ogami M, Ikura Y, Ohsawa M, Matsuo T, Kayo S, Yoshimi N, Hai E, Shirai N, Ehara S, Komatsu R, Naruko T, Ueda M. Telomere shortening in human coronary artery diseases. Arterioscler Thromb Vasc Biol. 2004;24:546–550. doi: 10.1161/01.ATV.0000117200.46938.e7. [DOI] [PubMed] [Google Scholar]
- 52.Chang E, Harley CB. Telomere length and replicative aging in human vascular tissues. Proc Natl Acad Sci U S A. 1995;92:11190–11194. doi: 10.1073/pnas.92.24.11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brouilette SW, Moore JS, McMahon AD, Thompson JR, Ford I, Shepherd J, Packard CJ, Samani NJ, West of Scotland Coronary Prevention Study Group Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. Lancet. 2007;369:107–114. doi: 10.1016/S0140-6736(07)60071-3. [DOI] [PubMed] [Google Scholar]
- 54.Fitzpatrick AL, Kronmal RA, Gardner JP, Psaty BM, Jenny NS, Tracy RP, Walston J, Kimura M, Aviv A. Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am J Epidemiol. 2007;165:14–21. doi: 10.1093/aje/kwj346. [DOI] [PubMed] [Google Scholar]
- 55.Basso N, Cini R, Pietrelli A, Ferder L, Terragno NA, Inserra F. Protective effect of long-term angiotensin II inhibition. Amer J Physiol Heart Circ Physiol. 2007;293:1351–1358. doi: 10.1152/ajpheart.00393.2007. [DOI] [PubMed] [Google Scholar]
- 56.Touyz RM, Schiffrin EL. Ang II-stimulated superoxide production is mediated via phospholipase D in human vascular smooth muscle cells. Hypertension. 1999;34:976–982. doi: 10.1161/01.hyp.34.4.976. [DOI] [PubMed] [Google Scholar]
- 57.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.