

Abstract
Photodynamic therapy (PDT) is a procedure that has applications in the selective eradication of neoplasia where sites of malignant lesions are clearly delineated. It is a two-step process whereby cells are first sensitized to light and then photoirradiated. This results in the formation of singlet molecular oxygen and other reactive oxygen species that can cause photodamage at sites where the photosensitizing agent has localized. Photosensitizers found to be clinically useful show affinity for the endoplasmic reticulum (ER), mitochondria, lysosomes, or combinations of these sites. The induction of apoptosis and/or autophagy in photosensitized cells is a common outcome of PDT. This report explores the following issues: (1) Does the induction of autophagy in PDT protocols occur independent of, or in association with, apoptosis? (2) Does the resulting autophagy play a prosurvival or prodeath role? (3) Do photosensitizers damage/inactivate specific proteins that are components of, or that modulate the autophagic process? (4) Can an autophagic response be mounted in cells in which lysosomes are specifically photodamaged? In brief, autophagy can occur independently of apoptosis in PDT protocols, and appears to play a prosurvival role in apoptosis competent cells, and a prodeath role in apoptosis incompetent cells. Mitochondrial and ER-localized sensitizers cause selective photodamage to some (i.e., Bcl-2, Bcl-xL, mTOR) proteins involved in the apoptotic/autophagic process. Finally, an aborted autophagic response occurs in cells with photodamaged lysosomes. Whereas autophagosomes form, digestion of their cargo is compromised because of the absence of functional lysosomes.
Keywords: apoptosis, autophagy, photodynamic therapy, lysosomes, endoplasmic reticulum, mitochondria
Introduction
Photodynamic therapy is a process involving the selective photosensitization of malignant cell types, usually involving porphyrins, porphyrin analogs or other agents with suitable photophysical properties. Subsequent photoirradiation leads to both direct tumor cell kill and the shutdown of the vascular supply.1 The initial step in the photodynamic process involves localization of the photosensitizing agent at subcellular loci. These can be highly specific or quite broad, and have been reported to include the endoplasmic reticulum (ER), mitochondria, Golgi, lysosomes and plasma membrane.2,3 Since these are critical sites for the initiation of cell-death pathways, it is reasonable to assume that photosensitizing agents that show affinity for these targets would therefore mediate photodamage to sites where this would be optimally lethal. The affinity relationships responsible for targeting phenomena have yet to be explored. Most photosensitizers are relatively hydrophobic and will be attracted to membranes. There are some exceptions to this rule, e.g., the sulfonated porphyrins/phthalocyanines and N-aspartyl chlorin e6 (NPe6). Even these molecules, although having substituents that render them water-soluble, bind to membranes because of their hydrophobic ring systems. The nucleus is not a site of localization for photosensitizers currently being used in clinical or preclinical trials. The explanation for this is unknown, but it is likely that potentially mutagenic effects of PDT are thereby minimized. Exposure of a photosensitizing agent to light at a wavelength corresponding to an absorbance band leads to a photophysical reaction resulting in the release of various reactive oxygen species (ROS). The most unstable of these, singlet molecular oxygen, will not migrate more than a fraction of a micron from the site of formation. As a result, photodamage can be quite specific. Other ROS formed downstream from singlet oxygen may, however, migrate longer distances from the site of formation. Notable among these is hydrogen peroxide, a product that can both evoke autophagy4 and be converted to the more reactive hydroxyl radical.5
Because of their high reactivity with virtually all macromolecular constituents, such as lipids, DNA and proteins, ROS represent a source of cytotoxicity and are therefore reduced by cellular detoxifying and antioxidant enzymes or agents. This first line of defense against ROS can be rapidly overwhelmed during PDT, leading to oxidative stress and progressive failure of cellular machinery. In mammalian cells, the autophagy-lysosomal system represents a major proteolytic system for the clearance of ROS-damaged organelles and irreversibly oxidized cytosolic proteins, which are prone to cross-linking and formation of protein aggregates.6 Consistent with this concept, accumulating evidence indicates that ROS can stimulate autophagy with functional consequences varying from cytoprotection to the activation of autophagic cell death.7 Although the molecular mechanisms by which ROS modulate autophagy are not fully understood, the type of ROS, degree of oxidative injury and the molecular targets involved can all affect the outcome of PDT.
The role of autophagy as a factor in PDT-induced cell death is not yet clear, but there are several opportunities for autophagy to play a role in the photodynamic process. The pro-autophagic protein Beclin 1 is known to bind to Bcl-2.8 Bcl-2 is photodamaged by many photosensitizers commonly used in investigational and clinical PDT studies.9–11 Loss of Bcl-2 function could therefore lead to the initiation of autophagy. There may also be an autophagic response to the photodamage of ER and/or mitochondria, in an attempt to recycle injured organelles. In the murine leukemia L1210 cell line, an early wave of autophagy was found to precede apoptosis.12 Prevention of autophagy by silencing the Atg7 gene promoted photokilling at low light doses, reducing the ‘shoulder’ on the dose-response curve. This result is consistent with the proposal that autophagy can serve as a protective mechanism.12,13 A similar finding has been made in the context of ionizing radiation.14
A different scenario emerges when considering the effects of PDT in apoptosis-deficient cells, such as murine embryonic fibroblasts (MEFs) doubly deficient for Bax and Bak. In these cells PDT stimulates a form of caspase-independent cell death, with a necrotic morphotype, for which aberrant autophagy stimulation is required.15 In the DU-145 cell line, the absence of Bax appears to offer protection from PDT-induced cell death, but large numbers of autophagic vacuoles and enhanced processing of the LC3 protein after PDT suggest that there may be a ‘cause-and-effect’ association between autophagy and cell death.16 In the human breast cancer-derived MCF-7 cell line, which is genetically null for procaspase-3 and inefficient in the induction of apoptosis following PDT, autophagy is an early and prominent feature of the post-PDT response, as evidenced by the presence of large quantities of autophagic vacuoles in the cell cytoplasm and extensive accumulation of the processed LC3-II protein.17 Overexpression of procaspase-3 restores the induction of apoptosis by PDT but does not change their overall sensitivity to PDT, as defined by loss of clonogenicity. However, interference with autophagy by chemical inhibitors or by knockdown of the autophagy-associated protein Atg7 makes MCF-7 cells more resistant to PDT, increasing the shoulder on the clonogenic dose-response curve. This suggests that in cells with defective apoptosis, autophagy is crucial for cell sensitivity to PDT18 (Oleinick et al. unpublished results). It therefore appears that during PDT the ultimate role of autophagy may be modulated by the availability of a functional apoptotic machinery.
Characterization of autophagy as a response to photodamage can utilize conventional techniques including electron microscopy for characterization of cytosolic vacuoles, processing of the LC3 protein, clustering of GFP-LC3 chimeric proteins, effects of gene-silencing protocols and use of inhibitors of autophagy. In a summary of methodology appropriate to the study of autophagy it is concluded that “…no individual assay is guaranteed to be the most appropriate one in every situation, and we strongly recommend the use of multiple assays to verify an autophagic response.”19 Many of these procedures have been used in the context of PDT, but there are additional factors to be dealt with. Principal among these is the need to determine that enzymes and pathways involved in the process are not adversely affected by photodamage. Moreover, because lysosomes are both an element of autophagy and a target for some photosensitizing agents,20–22 the potential effects of a significant loss of lysosomes and interruption of autophagosome processing must also be considered.
Results
PDT induction of autophagy
A variety of cell types initiate an autophagic response following photoirradiation in PDT protocols. For example, mTHPC is an amphiphilic photosensitizer that localizes primarily to mitochondria, and to a lesser extent the ER (reviewed in ref. 23). Whereas neither light nor mTHPC alone are cytostatic or cytotoxic to murine hepatoma 1c1c7 cells, combined treatment suppresses 1c1c7 proliferation in a light dose-dependent fashion (Fig. 1A). At longer illumination times the antiproliferative effects of PDT reflect cytotoxicity (Fig. 1B) and correlate with the induction of apoptosis, as monitored by measurements of DEVDase activities (an indicator of procaspase-3/7 activations; Fig. 1C). Photoirradiation of sensitized cultures also resulted in the induction of autophagy, as assessed by monitoring the conversion of LC3-I to LC3-II by western blot (Fig. 1D), or the aggregation of the cytoplasmic chimeric GFP-LC3 protein into punctate spots that can be monitored by fluorescence microscopy (Fig. 1E). Both techniques indicated that the induction of autophagy occurred earlier in 1c1c7 cultures treated with the higher light dose (Fig. 1D and E). At the higher light dose procaspase-3/7 activation preceded autophagosome formation, whereas at the lower light dose procaspase activation paralleled autophagosome formation (compare Fig. 1C and E).
Figure 1.

Kinetics of induction of autophagy and apoptosis in murine hepatoma 1c1c7 cultures following mTHPC-PDT. (A–D) Cultures of 1c1c7 cells were treated with nothing or 500 nM mTHPC (time zero in A and B) for ~20 h prior to being washed and refed with mTHPC-free medium. Some cultures were subsequently photoirradiated (time indicated by arrow) and harvested at varied times after light exposure to determine cell number (A), resistance to trypan blue permeability (B), activation of procaspase-3/7 dependent DEVDase activity (C), or conversion of LC3-I to LC3-II (D). The treatment conditions and times of harvest for the samples depicted in (A–D) are noted in the figure. Data in (A and B) represent means ± SD of analyses of three plates. Data in (C) represent means ± SD of triplicate analyses on single lysates. The results presented in (D) were replicated in a second independent study. (E) Similar treatments were performed with a derivative of the 1c1c7 cell line that stably expresses a chimeric GFP-LC3 fusion protein. These cells were analyzed by fluorescence microscopy for the appearance of autophagosomes (punctate fluorescent spots) following treatment with light, mTHPC or both. White bar in top left panel represents 20 microns.
The induction of autophagy in PDT protocols can occur independent of an apoptotic outcome. The human mammary carcinoma cell line MCF7 does not undergo a prototypical apoptotic response in PDT protocols because of a deletion rendering it caspase-3 null.24 As shown in Figure 2 and ref. 17, large amounts of LC3-II accumulate in a time- and dose-dependent manner in irradiated apoptosis-deficient MCF-7v cells, as well as in an apoptosis-competent MCF7 derivative that has been engineered to express procaspase-3 (i.e., MCF-7c3 cells). Cultured murine embryonic fibroblasts sensitized with the ER photosensitizer hypericin exhibit procaspase-3 activation following irradiation, whereas Bax−/−Bak−/− double knockout MEFs do not (Fig. 3). However, both wild-type and Bax−/−Bak−/− double knockout MEFs exhibit the accumulation of LC3-II following photoirradiation (Fig. 3). Indeed, LC3-II accumulated faster in the apoptosis-deficient double knockout MEFs. Similarly, Bax-deficient human prostate carcinoma DU145 cells undergo autophagy, and not apoptosis, following sensitization with CPO or Pc 4 and subsequent illumination.16,25
Figure 2.
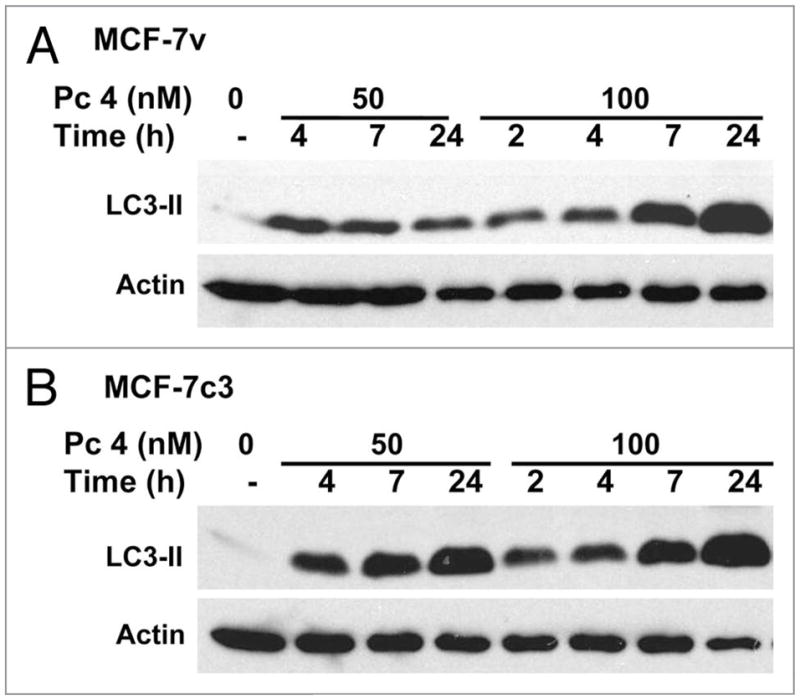
Time-course of accumulation of LC3-II in MCF-7 cells in response to two doses of PDT sensitized by Pc 4. Cultures of MCF-7v (A) and MCF-7c3 (B) cells were exposed to nothing or 50 or 100 nM Pc 4 for 18 h, after which all cultures were photoirradiated with 200 mJ/cm2 red light, as described in Methods. Cultures were returned to the incubator for various times up to 24 h after which the cells were collected and processed for western blot analysis with antibodies to LC3-II and actin as a loading control.
Figure 3.
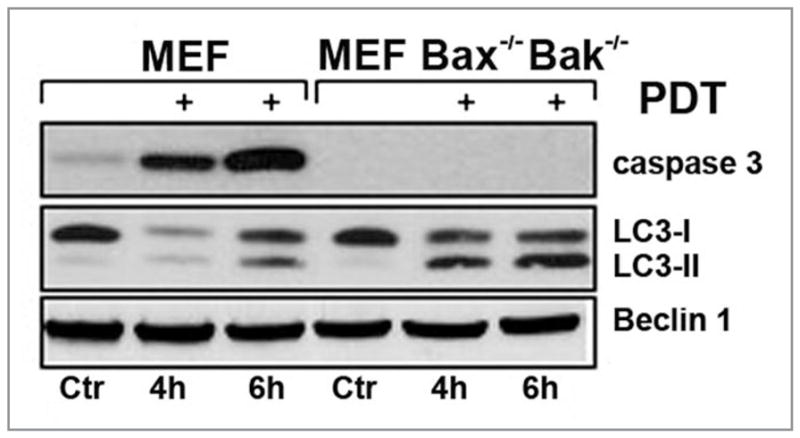
Time-dependent detection of LC3-conversion and caspase-3 processing by western blots of whole cell lysates of wild-type (WT) MEFs or Bax/Bak-deficient MEFs after PDT using hypericin. The western blot shows that the level of Beclin 1 remains unaltered during the time course of this experiment.
Cytoplasmic vacuolization commonly occurs in PDT protocols. Indeed, cytoplasmic vacuoles can be easily observed in photoirradiated cultures of 1c1c7 cells sensitized with mTHPC (Fig. 1E), rat L1210 leukemia cultures sensitized with the ER photosensitizer CPO (Fig. 4), and HeLa, wild-type MEF, and Bax−/−Bak−/− double knockout MEFs that have been sensitized with hypericin (Fig. 5). At issue is whether such vacuoles represent autophagosomes, or in some manner contribute to the development of autophagosomes. In the case of photoirradiated 1c1c7 cultures sensitized with mTHPC, the absence of vacuole labeling with fluorescent chimeric GFP-LC3 argues against the vacuoles being autophagosomes (Fig. 1E). However, in the case of L1210 cells, co-incubation with the PI-3-kinase inhibitor wortmannin suppressed vacuole development (Fig. 4). Given the role of PI-3-kinase in the development of the autophagosome,26 the wortmannin effects are consistent with CPO-PDT induced vacuoles being related to the autophagic process.
Figure 4.
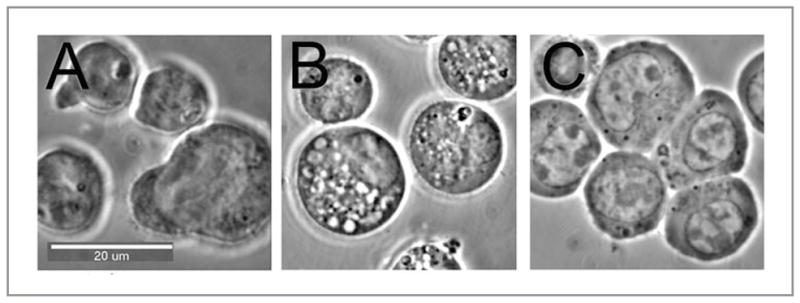
Vacuoles formed following PDT of L1210 cells: (A) control cells; (B) 15 min after an LD50 PDT dose using CPO as the photosensitizing agent; (C) same as (B) but with 10 nM wortmannin present.
Figure 5.

(A) Transmission electron microscopic (TEM) analysis of untreated (incubated with hypericin in the dark) HeLa cells and HeLa cells 6 h after PDT using hypericin as the photosensitizer and a light dose that will reduce viability by 70% 24 h later. (B) TEM analysis of wild-type murine embryonic fibroblasts (WT MEFs) or MEF Bax−/− Bak−/− before (untreated) or 6 h after hypericin-PDT. Arrows indicate vacuoles with detectable content in the treated cells. Magnification is indicated in the lower right corner. Representative EM photomicrographs are shown.
Electron microscopy (EM) remains one of the most sensitive methods to detect the accumulation of autophagic compartments in mammalian cells. Autophagosomes are unique vesicular double membrane-bound structures that contain regions of the cytoplasm, and possibly organelles. The images shown in Figure 6 illustrate the use of electron microscopy, using lead citrate + uranium acetate staining to help reveal membrane structures, to monitor the development of autophagosomes. These images represent a CPO-sensitized L1210 cell at successively higher degrees PDT dose. It can readily be of magnification 15 min after an LD50 seen that the resulting vacuoles are not empty, and are bounded by the double-membrane characteristic of the autophagosome.
Figure 6.
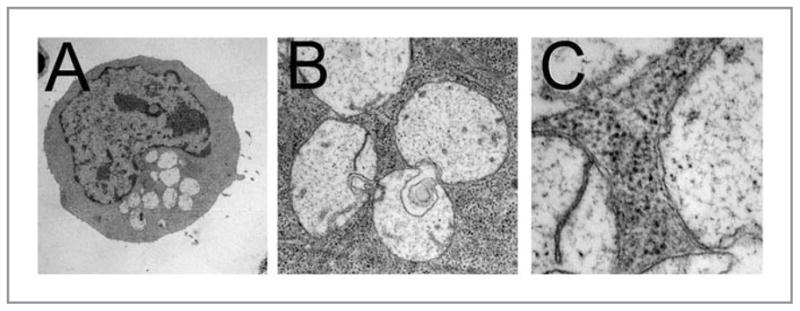
Progressive magnification of autophagic L1210 cells after PDT, showing double-membrane surrounding vacuoles along with evidence of recycled material. Conditions are indicated in the legend to Figure 4.
Autophagy as a factor in phototoxicity
The issue of whether autophagy plays a prosurvival or prodeath role after toxicant exposure is hotly contested.27,28 A cytoprotective role for autophagy in PDT is supported by recent findings with the ER-associated photosensitizer hypericin in apoptosis-competent cells (Dewaels and Agostinis, unpublished results). This photosensitizing agent provokes immediate and direct ROS-mediated damage to the sarco/endoplasmic Ca2+ ATPase (SERCA)2 pump, followed by ER-Ca2+ depletion, while sparing inositol trisphosphate (IP)3 receptor or Bcl-2 from direct photodamage.15 In this paradigm, siRNA-mediated knock down of Atg5 or pharmacological inhibition of autophagy by 3-methyladenine (3-MA) increases the cytotoxic effects of PDT (Dewaels and Agostinis, unpublished results). Similarly, a distinct shoulder is observed in the light-dose response survival curve corresponding to photoirradiated CPO-sensitized L1210 leukemia cells (Fig. 7A). This shoulder is absent in an Atg7 knockdown derivative of the L1210 cell line that is incapable of forming autophagosomes.
Figure 7.

(A) PDT efficacy in wild-type(●) L1210 cells and an Atg7 knockdown derivative of L1210 cells (○). Cultures of L1210 cells or a L1210 derivative whose expression of Atg7 had been stably knocked down with shRNA were sensitized with CPO and subsequently exposed to light. After photoirradiation cells were plated for assessment of colony formation. Data represent means ± SD of three plates per treatment group. (B) PDT efficacy in wild-type MCF-7 cells (black bars) and in cells whose expression of Atg7 has been stably knocked down (gray bars). Exponentially growing cultures of each cell line were exposed to the indicated concentrations of Pc 4 in complete growth medium for 18 h followed by 200 mJ/cm2 red light. Immediately after photoirradiation, the cells from each dish were collected by trypsinization, and aliquots were replated in 60-mm dishes in sufficient number to result in 50–100 colonies per dish. After 10–14 days, colonies were stained with 1% crystal violet, and colonies having at least 50 cells were counted by eye and normalized to the number of cells plated and the plating efficiency of the control untreated cells. Data for the two cell lines were compared by Student’s t test and found to be significantly different at all doses (p < 0.01).
Although the above studies suggest that stimulation of autophagy in apoptosis-competent cells increases cellular resistance to photokilling in PDT protocols, the same may not hold for cells incapable of mounting an apoptotic response. For example, knockdown of the autophagy-associated protein Atg7 increases the shoulder (instead of eliminating it) on clonogenic dose-response curves for irradiated apoptosis-resistant MCF7 cells that had been preloaded with the ER/mitochondrial sensitizer Pc 4 (Fig. 7B). Similarly, illuminated hypericin-sensitized Bax−/−Bak−/− double knockout MEFs develop a much stronger autophagic response than their apoptosis-competent wild-type counterparts (Figs. 3 and 5), and undergo a type of death with features consistent with necrosis.15,29 Suppression of PDT-induced autophagy in the Bax−/−Bak−/− double knockout MEFs inhibits cell death.15 Collectively, these studies suggest that autophagy plays a prodeath role in PDT-mediated killing of apoptosis-defective cells.
Protein targets of PDT
Singlet oxygen is a potent oxidant. An ever-increasing number of proteins appear to be targets for the singlet oxygen generated in PDT protocols. Some of these proteins are direct regulators of, or components of, the autophagic process. For example, several ER and/or mitochondrial photosensitizers (i.e., CPO, SnET2, BPD and Pc 4),9–11,30 but not hypericin15 or the endosomal/lysosomal sensitizer NPe6,31 photo-damage Bcl-2 and/or Bcl-xL. The extent to which these anti-apoptotic/anti-autophagic proteins are damaged is light-dose and cell-context dependent. Similarly, it was recently shown that a primary target of photosensitization by hypericin is the SERCA2 pump which is rapidly damaged and inactivated in a 1O2-dependent fashion after PDT in MEFs.15,29 Recently, another ER-associated protein, the IP3 receptor, was found to be a key regulator of autophagy.32 Photodamage elicited by hypericin does not affect IP3 receptor levels or activity in MEFs.15 However, PDT with the phthalocyanine photosensitizer Pc 4, which localizes to both the mitochondria and ER, photodamages the IP3 receptor (IP3R2 and IP3R3) and the SERCA2 pump in MCF-7 cells (Fig. 8). It is currently not known if the differential effects of Pc 4 and hypericin on the IP3 receptor in the above studies reflect cell type differences or an inherent difference between the two sensitizers.
Figure 8.
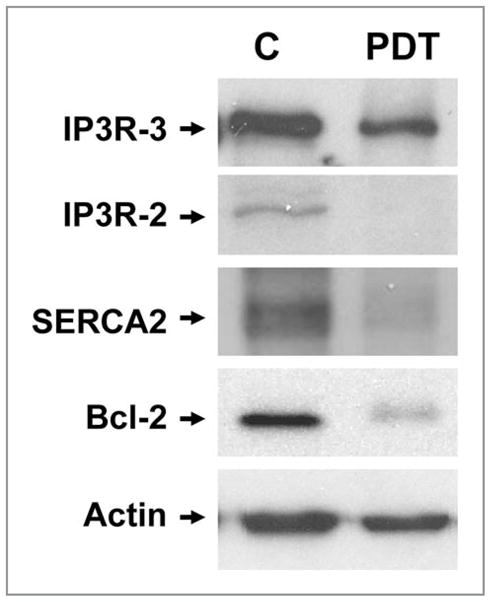
Photodamage to proteins of MCF-7c3 cells by Pc 4-PDT. Exponentially growing cultures of MCF-7c3 cells were incubated with nothing (labeled C) or 200 nM Pc 4 for 18 h, then irradiated with 200 mJ/cm2 red light, as described in Methods. Immediately after PDT, cells were collected from the monolayer by scraping and processed for western blot analysis, using antibodies to SERCA2, IP3-R2, IP3-R3, Bcl-2 and actin.
The mammalian target of rapamycin (mTOR) complex is a critical regulator of cell growth and proliferation. In some cases, pharmacological inhibition of mTOR can induce autophagy outright, sensitize cells to conditions causing autophagy, or enhance the autophagic response induced by a stressor. A recent study by Weyergang et al.33 suggests that the amphiphilic endolysosome-localizing photosensitizer AlPcS(2a) directly targets the mTOR signaling network in PDT protocols with a colon adenocarcinoma cell line. In particular, AlPcS(2a)-PDT causes a rapid, partial loss of both total mTOR and the Ser(2448) phosphorylated form of the enzyme in both cultured cells and tumor xenografts.
It should be noted that not all autophagy-related proteins undergo photodamage in PDT protocols. An examination of the effects of a LD90 PDT dose on Beclin 1, Atg5 and Atg7 revealed no significant photodamage (Fig. 9). These results were obtained with L1210 cells and the photosensitizer benzoporphyrin derivative monoacid ring A (BPD). BPD localizes preferentially to the mitochondria.34 It does not accumulate in lysosomes. Likewise, neither Beclin 1 (Fig. 3) nor Atg5 (Dewaels and Agostinis, unpublished results) were photodamaged in PDT protocols utilizing hypericin-photosensitized HeLa cells and MEFs.
Figure 9.
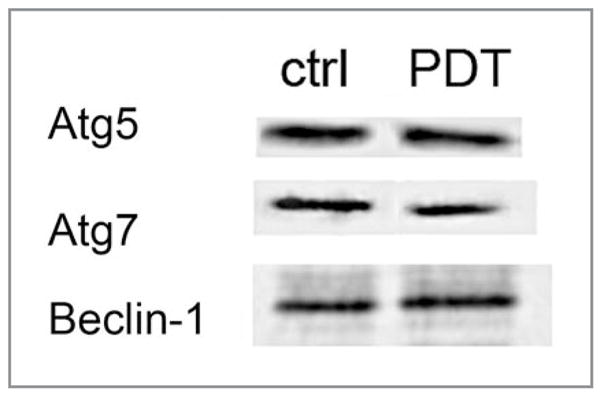
Lack of photodamage to three proteins involved in autophagy. L1210 cells were photosensitized with BPD and irradiated so as to produce 90% cell kill as determined by a clonogenic assay. Cells were prepared for western blot analysis 30 min after irradiation.
PDT damage to organelles involved in autophagy
Lysosomes and endosomes are the targets of several photosensitizing agents in common use, e.g., NPe6,21 TPPS1–4,35 AlPcS2–4,36,37 lysyl chlorin p6,38 and etiobenzochlorin.39 Some of these sensitizers preferentially localize to the membranes of the target organelles, whereas others concentrate in the organelle’s matrix. Tetra(4-sulphonatophenyl)-porphine (TPPS4) is an example of the latter class. Irradiation of cells sensitized with TPPS4 results in photo-oxidation of matrix components including the hydrolytic enzymes.35,36 Because of the short half-life and diffusion limit of singlet oxygen in biological environments,40 and perhaps as a result of the polysaccharide coating of the inner surface of the lysosomes, lysosomal enzymes are typically photoinactivated before the endocytic membranes are targeted in TPPS4-PDT protocols (Fig. 10). Photodamage to the latter is expected to result in loss of membrane integrity and release of the contents of the endocytic vesicles into the cytoplasm (Fig. 10). In this way, most if not all of the lysosomal enzymes that are released into the cytosol upon membrane disruption in TPPS4 protocols will be inactivated. Because lysosomal proteases are inactivated prior to membrane rupture, TPPS4-PDT theoretically provides a mechanism for selective destruction of endosomes/lysosomes without collateral damage to other organelles. Indeed, TPPS4-PDT-mediated destruction of endosomes/lysosomes has been used as a method for selective enrichment of autophagosomes.41
Figure 10.
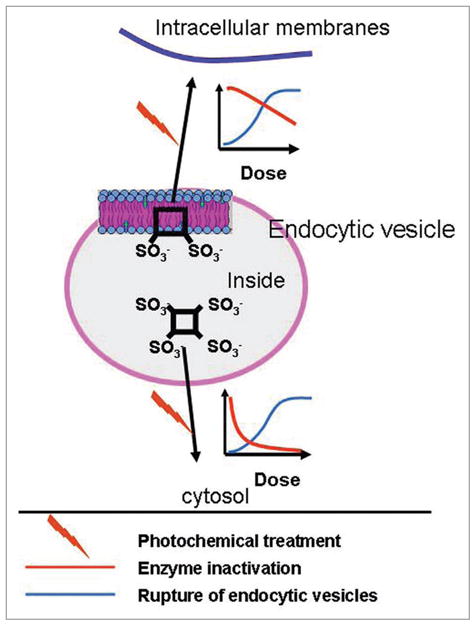
Proposed localization and photochemical effect of sulfonated photosensitizers in endocytic vesicles. The figure shows a schematic drawing of an endocytic vesicle and the main location of a hydrophilic tetrasulfonated and an amphiphilic disulfonated photosensitizer. The photochemical effect leads to relocalization of the photosensitizers as indicated on the figure with the indicated relation between breakage of endocytic vesicles and lysosomal enzyme inactivation.
NPe6 and TPPS2a are amphiphilic photosensitizers that bind to endosomal/lysosomal membranes. Upon exposure to light, such membranes will be damaged and become leaky before significant inactivation of lysosomal enzymes can occur (Fig. 10). The released proteases, if not neutralized by endogenous protease inhibitors, can have detrimental effects, including cathepsin cleavage of Bid and subsequent induction of the mitochondrial apoptotic program by a Bid cleavage product.21
Some amphiphilic photosensitizers enter the cell via the process of endocytosis.42,43 Subsequently, such sensitizers may be found in multiple vesicle types of the endocytic pathway (Fig. 11). This association of amphiphilic photosensitizers with endocytic vesicles, and the permeabilization of such vesicles following photoirradiation, is the basis for a therapeutic delivery system termed “photochemical internalization” (PCI).42,43 In PCI, endocytic vesicles containing both the endocytosed sensitizer and a therapeutic agent are photoirradiated so as to activate the sensitizer. This facilitates the release of the therapeutic agent via photooxidation of the vesicle membrane. The PCI procedure has been effective at facilitating delivery and intracellular release of several classes of therapeutics into the cytosol.43
Figure 11.
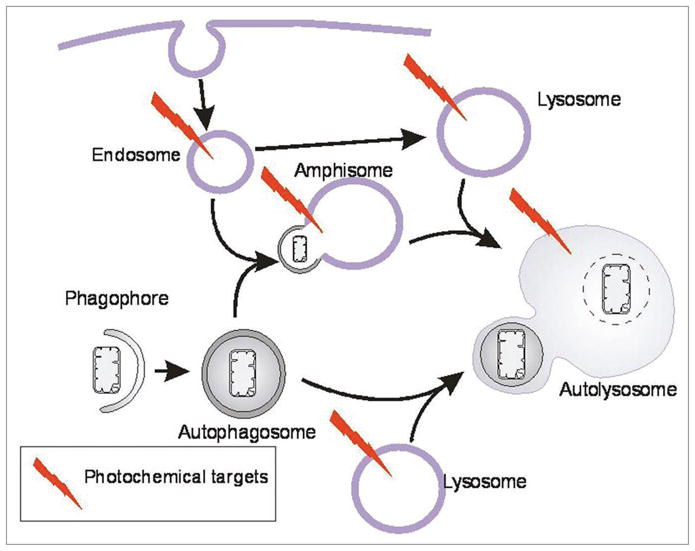
Schematic model of the formation of autophagosomes and endocytic vesicles and interaction between these pathways. The model also indicates the photochemical targets of photosensitizers taken up by endocytosis.
The autophagic sequestration of cytoplasm begins with the formation of phagophores resulting in double-membrane autophagosomes surrounding a fraction of the cytoplasm (Fig. 11). The autophagosomes may fuse with endosomes forming amphisomes, followed by fusion with lysosomes to form autolysosomes.44,45 Alternatively, autophagosomes may fuse directly with the lysosomes to form autolysosomes. In either case, the cytoplasmic contents of the autophagosomes become degraded by the hydrolytic enzymes of the lysosomes. In cells treated with photosensitizers taken up by endocytosis, endosomes, lysosomes, autolysosomes and most likely amphisomes will contain the photosensitizing agent.35,41,46,47 Upon irradiation of cells treated with hydrophilic photosensitizers such as TPPS4, the contents of these vesicles will be targets for photo-oxidation and their membranes will break.35,41
Effects of lysosomal photodamage on the autophagic process: measuring the autophagic flux
As described recently by Klionsky et al.19 an increased LC3-I to LC3-II conversion seen on western blots in response to a given cellular stress could reflect stimulation of autophagy (i.e., increased ON-rate) or the inhibition of the degradation/processing of autophagosomes (i.e., decreased OFF-rate). Hence, in PDT-treated cells it is highly recommended to carry out a thorough evaluation of “autophagic flux,” i.e., the complete degradation of autophagosomes and their cargo, to distinguish between stimulation of the ON-rate or decreased OFF-rate.
Bafilomycin A1 (BafA1), an inhibitor of the vacuolar H+ ATPase (V-ATPase), inhibits lysosome-autophagosome fusion and/or the degradation of autolysosomal LC3-II.48 Hence, BafA1 is often used to assess autophagic flux. In cells transfected with a GFP-LC3 expression plasmid, the chimeric fusion protein can be incorporated into autophagosomes. Upon fusion with lysosomes the chimeric protein undergoes proteolysis. However, unlike the LC3 portion of the molecule, the GFP peptide is rather resistant to mammalian hydrolases. The accumulation of “free GFP” after PDT as a degradation product generated during autophagosome processing can be monitored by western blotting. Comparison of MEFs subjected to hypericin-PDT, in the absence or presence of BafA1, revealed a distinct absence of GFP-LC3 processing in BafA1-treated cells (Fig. 12). From these experiments it is clear that initial photosensitization of the ER by hypericin stimulates the ON-rate of autophagy, thus indicating that in this PDT paradigm lysosomes can complete autophagosome processing.
Figure 12.
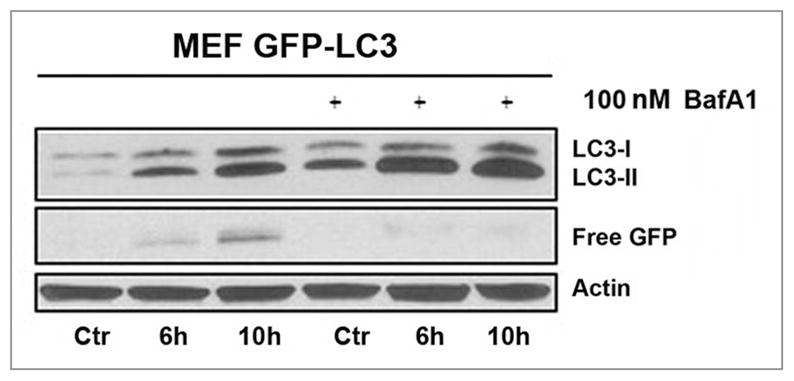
Flux analysis in MEFs stably expressing GFP-LC3. Whole cell lysates were made at the indicated time points and analyzed by western blots. The increased amount of LC3-II and the absence of ‘free GFP’ when cells were pre-treated with 100 nM Bafilomycin A1 1 h before PDT (with hypericin as the photosensitizer) indicates the autophagic flux. All controls represent cells incubated with hypericin in the dark.
As an alternate approach for monitoring autophagic flux, the effect of the lysosomal protease inhibitors E64d + acetylpepstatin A on LC3-II levels was examined in L1210 cells before and after photodamage.49 The effect of ammonium chloride, an agent known to prevent the acidification step necessary for further processing,50 was also examined. Photoirradiation of L1210 cultures preloaded with the ER/mitochondrial sensitizer CPO resulted in the accumulation of LC3-II (Fig. 13). Pretreatment with either the cathepsin inhibitors or ammonium chloride was equally effective in enhancing the accumulation of LC3-II (Fig. 13). Hence, as occurred with hypericin, CPO stimulated the ON-rate of autophagy. Note that the L1210 cell line shows a detectable level of “background” autophagy, a likely factor in the loss of protection from phototoxicity when autophagy is impaired (Fig. 7A).
Figure 13.
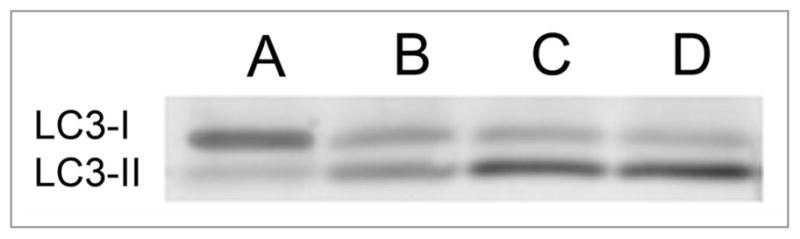
Effect of E64d + acetylpepstatin A vs. NH4Cl on processing of autophagosomes as indicated by conversion of LC3-I to LC3-II after PDT. (A) controls; (B) cells given an LD50 PDT dose (CPO); (C) same as (B) with E64d + acetylpepstatin A present; (D) as in (B) but with 15 mM NH4Cl present.
Neither hypericin nor CPO targets endosomes/lysosomes in PDT protocols. Lysosomes are known to be essential elements of autophagy, thus raising the possibility that substantial lysosomal photodamage might impair the autophagic process. The photosensitizer NPe6 localizes to lysosomes and causes their permeabilization and release of lysosomal enzymes following light exposure.21 In the following study murine hepatoma 1c1c7 cultures were photosensitized with NPe6 and exposed to a LD90 light dose. Exposure to light alone or NPe6 alone had no effect on acridine orange (AO) staining of acidic organelles (i.e., endosomes/lysosomes) or autophagosome formation (as monitored by the clustering of the GFP-LC3 chimeric protein; Fig. 14). However, within an hour of PDT, AO staining was totally lost (Fig. 14, top row). Autophagosome accumulation was obvious within 2 h of irradiation, and very prominent by 4 h (Fig. 14, second and third rows). It should be noted that under the above conditions pro-caspases-3 and -9 begin to undergo activation within 2–3 h of light exposure and reach their maximum by 6 h.51 Collectively these data indicate that autophagosome formation can occur under conditions in which late endosomes/lysosomes are destroyed in PDT protocols. However, completion of the autophagic process will most likely be compromised in PDT protocols involving lysosomal sensitizers. In such protocols there may be no lysosomes with which the autophagosomes can fuse. Alternately, the lysosomes may be dysfunctional because of photoinactivation of their enzymatic contents.
Figure 14.

Disruption of lysosomes in PDT protocols does not inhibit autophagosome formation. A variant of the murine hepatoma 1c1c7 cell line that stably expressed a GFP-LC3 fusion protein either received no treatment, or was incubated with 33 μM NPe6 for 1 h prior to being washed and refed. Cultures were subsequently photoirradiated for 100 s. Some cultures were incubated with 500 nM acridine orange (AO) for 10 minutes prior to being analyzed for the presence of AO fluorescent punctate spots (indicative of intact acidic vesicles; top row). Other cultures were analyzed for the development of green fluorescent GFP punctate spots indicative of autophagosomes (middle and bottom rows). In the top row AO fluorescence in the NPe6 + PDT treatment group was analyzed 1h after photoirradiation. In the middle row GFP fluorescence was analyzed 6 h after photoirradiation, or after 7 h of incubation with NPe6.
Discussion
Based on studies carried out with different cell lines and photosensitizing agents, we can draw several conclusions relating to interactions between photodynamic therapy and autophagy. First, the induction of autophagy is a common outcome in PDT protocols. It occurs in a variety of cell types, and is not limited to photosensitizers that accumulate in specific organelles. As documented herein, photosensitizers that preferentially accumulate in late endosomes/lysosomes (i.e., NPe6), ER (i.e., hypericin, CPO), mitochondria (i.e., mTHPC, BPD), or ER + mitochondria (i.e., Pc 4) all induced autophagy following irradiation. Second, apoptosis is also a common outcome of PDT, and often occurs in cells undergoing autophagy. The initiation and development of autophagy can precede, parallel, or follow the development apoptosis. Cell type, photosensitizer type and its concentration, and light dose all influence the kinetics at which apoptotic/autophagic responses develop in PDT protocols. Third, the autophagic response occurring after PDT may have different roles in apoptosis competent and deficient cell types. In the case of ionizing radiation,14 the lethal effects of PDT can be partially alleviated by autophagy, at least in cells competent for apoptosis. In contrast, relative to their apoptosis-competent counterparts, apoptosis-defective cells exhibit enhanced autophagic responses and sensitivity to killing in PDT protocols employing ER and mitochondrial photosensitizers. In other words, PDT-induced autophagy appears to play a prosurvival role in apoptosis competent cells, and possibly a prodeath role in apoptosis defective cells.
Because the induction of autophagy is a common response in PDT protocols, it seems unlikely that proteins responsible for the assembly of autophagosomes are PDT targets. Indeed, as reported herein, key autophagic proteins such as Beclin 1, Atg5 and Atg7 appear to be unaffected in PDT protocols employing ER and mitochondrial sensitizers. On the other hand, PDT may facilitate the development of autophagy by specifically photo-oxidizing/inactivating proteins that function as negative regulators of autophagy. For example, both molecular and pharmacological approaches indicate that Bcl-2 and Bcl-xL are negative regulators of autophagy.8,53,55 Interestingly, several laboratories have demonstrated that ER and mitochondrial photosensitizers, but not a lysosomal sensitizer (i.e., NPe6), photodamage Bcl-2 and Bcl-xL.10,11,30,31 Similarly, in some cell types, pharmacological inhibition of mTOR activity has been shown to trigger or sensitize cells to the development of autophagy.54,55 Very recent studies suggest that mTOR is photodamaged in PDT protocols utilizing the sensitizer AlPcS(2a).33 Although this discussion has focused on the immediate protein targets of PDT that may be relevant to autophagy, it should be emphasized that PDT may also induce the synthesis and translation of mRNAs that encode proteins that are also relevant to the process.56 This latter issue, as well as the further identification of PDT protein targets, needs to be considered in future studies.
Photosensitizers that accumulate in lysosomes, ER or mitochondria are all capable of inducing the formation of autophagosomes in PDT protocols. However, the subsequent processing of autophagosomes most likely differs for the different classes of sensitizers. Specifically, “autophagic flux” studies indicate that the autophagosomes formed in ER/mitochondrial PDT protocols fuse with lysosomes, and eventually (along with their cargo) undergo degradation. In contrast, autophagosome processing is expected to be compromised in PDT protocols employing lysosomal sensitizers because of profound changes to the lysosomes. In particular, depending upon the specific lysosomal photosensitizer employed and the light dose applied, lysosomal luminal proteases may undergo irreversible inactivation without profound effects on lysosomal membrane permeability.35,36 Alternatively, lysosomal membranes may be damaged and permeabilized, leading to the release of proteases and alkalinization of the organelle.21 Such effects are expected to markedly affect either autophagosome-lysosome fusion, and/or degradation of the cargo contained in the autolysosomes. In effect, PDT protocols with lysosomal sensitizers most likely induce an aborted form of autophagy. The extent to which this disruption of the autophagic program affects the relative cytotoxicity of lysosomal photosensitizers is not known. However, it is anticipated that PDT with lysosomal sensitizers might synergize with other agents/protocols whose therapeutic efficacy is muted by the autophagic response that normally occurs following their administration. This issue is currently under investigation.
In whole animal studies photosensitizers accumulate in both tumor cells and tumor vasculature. Tumor eradication in PDT protocols is a consequence of direct tumor cell kill, as well as tumor vascular shutdown.1 The latter will cause nutrient depletion and hypoxia, well-characterized inducers of autophagy.57,58 Given the prosurvival role of autophagy, PDT-mediated vascular damage might contribute to tumor cell adaptation in the face of a hostile hypoxic environment, and enhance the resistance of surviving cancer cells to subsequent PDT. Although this has not yet been validated experimentally in vivo, it seems possible that combining PDT with an inhibitor of autophagy might result in a better therapeutic outcome.
Materials and Methods
Studies with murine leukemia cells in suspension culture
Murine leukemia L1210 cells are grown in suspension culture in a modified α-MEM growth medium + 10% horse serum.59 In this cell line, both apoptosis and autophagy can readily be induced when Bcl-2 is inactivated.59 For studies involving the photosensitizing agent CPO, cells were loaded by exposing them to a 2 μM concentration of this agent for 30 min, then irradiated with a sufficient dose of red light (590–630 nm) to produce the desired level of photokilling as determined by clonogenic assays. Other procedures are described elsewhere.12,13,16 In recent experiments, it was found that the more commonly used photosensitizer BPD60 yields essentially the same results that are obtained with CPO. BPD shows a localization pattern favoring mitochondria34 and targets Bcl-2 for photodamage (Kessel D, unpublished data). Preparation of Atg7 knockdowns and western blotting techniques are described in ref. 61. A highly selective LC3 antibody was obtained from Proteintech Inc., Chicago IL.
Studies with cultured HeLa cells and MEFs using hypericin
Experiments to monitor autophagy and apoptosis induction following the light activation of the naturally occurring photosensitizer hypericin, which accumulates preferentially in ER membranes, have been described in recent work.15,29 Briefly, HeLa cells or MEFs are incubated with hypericin for 16 h in fetal bovine serum-containing DMEM at a concentration of 150 nM or 750 nM, respectively, to allow similar uptake of the dye (measured by FACS analysis) and subcellular distribution (measured by confocal laser microscopy). Irradiation is performed by placing the cell culture plates on a plastic diffuser sheet above a set of seven L18W30 fluorescent lamps (Osram) (fluence rate, 4.5 mW/cm2). The cells are exposed to a light dose which produces 70% cell death 24 h after irradiation, as determined by trypan blue exclusion. Other procedures, such as preparation of cell lysates for western blotting, TEM analysis and GFP-LC3 transfection, have been described elsewhere.15,29
Studies with murine hepatoma 1c1c7 cells and the photosensitizers mTHPC and NPe6
Murine hepatoma 1c1c7 cells were grown in αMEM supplemented with 5% fetal bovine serum and antibiotics in a 5% CO2 atmosphere at 37°C. A 1c1c7 derivative cell line that stably expresses a GFP-LC3 fusion protein was maintained in the above medium containing 500 ng/ml Gentamycin. For the studies reported herein, LC3-GFP 1c1c7 cells were not plated in Gentamycin. The conditions used for photosensitization of 1c1c7 cells with NPe6 (dissolved in water) and photoirradiation have been described in detail.51 For studies involving mTHPC (dissolved in water), 1c1c7 cultures were preloaded with the photosensitizer ~18 h prior to being washed, refed with complete medium lacking mTHPC, and photoirradiated at room temperature using a 600 W quartz-halogen lamp with IR radiation attenuated by a 10 cm layer of water. The bandwidth was further defined by using a 630 nm high pass filter and an 850 nm cutoff filter. In order to label acidic organelles, cultures were incubated with 500 nM acridine orange (AO) for 10 minutes prior to being washed, refed and immediately imaged. AO red fluorescence (indicative of an acidic environment) was acquired using excitation at 480 nm and emission at 630 nm. GFP fluorescence was acquired using excitation at 450–490 nm and emission at 515–565 nm. Images were captured using an Axioplan 2 Fluorescence Imaging Microsocope (Carl Zeiss, Cologne, Germany). Activation of procaspase-3/7 was monitored by measuring DEVDase activities. The procedure used for measuring DEVDase activities has been described in detail.51 DEVDase specific activities are presented as nmol of product generated per minute per mg of whole cell extract. The western blot procedures, and sources of antibodies used for the detection of GAPDH and LC3-I/II have been described in detail.62
Studies with human breast cancer MCF-7 cells with the photosensitizer Pc 4
The cells were cultured in RPMI medium with 10% fetal bovine serum and preloaded with Pc 4 by addition of a small aliquot of a stock solution to the complete medium overlying the monolayer cultures. Pc 4 enters the cells rapidly, reaching maximum uptake by 1–2 h, as judged by fluorescence measurements on Pc 4 extracted from cells in ethyl acetate or by flow cytometry,63 and localizing in mitochondria and ER/Golgi membranes, as judged by confocal microscopy.63,64 Photoirradiation was accomplished by placing the culture plates on a glass plate above a light-emitting diode array (EFOS, Mississauga, ONT, Canada; λmax ~675 nm; fluence rate 200 mW/cm2). The cells were exposed to a range of light doses to produce up to 99% cell death, as determined by clonogenic assay. Procedures for western blotting, electron or light microscopy, and the addition of inhibitors or other probes have been published.11
Acknowledgments
Studies in Dr. Kessel’s laboratory have been supported by CA 23378 from the NIH since 1983. Research in Dr. Agostinis’ lab is supported by the Fund for Scientific Research (FWO)-Flanders (Belgium) [grant number G.0661.09]; the Interuniversity attraction pole IAP6/18 of the Belgian Federal Government, and the Catholic University of Leuven [OT/06/49]. We would like to thank Dr. W. Martinet (University of Antwerp) for excellent help with TEM analysis. Studies in Dr. Oleinick’s laboratory were supported by CA 83917 and CA 106491 from the NIH and by the Wearn Endowment, Case Western Reserve University, and used the services of the Flow Cytometry and Confocal Microscopy Core Facilities of the Case Comprehensive Cancer Center and the Electron Microscopy Core Facility of the CWRU School of Medicine. Studies in the Reiners laboratory utilized the services of the Cell Imaging and Cytometry Facility Core at Wayne State University, which is supported by P30 ES06639 from the NIH.
Abbreviations
- AlPcS2 or 4
di- or tetrasulfonated aluminium phthalocyanines
- BPD
benzoporphyrin-derivative monoacid ring A
- CPO
9-capronyloxytetrakis (methoxyethyl)porphycene
- GFP
green fluorescent protein
- LC3
microtubule-associated protein light chain 3
- 3-MA
3-methyladenine
- MEF
murine embryonic fibroblast
- mTHPC
meso-tetra-hydroxyphenyl-chlorin
- NPe6
N-aspartyl chlorin e6
- Pc 4
the silicon phthalocyanine HOSiPcOSi(CH3)2(CH2)3N(CH3)2
- PCI
photochemical internalization
- PDT
photodynamic therapy
- ROS
reactive oxygen species
- SnET2
tin etiopurpurin dichloride
- TPPS2 or 4
di- or tetrasulfonated tetraphenylporphines
References
- 1.Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, et al. Photodynamic therapy. J Natl Cancer Inst. 1998;90:889–905. doi: 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kessel D, Luo Y, Deng Y, Chang CK. The role of sub-cellular localization in initiation of apoptosis by photodynamic therapy. Photochem Photobiol. 1997;65:422–6. doi: 10.1111/j.1751-1097.1997.tb08581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kessel D. Correlation between subcellular localization and photodynamic efficacy. J Porph Phthalo. 2004;8:1009–14. [Google Scholar]
- 4.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertling CJ, Lin F, Girotti AW. Role of hydrogen peroxide in the cytotoxic effects of UVA/B radiation on mammalian cells. Photochem Photobiol. 1996;64:137–42. doi: 10.1111/j.1751-1097.1996.tb02433.x. [DOI] [PubMed] [Google Scholar]
- 6.Nyström T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005;24:1311–7. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends in Cell Biology. 2007;17:422–7. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Kim HRC, Luo Y, Li G, Kessel D. Enhanced apoptotic response to photodynamic therapy after Bcl-2 transfection. Cancer Res. 1999;59:3429–32. [PMC free article] [PubMed] [Google Scholar]
- 10.Kessel D, Castelli M. Evidence that Bcl-2 is the target of three photosensitizers that induce a rapid apoptotic response. Photochem Photobiol. 2001;74:318–22. doi: 10.1562/0031-8655(2001)074<0318:etbitt>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 11.Xue LY, Chiu SM, Oleinick NL. Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene. 2001;20:3420–7. doi: 10.1038/sj.onc.1204441. [DOI] [PubMed] [Google Scholar]
- 12.Kessel D, Arroyo AS. Apoptotic and autophagic responses to Bcl-2 inhibition and photodamage. Photochem Photobiol Sci. 2007;6:1290–5. doi: 10.1039/b707953b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kessel D, Reiners JJ., Jr Apoptosis and autophagy after mitochondrial or endoplasmic reticulum photodamage. Photochem Photobiol. 2007;83:1024–8. doi: 10.1111/j.1751-1097.2007.00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485–94. doi: 10.1158/0008-5472.CAN-07-0562. [DOI] [PubMed] [Google Scholar]
- 15.Buytaert E, Callewaert G, Hendrickx N, Scorrano L, Hartmann D, Missiaen L, et al. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J. 2006;20:756–8. doi: 10.1096/fj.05-4305fje. [DOI] [PubMed] [Google Scholar]
- 16.Kessel D, Vicente MG, Reiners JJ., Jr Initiation of apoptosis and autophagy by photodynamic therapy. Lasers Surg Med. 2006;38:482–8. doi: 10.1002/lsm.20334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue LY, Chiu SM, Azizuddin K, Joseph S, Oleinick NL. The death of human cancer cells following photodynamic therapy: Apoptosis competence is necessary for Bcl-2 protection but not for induction of autophagy. Photochem Photobiol. 2007;83:1016–23. doi: 10.1111/j.1751-1097.2007.00159.x. [DOI] [PubMed] [Google Scholar]
- 18.Oleinick NL, Xue LY, Chiu SM, Joseph S. Autophagy in response to photodynamic therapy: Cell survival vs. cell death. Proc SPIE; 2009. p. 7164. in press. [Google Scholar]
- 19.Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:139–40. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kessel D, Luo Y, Mathieu P, Reiners JJ., Jr Determinants of the apoptotic response to lysosomal photodamage. Photochem Photobiol. 2000;71:196–200. doi: 10.1562/0031-8655(2000)071<0196:dotart>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 21.Reiners JJ, Jr, Caruso JA, Mathieu P, Chelladurai B, Yin XM, Kessel D. Release of cytochrome c and activation of pro-caspase-9 following lysosomal photodamage involves Bid cleavage. Cell Death Differ. 2002;9:934–44. doi: 10.1038/sj.cdd.4401048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez ME, Azizuddin K, Chiu SM, Xue LY, Zhang P, Lam M, et al. Structural factors and mechanisms underlying the improved photodynamic cell killing with silicon phthalocyanine photosensitizers directed to lysosomes vs. mitochondria. Photochem Photobiol. 2009;85:1189–200. doi: 10.1111/j.1751-1097.2009.00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sasnauskiene A, Kadziauskas J, Vezelyte N, Jonusiene V, Kirveliene V. Apoptosis, autophagy and cell cycle arrest following photodamage to mitochondrial interior. Apoptosis. 2009;14:276–86. doi: 10.1007/s10495-008-0292-8. [DOI] [PubMed] [Google Scholar]
- 24.Li F, Srinivasan A, Wang Y, Armstrong RC, Tomaselli KJ, Fritz LC. Cell specific induction of apoptosis by microinjection of cytochrome c. Bcl-xL has activity independent of cytochrome c release. J Biol Chem. 1997;272:30299–305. doi: 10.1074/jbc.272.48.30299. [DOI] [PubMed] [Google Scholar]
- 25.Chiu SM, Xue LY, Usuda J, Azizuddin K, Oleinick NL. Bax is essential for mitochondrion-mediated apoptosis but not for cell death caused by photodynamic therapy. Br J Cancer. 2003;89:1590–7. doi: 10.1038/sj.bjc.6601298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, Carnuccio R, et al. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009;16:87–93. doi: 10.1038/cdd.2008.131. [DOI] [PubMed] [Google Scholar]
- 27.Scarlatti F, Granata R, Meijer AJ, Codogno P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2009;16:12–20. doi: 10.1038/cdd.2008.101. [DOI] [PubMed] [Google Scholar]
- 28.Levine B, Kroemer G. Autophagy in aging, disease and death: the true identity of a cell death imposter. Cell Death Different. 2009;16:1–2. doi: 10.1038/cdd.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buytaert E, Callewaert G, Vandenheede JR, Agostinis P. Deficiency in apoptotic effectors Bax and Bak reveals an autophagic cell death pathway initiated by photodamage to the endoplasmic reticulum. Autophagy. 2006;2:238–40. doi: 10.4161/auto.2730. [DOI] [PubMed] [Google Scholar]
- 30.Xue LY, Chiu SM, Fiebig A, Andrews DW, Oleinick NL. Photodamage to multiple Bcl-xL isoforms by photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene. 2003;22:9197–204. doi: 10.1038/sj.onc.1207019. [DOI] [PubMed] [Google Scholar]
- 31.Saeki K, You A, Okuma SA, Susin SA, Kroemer G, Takaku GF. Bcl-2 downregulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ. 2000;7:1263–9. doi: 10.1038/sj.cdd.4400759. [DOI] [PubMed] [Google Scholar]
- 32.Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14:1029–39. doi: 10.1038/sj.cdd.4402099. [DOI] [PubMed] [Google Scholar]
- 33.Weyergang A, Berg K, Kaalhus O, Selbo PK. Photodynamic therapy targets the mTOR signaling network in vitro and in vivo. Mol Pharm. 2009;6:255–64. doi: 10.1021/mp800156e. [DOI] [PubMed] [Google Scholar]
- 34.Peng TI, Chang CJ, Guo MJ, Wang YH, Yu JS, Wu HY, et al. Mitochondrion-targeted photosensitizer enhances the photodynamic effect-induced mitochondrial dysfunction and apoptosis. Ann N Y Acad Sci. 2005;1042:419–28. doi: 10.1196/annals.1338.035. [DOI] [PubMed] [Google Scholar]
- 35.Berg K, Moan J. Lysosomes as photochemical targets. Int J Cancer. 1994;59:814–22. doi: 10.1002/ijc.2910590618. [DOI] [PubMed] [Google Scholar]
- 36.Moan J, Berg K, Bommer JC, Western A. Action spectra of phthalocyanines with respect to photosensitization of cells. Photochem Photobiol. 1992;56:171–5. doi: 10.1111/j.1751-1097.1992.tb02144.x. [DOI] [PubMed] [Google Scholar]
- 37.Ambroz M, MacRobert AJ, Morgan J, Rumbles G, Foley MS, Phillips D. Time-resolved fluorescence spectroscopy and intracellular imaging of disulphonated aluminium phthalocyanine. J Photochem Photobiol B. 1994;22:105–17. doi: 10.1016/1011-1344(93)06955-3. [DOI] [PubMed] [Google Scholar]
- 38.Leach MW, Higgins RJ, Autry SA, Boggan JE, Lee SJ, Smith KM. In vitro photodynamic effects of lysyl chlorin p6: cell survival, localization and ultrastructural changes. Photochem Photobiol. 1993;58:653–60. doi: 10.1111/j.1751-1097.1993.tb04948.x. [DOI] [PubMed] [Google Scholar]
- 39.Kessel D, Morgan A. Photosensitization with etiobenzochlorins and octaethyl-benzochlorins. Photochem Photobiol. 1993;58:521–6. doi: 10.1111/j.1751-1097.1993.tb04925.x. [DOI] [PubMed] [Google Scholar]
- 40.Moan J, Berg K. The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen. Photochem Photobiol. 1991;53:549–53. doi: 10.1111/j.1751-1097.1991.tb03669.x. [DOI] [PubMed] [Google Scholar]
- 41.Strømhaug PE, Berg TO, Berg K, Seglen PO. A novel method for the study of autophagy: Destruction of hepatocytic lysosomes, but not autophagosomes, by the photosensitizing porphyrin tetra(4-sulfonatophenyl)porhine (TPPS4) Biochem J. 1997;321:217–25. doi: 10.1042/bj3210217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berg K, Folini M, Prasmickaite L, Selbo PK, Bonsted A, Engesaeter BØ, et al. Photochemical internalization: A new tool for drug delivery. Curr Pharm Biotechnol. 1991;8:362–72. doi: 10.2174/138920107783018354. [DOI] [PubMed] [Google Scholar]
- 43.Hogset A, Prasmickaite L, Selbo PK, Hellum M, Engesaeter BØ, Bonsted A, et al. Photochemical internalization in drug and gene delivery. Adv Drug Deliv Rev. 2004;56:95–115. doi: 10.1016/j.addr.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 44.Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem. 1998;273:21883–92. doi: 10.1074/jbc.273.34.21883. [DOI] [PubMed] [Google Scholar]
- 45.Fader CM, Colombo MI. Autophagy and multi-vesicular bodies: two closely related partners. Cell Death Differ. 2009;16:70–8. doi: 10.1038/cdd.2008.168. [DOI] [PubMed] [Google Scholar]
- 46.Berg K, Moan J. Lysosomes and microtubules as targets in photochemotherapy of cancer. Photochem Photobol. 1997;65:403–9. doi: 10.1111/j.1751-1097.1997.tb08578.x. [DOI] [PubMed] [Google Scholar]
- 47.Berg K, Prydz K, Moan J. Photochemical treatment with the lysosomally localized dye tetra(4-sulfonatophenyl)porphine results in lysosomal release of the dye but not of N-acetyl-D-glucosaminidase activity. Biochim Biophys Acta. 1993;1158:300–6. doi: 10.1016/0304-4165(93)90029-8. [DOI] [PubMed] [Google Scholar]
- 48.Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008;4:849–50. doi: 10.4161/auto.6845. [DOI] [PubMed] [Google Scholar]
- 49.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 50.Amenta JS, Hlivko TJ, McBee AG, Shinozuka H, Brocher S. Specific inhibition by NH4Cl of autophagy-associated proteloysis in cultured fibroblasts. Exp Cell Res. 1978;115:357–66. doi: 10.1016/0014-4827(78)90289-6. [DOI] [PubMed] [Google Scholar]
- 51.Caruso JA, Mathieu PA, Joiakim A, Leeson B, Kessel D, Sloane B, et al. Differential susceptibilities of murine hepatoma 1c1c7 and Tao cells to the lysosomal photosensitizer NPe6: influence of aryl hydrocarbon receptor on lysosomal fragility and protease contents. Mol Pharmacol. 2004;65:1016–28. doi: 10.1124/mol.65.4.1016. [DOI] [PubMed] [Google Scholar]
- 52.Kessel D, Reiners JJ., Jr Initiation of apoptosis and autophagy by the Bcl-2 antagonist HA14–1. Cancer Lett. 2007;249:294–9. doi: 10.1016/j.canlet.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castelli M, Reiners JJ, Jr, Kessel D. Mechanism for the proapoptotic activity of ursodeoxycholic acid: effects on Bcl-2 conformation. Cell Death Differ. 2004;11:906–14. doi: 10.1038/sj.cdd.4401433. [DOI] [PubMed] [Google Scholar]
- 54.Hall MN. mTOR-What does it Do? Transplant Proc. 2008;40:5–8. doi: 10.1016/j.transproceed.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 55.Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–65. doi: 10.4161/auto.6555. [DOI] [PubMed] [Google Scholar]
- 56.Buytaert E, Matroule JY, Durinck S, Close P, Kocanova S, Vandenheede JR, et al. Molecular effectors and modulators of hypericin-mediated cell death in bladder cancer cells. Oncogene. 2008;27:1916–29. doi: 10.1038/sj.onc.1210825. [DOI] [PubMed] [Google Scholar]
- 57.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: Cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–48. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 58.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Kessel D, Reiners JJ., Jr Initiation of apoptosis and autophagy by the Bcl-2 antagonist HA14–1. Cancer Lett. 2007;249:294–9. doi: 10.1016/j.canlet.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Granville DJ, Levy JG, Hunt DW. Photodynamic therapy induces caspase-3 activation in HL-60 cells. Cell Death Differ. 1997;4:623–8. doi: 10.1038/sj.cdd.4400286. [DOI] [PubMed] [Google Scholar]
- 61.Kessel D, Reiners JJ, Jr, Hazeldine ST, Polin L, Horwitz JP. The role of autophagy in the death of L1210 leukemia cells initiated by the new antitumor agents, XK469 and SH80. Mol Cancer Ther. 2007;6:370–9. doi: 10.1158/1535-7163.MCT-05-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elliott A, Reiners JJ., Jr Suppression of autophagy enhances the cytotoxicity of the DNA damaging aromatic amine p-anilinoanaline. Toxicol Appl Pharmacol. 2008;232:169–79. doi: 10.1016/j.taap.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ke MS, Xue LY, Feyes DK, Azizuddin K, Baron ED, McCormick TS, et al. Apoptosis mechanisms related to the increased sensitivity of Jurkat T-cells versus A-431 epidermoid cells to photodynamic therapy with the phthalocyanine Pc 4. Photochem Photobiol. 2008;84:407–14. doi: 10.1111/j.1751-1097.2007.00278.x. [DOI] [PubMed] [Google Scholar]
- 64.Lam M, Oleinick NL, Nieminen AL. Photodynamic therapy-induced apoptosis in epidermoid carcinoma cells. Reactive oxygen species and mitochondrial inner membrane permeabilization. J Biol Chem. 2001;276:47379–86. doi: 10.1074/jbc.M107678200. [DOI] [PubMed] [Google Scholar]