

Abstract
The Sleeping Beauty (SB) transposon system has been used as an insertional mutagenesis tool to identify novel cancer genes. To identify glioma-associated genes, we evaluated tumor formation in brain tissue from 117 transgenic mice that had undergone constitutive SB-mediated transposition. Upon analysis, 21 samples (18%) contained neoplastic tissue with features of high grade astrocytomas. These tumors expressed glial markers and were histologically similar to human glioma. Genomic DNA from SB-induced astrocytoma tissue was extracted and transposon insertion sites were identified. Insertions in the growth factor gene Csf1 were found in 13 of the 21 tumors (62%), clustered in introns 5 and 8. Using RT-PCR, we documented increased Csf1 RNAs in tumor versus adjacent normal tissue, with identification of transposon-terminated Csf1 mRNAs in astrocytomas with SB insertions in intron 8. Analysis of human glioblastomas revealed increased levels of Csf1 RNA and protein. Together, these results indicate that SB-insertional mutagenesis can identify high-grade astrocytoma-associated genes, and they imply an important role for CSF1 in the development of these tumors.
Keywords: Astrocytoma, Sleeping Beauty, Mutagenesis, Colony stimulating factor-1
Introduction
Among glial brain tumors (gliomas), high-grade astrocytomas including glioblastoma multiforme (GBM) are the deadliest and most common subtype (1). Their invasiveness and heightened rate of recurrence makes the prognosis of these tumors particularly grim. Although much is known about the histopathology and clinical course of astrocytomas, knowledge of molecular mechanisms leading to their development is still lacking. Reverse genetic approaches have been successful in promoting glioma formation in mice (2). However, these techniques require a candidate gene approach and are of limited use with respect to novel tumor gene discovery. To address this, retroviral and transposon-based tools such as SB have been developed which randomly mutate the genome and allow for rapid target identification in genetic screens (3, 4).
SB has been developed as a gene discovery tool in mice (5, 6) and has been shown to promote cancer formation due to transposon-mediated alterations in gene structure and expression (7-10). In this report we histologically characterize a panel of SB-induced gliomas and describe the identification of candidate genes including Colony stimulating factor-1 (Csf1), which is insertionally mutated in the majority of the gliomas.
Materials and Methods
Mice
Rosa26-SB11 transgenic animals (8), and low-copy (lines 68 and 76) T2/onc animals used in this study were previously described (7) with the exception of animals RosaA33 and RosaD34 which harbor a slightly modified mutagenic element (T2oncATG) containing elements designed to enhance translation of transcripts initiating in the over-expression elements of T2/onc (16). p19Arf and Blm mutant mice were previously described (11, 12) Transgenic animals were sacrificed when they showed any signs of morbidity, including neurologic symptoms. Table S1 summarizes the neurologic symptom/necropsy findings of the animals that were found to have brain tumors.
DNA extraction and linker-mediated PCR
FFPE brain tissue was sectioned and an adjacent parallel slide stained with hematoxylin and eosin (H&E) was used as a reference for tumor-tissue macrodissection. Tissue was de-paraffinized in xylene and digested with proteinase K. DNA was extracted with phenol/chlorform followed by ethanol precipitation.
Linker-mediated PCR and CIS identification
Linker-mediated PCR was performed as described (13) with the following modifications: Primary PCR products were purified using the Genelute system (Sigma) and diluted 1:25 prior to secondary PCR. Nested PCR products were then “shot-gun cloned” into the TOPO PCR 2.1 vector (Invitrogen) and transformed into TOP10F competent cells (Invitrogen). Bacterial clones were sequenced by Agencourt Biosciences using the M13F primer. PCR products were also generated using “bar-coded” primers and subjected to 454-pyrosequencing. Transposon insertion site mapping was performed as described (14) using the mm9 genome build. In addition to eliminating local hops, we eliminated insertions which mapped to the En2 (n=54) or the Foxf2 (n=2) loci because the T2/onc transposon includes the splice acceptor and splice donor sequences (respectively) from these genes. CISs were determined as described (15) using an expected fraction (Efr) of .005 and a dataset of 1000 insertions. Using these criteria, a CIS was defined as 2 insertions within 13kb, 3 within 269kb and 4 within 878kb. Insertions from the same tumor were not allowed to exclusively define a CIS.
Mouse tumor RNA extraction and RT-PCR
FFPE tissue from tumor and adjacent normal tissue was de-paraffinized and RNA was extracted using the HighPure RNA paraffin system (Roche). cDNA was generated using the Superscript III cDNA synthesis kit (Invitrogen) and primers spanning the junction between exons 2 and 3 (mmCSF1 Exon2 For and mmCSF1 Exon3 Rev) of Csf1 as well as primers to β-actin (mmB-actinRTf and mmB-actinRTr). See Table S2 for primer sequences. Fluorescence was measured (using SYBRR® Green intercalation) on an Applied Biosystems AB7900HT PCR machine.
Fusion transcript identification
RNA derived from tumor and adjacent normal FFPE mouse tissue was subjected to whole transcriptome amplification using the WT-Ovation FFPE system (NuGEN). Primers “mmCSF1 exon 8” and “Univ Rev T2/onc for fusion” were then used to amplify fusion products between the 8th exon of the Csf1 gene and the transposon splice acceptor. The products were then sequenced using primer “En2 SA RTR2 nested”. See Table S2 for primer sequences.
Human RNA extraction and RT-PCR
RNA from thin-sectioned frozen GBM tissue was extracted in Trizol (Invitrogen), column purified (Qiagen) and converted to cDNA using the Superscript III cDNA synthesis kit (Invitrogen). Custom Taqman (Applied Biosystems) qRT-PCR assays were designed to assess CSF1 and β-ACTIN expression on an AB7900HT PCR machine. See Table S2 for primer sequences.
IHC
Stains were performed on a DAKO Autostainer (Dako North America) using the Dual Link Envision+ (Dako) detection system, with EDTA or citrate for antigen retrieval and DAB as the chromogen. All antibodies (Table S3) were optimized with serial dilutions. Stains for CSF1 and CSF1R were scored using the following semiquantitative four tiered scale: 0=negative; 1=reactivity in <10% of the cells; 2=strong reactivity<50% of cells or weak reactivity in >50% of cells; 3=strong reactivity in >50% of cells. For the human TMA, the median of all scorable cores was used for a summary score.
Western Blotting
Frozen GBM tissue was lysed in RIPA buffer. Lysates from normal brain tissue were purchased from Protein Biotechnologies. Samples were boiled in reducing Laemmli buffer, run on 4-15% precast SDS-PAGE gels, transferred to PVDF membranes, blocked in TBST buffer + 5% NFDM, and probed with antibodies to CSF1, (Table S3). The CSF1 blots were then stripped and reprobed for GAPDH. For the each blot, the “ImageJ” program was used to quantify relative band intensity.
Biospecimens Collection
Human tissue used in this study was obtained according to Mayo Clinic Institutional Review Board (IRB) policies.
Results
Mice doubly transgenic for a constitutively expressed SB transposase as well as the mutagenic transposon (T2/onc) develop high grade astrocytomas
To evaluate whether SB mutagenesis can promote brain tumor formation, brain tissue was isolated and analyzed from animals that had mobilizing transposons due to being doubly transgenic for both a constitutively expressed SB transposase (Rosa26-SB11) (6) as well as a chromosomal concatemer harboring T2/onc mutagenic transposons (7). Brains from mice with mobilizing transposons on an otherwise wild-type genetic background as well as mice with mobilizing transposons on p19Arf+/-; p19Arf-/- or Blm mutant tumor pre-disposed genetic backgrounds were examined (11, 12). Although some gliomas identified have been briefly described in a prior publication (16), a careful histologic characterization and extensive glioma transposon insertion site analysis was not performed. In addition, gliomas on tumor pre-disposed genetic backgrounds have not previously been described.
Formalin-fixed paraffin embedded (FFPE) brain tissue from 117 mice with mobilizing transposons on wild-type and tumor-predisposed genetic backgrounds was examined for tumors. We identified 21 (18%) gliomas (Table 1 and Table S4). In addition to FFPE, frozen tissue obtained at necropsy was also available for 1 glioma (76Rosa164). Among the 91 brains examined from mice on an otherwise wild-type genetic background, 13 (14%) SB-induced gliomas were identified. Of the specimens taken from p19Arf-/- and p19Arf+/- animals, 1 of 11 (9%) and 6 of 12 (50%) contained glioma tissue, respectively. Finally, 1 of 3 Blm mutant specimens had an identifiable glioma (Table S4). No gliomas were identified in 42 control samples (Table S4) without mobilizing transposons, suggesting that these tumors were SB-induced. A single brain tumor was identified in a Rosa26-SB11(-); T2/onc(+); p19Arf-/- animal. However, this lesion was classified as a primitive neuroectodermal tumor (PNET), based on histology and expression of the neuronal marker synaptophysin (Fig. S1).
Table 1.
Comprehensive list of SB-induced astrocytomas
| Mouse | Type | Genetic Background | Intracereberal Location | #unique insertions† |
|---|---|---|---|---|
| 68;Rosa 438 | AA | Hemispheric | 7 | |
| 68;Rosa 439 | AA | Hemispheric | 5 | |
| 68;Rosa 447 | GBM | Posterior fossa | 28 | |
| 68;Rosa 544 | AA | Hemispheric | 32 | |
| p19 68;Rosa 117 | AA | p19 +/- | Hemis./Post. Fossa | 18 |
| 76;Rosa 616 | GBM | Posterior fossa | 47 | |
| 76;Rosa 164 | AA | Posterior fossa | 131 | |
| 76;Rosa 190 | GBM | Hemispheric | 1 | |
| 76;Rosa 415 | AA | Hemispheric | 7 | |
| 76;Rosa 453 | AA | Posterior fossa | 1 | |
| 76;Rosa 520 | AA | Hemispheric | 21 | |
| 76;Rosa 582 | AA | Hemispheric | 157 | |
| BL76;Rosa 150 | AA | Blm -/- | Posterior fossa | 3 |
| p19 76;Rosa 281 | AA | p19 +/- | Posterior fossa | 36 |
| p19 76;Rosa 292 | GBM | p19 +/- | Posterior fossa | 71 |
| p19 76;Rosa 298 | AA | p19 +/- | Posterior fossa | 67 |
| p19 76;Rosa 327 | GBM | p19 -/- | Posterior fossa | 17 |
| p19 76;Rosa 333 | GBM | p19 +/- | Hemispheric | 90 |
| p19 76;Rosa 339 | AA | p19 +/- | Hemis./Post. fossa | 89 |
| T2/oncATG;Rosa A33 | AA | Posterior fossa | 32 | |
| T2/oncATG;Rosa D34 | AA | Posterior fossa | 27 |
Represents unique insertions after removal of local hops
Upon extensive histologic examination, all 21 gliomas showed characteristics of high grade astrocytomas including an invasive phenotype (Fig. 1A). A subset of these tumors (n=6, 29%) were analogous to grade IV glioblastoma multiforme (GBM) due to high mitotic activity and pseudopalisading necrosis (Fig. 1A). Tumors lacking necrosis (n=15, 71%) were classified as grade III (anaplastic) astrocytomas (Table 1). The majority of the tumors were large and involved entire lobes, brain hemispheres or both hemispheres and posterior fossa (Table 1). Eleven cases (52%) involved the posterior fossa. Eight of these tumors in the posterior fossa showed predominant involvement of the brainstem, while three were limited to the cerebellum. By immunohistochemical (IHC) analysis, the tumors stained positive in a subset of cells for the glial markers GFAP and S100 (Fig. 1B, C) but were negative for the neuronal markers Map2 and synaptophysin (Fig. S1). Several of the SB-induced gliomas were examined by IHC for Olig1 and Olig2 expression because these tumor types have been shown to express variable levels of these bHLH transcription factors (17). In 7 of 7 tumors, variable positive staining was seen for Olig2 (Fig. 1D), whereas Olig1 was weakly positive in only 2 of 7 (29%). Many of the mice developed other cancers (mainly hematopoietic, (16)) due to the body-wide transposition (see Table S1). To rule out malignancies of hematopoietic/histiocytic origin, IHC was conducted for CD45, CD68 and lysozyme in a subset of tumors. Positive staining was seen in macrophages/microglia, but tumor cells lacked expression of these markers (Fig. S2). Together, these results strongly support a glial origin of these tumors.
Figure 1. SB-Induced Brain Tumors Display Characteristics of High Grade Astrocytomas.
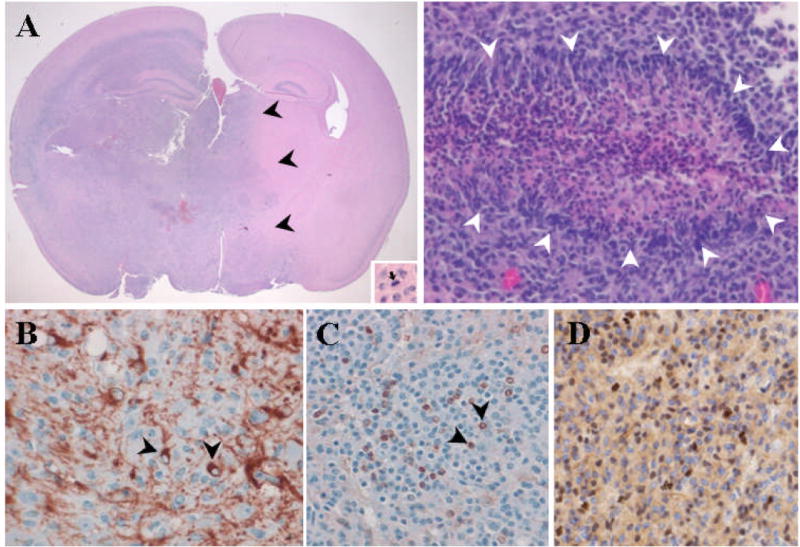
(A) Left: Low power image of large, SB-induced astrocytoma including mitotic figure (inset). Note invasive edge (black arrowheads). Right: Image depicting pseudopallisading necrosis (white arrowheads), a classic histologic feature of human GBM. (B, C, D,) A subset of tumor cells stain positive for glial markers GFAP (black arrowheads, B), S100 (Black arrowheads, C), and Olig2 (D).
Identification of genes tagged with T2/onc
Using sections stained with hemotoxylin and eosin (H&E) as a reference, adjacent unstained sections were micro-dissected in order to enrich for tumor cells. Genomic DNA was extracted and subjected to a previously described linker-mediated PCR approach (7) to amplify genomic fragments containing transposon insertions. These products were then cloned and sequenced. PCR amplified DNA from a subset of the tumors was also subjected to 454 pyrosequencing. Upon analysis of the sequencing data, 1375 unique transposon insertions were identified among the 21 gliomas. Since SB transposons frequently re-integrate into regions adjacent to the donor locus (18), chromosome 1 insertions from 76Rosa (458 of 1196, 38%) mice and chromosome 15 insertions from 68Rosa (30 of 120, 25%) mice were eliminated from the dataset. The locations of the transposon concatemers in the T2/oncATG lines are currently unknown. Therefore all unique insertions from the two gliomas which developed in these animals (n=59) were retained for analysis. After eliminating known local transposition events (local hops), a collection of 887 unique insertions was analyzed (Table S5).
Upon analysis of the dataset, 27 distinct genes had transposon insertions in two or more tumors. Using a previously described method for assigning non-random clusters of proviral insertions, 5 of these genes met the criteria for Common Insertion Sites (CISs) (15) (Table 2). These insertion sites map to distinct locations in and around Csf1, Fli1, Mkln1, Vps13a, and Sfi1.
Table 2.
Common insertion sites (CISs)‡
| Tumor | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse Gene | 76;Rosa582 | 76;Rosa164 | p19 76; Rosa333 | p19 76; Rosa339 | p19 76; Rosa292 | 76;Rosa616 | p19 76; Rosa281 | 68;Rosa544 | T2/oncATG;R osaA33 | 68;Rosa447 | 76;Rosa520 | p19 68; Rosa117 | p19 76; Rosa327 | 68;Rosa438 | 76;Rosa415 | #Tumors |
| Csf1 | 1 | 26 | 26 | 25 | 1 | 9 | 19 | 19 | 55 | 1 | 6 | 8 | 1 | 13 | ||
| Fli1 | 1 | 20 | 3 | 1, 12§ | 4 | |||||||||||
| Mkln1 | 2 | 1 | 2 | 3 | ||||||||||||
| Vps13a | 1 | 2 | 2 | |||||||||||||
| Sfi1 | 1 | 4 | 2 | |||||||||||||
Numbers represent times insertion was identified within a given tumor
CIS as defined by Mikkers et al., 2002
2 Unique insertions were identified at the Fli1 locus for tumor 544. See Table S2
The most commonly identified insertion site for the SB transposon in gliomas was the Csf1 locus. The majority of the 21 gliomas (n=13, 62%) contained identifiable T2/onc insertions in Csf1 (Table 2). Although they did not correlate with tumor location or grade, 2 distinct insertional sub-clusters were identified within Csf1 (Fig. 2A). Of the 13 insertions, 6 were identified in the 5th intron and 6 in the 8th intron. A 13th insertion was located ~9kb upstream of the Csf1 locus. With a single exception (68Rosa447, see Fig. 2A) all of the Csf1 transposon insertions were inversely oriented to the gene’s transcription. Csf1 has not been identified as a CIS in SB models of other tumors (7-10, 16, 19, 20), suggesting that transposon mutagenesis of Csf1 correlates with high-grade astrocytoma formation and not with the formation of other SB-induced cancers.
Figure 2. Clustering of SB insertions at the Csf1 locus as well as elevated expression suggests a functional connection to pathogenesis of high grade astrocytomas.
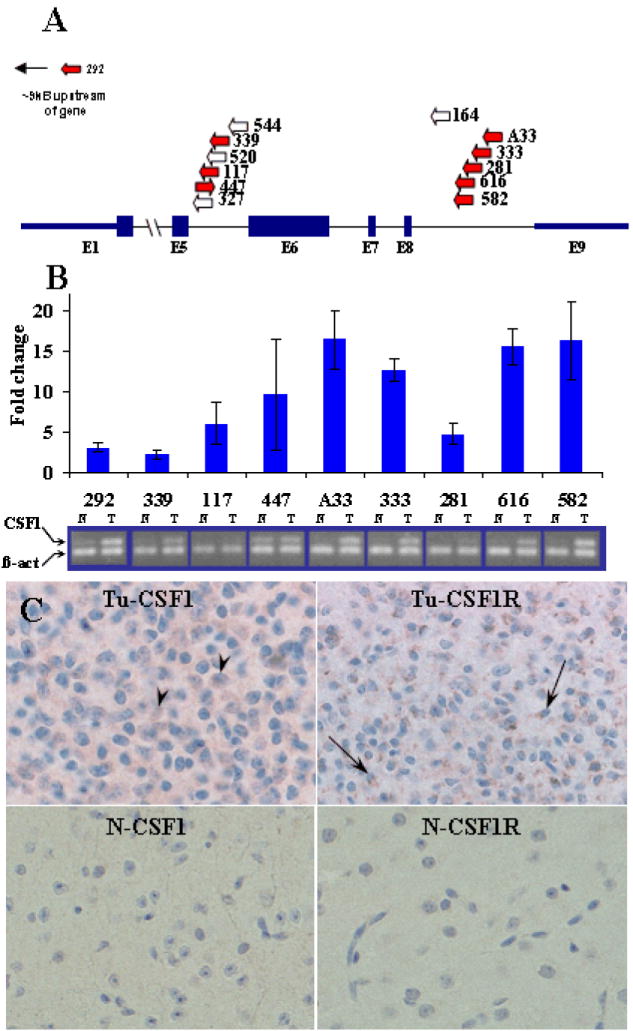
(A) SB insertions at the Csf1 locus cluster in the 5th and 8th introns and are largely oriented in the same direction as Csf1 transcription (arrows). Underlined insertions denote chimeric transcripts. Blue arrows indicate samples used for comparative PCR analysis in B. (B) qRT (above) and semi-quantitative (below) PCR analysis of primary murine GBM samples showing elevation of Csf1 transcript in tumor (T) vs. adjacent normal (N) tissue. Error bars represent standard deviation of fold-change. (C) Top: Neoplastic cells from mouse tumor 333 display immunohistochemical staining of Csf1 (arrowheads) and its receptor CSF1R (arrows) respectively. Bottom: In contrast, adjacent normal tissue from the same specimen did not show appreciable staining for either Csf1 or Csf1R.
Transposon insertions in the murine CSF1 locus correlate with mRNA over-expression
To assess whether expression was altered in tumors with Csf1 insertions, semi-quantitative and quantitative RT-PCR was performed. In 9 of the 9 samples evaluated, the levels of Csf1 transcript were elevated compared to adjacent normal tissue (Fig. 2B). These included 3 specimens harboring intron 5 insertions, 5 specimens with intron 8 insertions, and the one sample for which there was an upstream insertion (Fig. 2A). In addition, IHC for Csf1 as well as the Csf1 receptor (Csf1R) was performed on a subset of the SB-induced gliomas (Fig. 2C). Positive Csf1 staining in scattered tumor cells was observed in 8 of 14 (57%) specimens, and positive Csf1R staining was seen in 11 of 13 (85%) specimens. However, neither Csf1 nor Csf1R staining correlated with Csf1 insertion status.
Altered mRNA splicing is one mechanism by which the T2/onc transposon is thought to modulate gene expression (7). We sought to determine whether chimeric Csf1:T2/onc transcripts were being produced in tumors harboring intragenic Csf1 insertions. To address this, RNA was extracted from parallel tissue sections and subjected to whole-transcriptome amplification (see Materials and Methods). Oligonucleotide primers were then designed to amplify putative T2/onc:Csf1 fusion transcripts by RT-PCR. Among tumors with insertions in the 8th intron, 5 of 5 tested yielded chimeric transcripts between exon 8 of Csf1 and the engrailed 2 (En2) splice acceptor sequence contained within the T2/onc transposon (Fig. 2A, and Fig. S3). Similar products were not identified for tumors harboring insertions in the 5th intron.
CSF1 and its receptor CSF1R are over-expressed in high-grade human astrocytomas
Based on SB mutagenesis and over-expression of CSF1 in murine astrocytomas, as well as a previous report correlating CSF1 expression and glioma grade (21), we performed IHC on human GBM tissue microarrays (TMAs). A semi-quantitative scoring strategy (0 to 3+), revealed that CSF1 was over-expressed (3+) in the majority of samples (64%, n=69) and in cells with histologic features of transformed astrocytes (Figs. 3A, S4). It has been shown that CSF1R is transcriptionally up-regulated in normal and neoplastic astrocytes (22). Furthermore, it has been suggested that CSF1 and CSF1R may act via an autocrine signaling loop to promote survival of cervical cancer cells (23). Therefore, we evaluated CSF1R expression in parallel sections of the same TMA. As with CSF1, CSF1R was over-expressed in the majority of cases (72%, n=68) (Figs. 3A, S4). Western blotting was performed to assess relative CSF1 levels in human GBMs. As illustrated in Fig. 3B, GBM lysates (T1-T15) contain increased levels (~5 fold) of CSF1 protein compared to non-tumor controls (N1-N5). We performed Taqman®-mediated qRT-PCR on human GBM samples as well as non-neoplastic (gliotic) brain tissue to evaluate CSF1 transcript levels. Our results indicate that GBM tissue contains ~4-fold more CSF1 transcript than non-neoplastic controls (Fig. 3C).
Figure 3. Over-expression of CSF1 in human GBM.
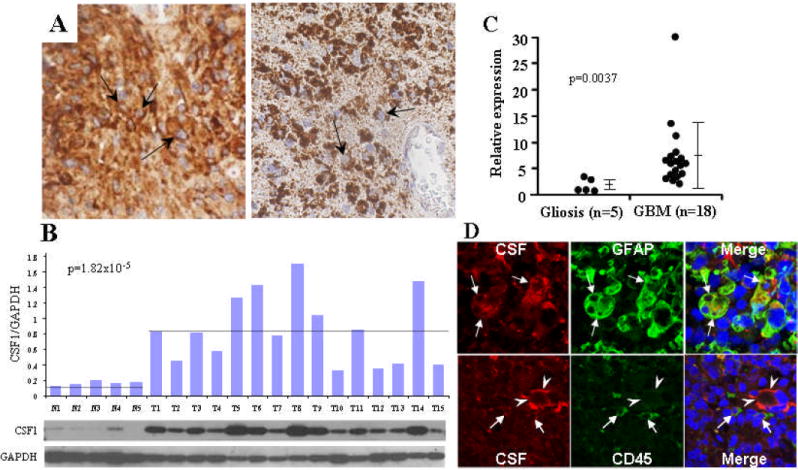
(A) Immunohistochemical staining showing CSF1 (Arrows, right) and CSF1R (Arrows, left) staining in human GBM tissue sections. (B) Western blot of whole cell lysates from normal (post-mortem) brain (N1-N5) as well as from primary human GBMs (T1-T15). Bars represent the ratio of CSF1 to GAPDH signal intensity. (C) qRT-PCR result showing elevated levels of CSF1 transcript in normal (n=5) vs. GBM (n=18) tissue (p-value generated using Wilcoxon rank-sum test). (D) Top: Dual label immunofluorescence images illustrating tumor cells (white arrows) expressing both CSF1 (Red) and the glial marker GFAP (Green). Bottom: Separate immunofluorescence experiment illustrating distinct CSF1 staining in (red) tumor cells (arrowheads) and CD45 staining in (green) infiltrating lymphoid/myeloid cells.
Neoplastic cells appear to be the source of CSF1 production
CSF1 over-expression has been observed in cell lines derived from neoplastic and non-neoplastic astrocytes (24). To determine whether tumor cells were producing CSF1 in the context of GBM, we conducted immunofluorescence (IF) experiments on human GBM sections using antibodies to GFAP), a marker commonly used to aid in the diagnosis of astrocytomas, as well as to CSF1. The staining pattern indicated a strong overlap between cells staining positive for both proteins (Fig. 3D, top). Because there is evidence that activated microglial cells express CSF1 (25), we co-stained GBM specimens with antibodies to the pan-hematolymphoid marker CD45, as well as to CSF1. These antibodies labeled distinct cell populations (Fig. 3D, bottom), suggesting tumor cells, not associated macrophages/microglia, are the primary source of the observed CSF1 expression.
Discussion
We have shown that mobilization of T2/onc transposons by a constitutively active SB transposase can promote high-grade astrocytoma formation on an otherwise wild-type genetic background as well as tumor-predisposed genetic backgrounds. Because deletions involving the ARF locus are common in human gliomas (11), we chose to perform mutagenesis on both p19Arf+/- and p19Arf-/- backgrounds. Although limited numbers of mice were examined, the penetrance of SB-induced gliomas appears higher on the p19Arf+/- background than the p19Arf-/- background. Because p19Arf-/- mice with mobilizing transposons die at a very young age of other tumors such as leukemias and lymphomas (16), we hypothesize that they do not live sufficient long enough to develop gliomas. Larger cohorts of p19Arf and Blm mutant animals with mobilizing transposons will be necessary to fully investigate if performing mutagenesis on these backgrounds influences glioma penetrance.
Irrespective of the genetic background on which they arose, the SB-induced gliomas displayed many of the histopathological features of human astrocytomas and were negative for neuronal and hematopoietic markers. Examination of genomic DNA from these tumors identified several loci that are non-randomly associated with glioma development. The most common insertionally mutated gene in the SB-induced gliomas was Csf1. The Csf1 locus produces alternatively spliced transcripts (in mice and humans) leading to the translation of secreted as well as cell-surface proteins (reviewed in (26)). Csf1 is a cytokine originally shown to facilitate growth of hematopoietic cells in mice (27). It has since been shown that CSF1 expression is up-regulated in breast cancer (28), female reproductive system cancers (29), tenosynovial giant cell tumor (30), and glioma (21). These studies suggest that increased expression of CSF1 may facilitate tumor growth by recruitment of receptor-bearing macrophages. Recent evidence suggests that glioma-associated microglia express membrane type 1 metalloprotease (MT1-MMP) which promotes tumor cell invasion by compromising the extracellular matrix (31). Although previous studies have demonstrated CSF1 over-expression in the context of cancer, this report provides the first forward mutagenesis-based evidence associating Csf1 with the formation of glioma.
As mentioned above, we identified two distinct transposon insertional clusters at the Csf1 locus, both of which correlated with up-regulation of the Csf1 message. For tumors harboring 8th intron insertions, chimeric transcripts were identified between exon 8 of Csf1 and the En2 splice acceptor sequence in T2/onc. As the translational stop for Csf1 resides within exon 8, these transcripts are not predicted to alter the Csf1 open reading frame, indicating that it is the level of CSF1 and not its altered structure that is involved in gliomagenesis. Interestingly, this 8th intron cluster of insertions seemed to correlate with the most robust transcriptional up-regulation (Fig. 2B). It has been suggested that the human CSF1 transcript may be unstable due to the presence of AU-rich elements (AREs) in the 3’ UTR (32). In addition, the miRanda algorithm (developed to identify potential micro-RNA (miRNA) targets) predicts multiple miRNA binding sites in the 3’UTR of human CSF1 (33). Among these is miR-128, which has been shown to inhibit glioma cell self renewal and to be under-expressed in glioma (34). If the 3’UTR is important for negative regulation of CSF1, downregulation of factors such as miR-128 may promote aberrant over-expression of CSF1. In addition, exclusion of 3’UTR regulatory elements due to premature splicing may be the mechanism by which the SB transposon promotes over-expression in murine tumors harboring 8th intron Csf1 insertions (Fig. S3C).
In addition to its ability to alter mRNA splicing, T2/onc contains a murine stem cell virus long terminal repeat (LTR) sequence designed to promote transcription of nearby genes (35). This may explain the observed Csf1 up-regulation in tumor p1976Rosa292 within which we identified an insertion upstream of Csf1 (Fig. S3A).
We were not able to detect fusion transcripts in FFPE tumors with insertions in the 5th intron of Csf1. This could be due to technical limitations, or it could be that these insertions promote up-regulation by an entirely different mechanism. For example, the intron 5 insertions may act as intragenic enhancers of full-length or truncated Csf1 gene products (Fig. S3B). Exon 6 of Csf1 undergoes alternative splicing (36), which may be altered due to the intron 5 cluster of insertions adjacent to this region. Regardless, clustering of the intron 5 transposon insertions implies functional significance. It should be noted that even though SB-insertion creates chimeric CSF1 transcript in murine glioma, these transcripts do not alter the CSF1 open reading frame, apparently highlighting that it is the level of CSF1 and not its altered structure, which is involved in gliomagenesis.
CSF1 is up-regulated in high-grade human astrocytomas (21). Based on the TCGA data and other sequencing studies, this upregulation is likely not a result of copy number alterations, exonic point mutations, or promoter methylation (37, 38). The question remains as to whether CSF1 is capable of enhancing tumor growth. There are multiple expressed isoforms of the CSF1 protein (36). Interestingly, exclusive expression of the cell surface CSF1 isoform is capable of inducing macrophage-mediated cell killing in an allograft model of neoplastic rat glioma (39). However, Komohara and colleagues propose a model in which secretion of CSF1 by glioma cells results in the recruitment of tumor associated macrophages (TAMs), provoking tumor growth by secreting molecules such as TGF-β, which suppresses normal immune response (21). Our immunostaining experiments suggest that CSF1 is produced by tumor cells and therefore support this model.
The second-most frequently mutated locus identified in the SB-induced gliomas was the ETS-like transcription factor Friend leukemia integration factor 1 (Fli1, Table 2). FLI1 has been shown to promote cancers including Ewing’s sarcoma (40) and has been identified (as a CIS) in other SB-induced tumor types, including hematopoetic tumors (8, 16) and hepatocellular carcinoma (9).
The remaining CIS (see Table 2), consisted of Muskelin 1 (Mkln1), Vacuolar protein-sorting 13a (Vps13A), and Suppressor of FIL1 (Sfi1). MKLN1, is a protein known to mediate cell adhesion (41), is expressed in the mouse brain (42), and was recently shown to be amplified in human GBM (43). Neither VPS13A nor SFI1 have been implicated in glioma formation. However, Vps13A is expressed in the mouse brain (44), and Sfi1 has been shown to induce G2 arrest in Saccharomyces cerevisiae (45).
Of note, we did not identify transposon insertions in the known glioma oncogenes Pdgf and Egfr. However, single insertions were found in the Pten, Akt2 and Akt3 genes (Table S1). Other genes known to be involved in human gliomas were also not insertionally mutated in the SB-induced gliomas examined so far. These differences could be due to intrinsic species-specific differences in brain tumor development, limitations of the T2/onc mutagenic elements, the timing of the cycles of SB insertion/deletion, and/or due to methodological differences related to using DNA obtained from fresh versus formalin-fixed specimens. The latter possibility is unlikely since the DNA used for linker-mediated PCR for tumor 76Rosa164 was derived from frozen material and we recovered Csf1 insertions from this tumor. SB screens are not absolutely unbiased. For example, the insertion of the SB T2/Onc vector may not be able to recapitulate the effects of certain point mutations, such as activating mutations in the IDH1 gene (38). Instead, we might expect SB insertional mutagenesis to reveal pathways and molecules that are upstream or downstream of genes altered directly in human tumors. Finally, it is possible that the SB-induced gliomas represent only one genetic subset of human gliomas. Human gliomas are heterogenous and the mutations that are present could be influenced by the inherited genetic polymorphisms (46). Additional screens in a variety of contexts will answer these questions definitively.
Recent work has shown that SB insertional mutagenesis promotes glioma formation on an otherwise wild-type genetic background (16). We have carefully histologically evaluated these and additional gliomas arising on tumor pre-disposed genetic backgrounds and have demonstrated that SB may have identified a novel candidate glioma gene. Through locating a high percentage of SB insertions at the mouse Csf1 locus, this study reinforces previous findings that CSF1 over-expression is associated with high-grade gliomas and provides the first forward-mutagenesis evidence that alterations in CSF1 may contribute directly to astrocytoma formation.
Supplementary Material
Acknowledgments
We acknowledge the Tissue and Cell Molecular Analysis (TACMA) core facility at Mayo Clinic, particularly Bridget Hoesley and Vivian Negron for assistance with tissue sectioning, IHC, and IF analysis. We also thank the Biostatistics and Informatics Shared Resource at the University of Minnesota Masonic Cancer Center for assistance with DNA sequence analysis. We thank Dr. Allan Bradley and Dr. Martine Roussel for the p19Arf and Blm mutant mice. Finally, we thank Drs. Michael Taylor and John Ohlfest for productive discussions regarding SB mutagenesis.
Funding: This work was funded by the Minnesota Partnership for Biotechnology and Medical Genomics grant L6526214101 (DL and RBJ); an NIH R01CA113636 (DL); a T32 National Research Service Award (NRSA) institutional training grant PA-06-468, an American Cancer Society pre-doctoral fellowship and by NIH K01CA122183 (LSC).
Footnotes
Potential Conflict of Interest: None
References
- 1.CBTRUS Statistical Report: Primary Brain Tumors in the United States, 1998-2002. Published by the Central Brain Tumor Registry of the United States. 2005 [Google Scholar]
- 2.Fomchenko EI, Holland EC. Mouse models of brain tumors and their applications in preclinical trials. Clin Cancer Res. 2006;12:5288–5297. doi: 10.1158/1078-0432.CCR-06-0438. [DOI] [PubMed] [Google Scholar]
- 3.Liu D, Yang X, Yang D, Songyang Z. Genetic screens in mammalian cells by enhanced retroviral mutagens. Oncogene. 2000;19:5964–5972. doi: 10.1038/sj.onc.1203992. [DOI] [PubMed] [Google Scholar]
- 4.Plasterk RH, Izsvak Z, Ivics Z. Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet. 1999;15:326–332. doi: 10.1016/s0168-9525(99)01777-1. [DOI] [PubMed] [Google Scholar]
- 5.Carlson CM, Dupuy AJ, Fritz S, Roberg-Perez KJ, Fletcher CF, Largaespada DA. Transposon mutagenesis of the mouse germline. Genetics. 2003;165:243–256. doi: 10.1093/genetics/165.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dupuy AJ, Fritz S, Largaespada DA. Transposition and gene disruption in the male germline of the mouse. Genesis. 2001;30:82–88. doi: 10.1002/gene.1037. [DOI] [PubMed] [Google Scholar]
- 7.Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–276. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- 8.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436:221–226. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- 9.Keng VW, Villanueva A, Chiang DY, et al. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat Biotechnol. 2009;27:264–274. doi: 10.1038/nbt.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Starr TK, Allaei R, Silverstein KA, et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747–1750. doi: 10.1126/science.1163040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–2222. [PubMed] [Google Scholar]
- 12.Luo G, Santoro IM, McDaniel LD, et al. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet. 2000;26:424–429. doi: 10.1038/82548. [DOI] [PubMed] [Google Scholar]
- 13.Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- 14.Akagi K, Suzuki T, Stephens RM, Jenkins NA, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32(Database issue):D523–527. doi: 10.1093/nar/gkh013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mikkers H, Allen J, Knipscheer P, et al. High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat Genet. 2002;32:153–159. doi: 10.1038/ng950. [DOI] [PubMed] [Google Scholar]
- 16.Collier LS, Adams DJ, Hackett CS, et al. Whole-body sleeping beauty mutagenesis can cause penetrant leukemia/lymphoma and rare high-grade glioma without associated embryonic lethality. Cancer Res. 2009;69:8429–8437. doi: 10.1158/0008-5472.CAN-09-1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riemenschneider MJ, Koy TH, Reifenberger G. Expression of oligodendrocyte lineage genes in oligodendroglial and astrocytic gliomas. Acta neuropathologica. 2004;107:277–282. doi: 10.1007/s00401-003-0809-8. [DOI] [PubMed] [Google Scholar]
- 18.Luo G, Ivics Z, Izsvak Z, Bradley A. Chromosomal transposition of a Tc1/mariner-like element in mouse embryonic stem cells. Proc Natl Acad Sci USA. 1998;95:10769–10773. doi: 10.1073/pnas.95.18.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rahrmann EP, Collier LS, Knutson TP, et al. Identification of PDE4D as a proliferation promoting factor in prostate cancer using a Sleeping Beauty transposon-based somatic mutagenesis screen. Cancer Res. 2009;69:4388–4397. doi: 10.1158/0008-5472.CAN-08-3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tolar J, Nauta AJ, Osborn MJ, et al. Sarcoma derived from cultured mesenchymal stem cells. Stem cells. 2007;25:371–379. doi: 10.1634/stemcells.2005-0620. [DOI] [PubMed] [Google Scholar]
- 21.Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. 2008;216:15–24. doi: 10.1002/path.2370. [DOI] [PubMed] [Google Scholar]
- 22.Tada M, Diserens AC, Desbaillets I, de Tribolet N. Analysis of cytokine receptor messenger RNA expression in human glioblastoma cells and normal astrocytes by reverse-transcription polymerase chain reaction. J Neurosurg. 1994;80:1063–1073. doi: 10.3171/jns.1994.80.6.1063. [DOI] [PubMed] [Google Scholar]
- 23.Kirma N, Hammes LS, Liu YG, et al. Elevated expression of the oncogene c-fms and its ligand, the macrophage colony-stimulating factor-1, in cervical cancer and the role of transforming growth factor-beta1 in inducing c-fms expression. Cancer Res. 2007;67:1918–1926. doi: 10.1158/0008-5472.CAN-06-1991. [DOI] [PubMed] [Google Scholar]
- 24.Alterman RL, Stanley ER. Colony stimulating factor-1 expression in human glioma. Molecular and chemical neuropathology / sponsored by the International Society for Neurochemistry and the World Federation of Neurology and research groups on neurochemistry and cerebrospinal fluid. 1994;21:177–188. doi: 10.1007/BF02815350. [DOI] [PubMed] [Google Scholar]
- 25.Hao AJ, Dheen ST, Ling EA. Expression of macrophage colony-stimulating factor and its receptor in microglia activation is linked to teratogen-induced neuronal damage. Neuroscience. 2002;112:889–900. doi: 10.1016/s0306-4522(02)00144-6. [DOI] [PubMed] [Google Scholar]
- 26.Douglass TG, Driggers L, Zhang JG, et al. Macrophage colony stimulating factor: not just for macrophages anymore! A gateway into complex biologies. Int Immunopharmacol. 2008;8:1354–1376. doi: 10.1016/j.intimp.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 27.Metcalf D, Stanley ER. Haematological effects in mice of partially purified colony stimulating factor (CSF) prepared from human urine. Br J Haematol. 1971;21:481–492. doi: 10.1111/j.1365-2141.1971.tb02709.x. [DOI] [PubMed] [Google Scholar]
- 28.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baiocchi G, Kavanagh JJ, Talpaz M, Wharton JT, Gutterman JU, Kurzrock R, et al. Expression of the macrophage colony-stimulating factor and its receptor in gynecologic malignancies. Cancer. 1991;67:990–996. doi: 10.1002/1097-0142(19910215)67:4<990::aid-cncr2820670422>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 30.West RB, Rubin BP, Miller MA, et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc Natl Acad Sci USA. 2006;103:690–695. doi: 10.1073/pnas.0507321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Markovic DS, Vinnakota K, Chirasani S, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A. 2009;106:12530–12535. doi: 10.1073/pnas.0804273106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y, Yi X, Stoffer JB, et al. The multifunctional protein glyceraldehyde-3-phosphate dehydrogenase is both regulated and controls colony-stimulating factor-1 messenger RNA stability in ovarian cancer. Mol Cancer Res. 2008;6:1375–1384. doi: 10.1158/1541-7786.MCR-07-2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS, et al. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Godlewski J, Nowicki MO, Bronisz A, et al. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008;68:9125–9130. doi: 10.1158/0008-5472.CAN-08-2629. [DOI] [PubMed] [Google Scholar]
- 35.Collier LS. Largaespada DA Hopping around the tumor genome: transposons for cancer gene discovery. Cancer Res. 2005;65:9607–9610. doi: 10.1158/0008-5472.CAN-05-3085. [DOI] [PubMed] [Google Scholar]
- 36.Stanley ER, Berg KL, Einstein DB, et al. Biology and action of colony--stimulating factor-1. Mol Reprod Dev. 1997;46:4–10. doi: 10.1002/(SICI)1098-2795(199701)46:1<4::AID-MRD2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 37.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jadus MR, Irwin MC, Irwin MR, et al. Macrophages can recognize and kill tumor cells bearing the membrane isoform of macrophage colony-stimulating factor. Blood. 1996;87:5232–5241. [PubMed] [Google Scholar]
- 40.Truong AH, Ben-David Y. The role of Fli-1 in normal cell function and malignant transformation. Oncogene. 2000;19:6482–6489. doi: 10.1038/sj.onc.1204042. [DOI] [PubMed] [Google Scholar]
- 41.Adams JC, Seed B, Lawler J. Muskelin, a novel intracellular mediator of cell adhesive and cytoskeletal responses to thrombospondin-1. Embo J. 1998;17:4964–4974. doi: 10.1093/emboj/17.17.4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tagnaouti N, Loebrich S, Heisler F, et al. Neuronal expression of muskelin in the rodent central nervous system. BMC Neurosci. 2007;8:28. doi: 10.1186/1471-2202-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nord H, Hartmann C, Andersson R, et al. Characterization of novel and complex genomic aberrations in glioblastoma using a 32K BAC array. Neuro Oncol. 2009;11:803–818. doi: 10.1215/15228517-2009-013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mizuno E, Nakamura M, Agemura A, et al. Brain-specific transcript variants of 5’ and 3’ ends of mouse VPS13A and VPS13C. Biochem Biophys Res Commun. 2007;353:902–907. doi: 10.1016/j.bbrc.2006.12.122. [DOI] [PubMed] [Google Scholar]
- 45.Ma P, Winderickx J, Nauwelaers D, et al. Deletion of SFI1, a novel suppressor of partial Ras-cAMP pathway deficiency in the yeast Saccharomyces cerevisiae, causes G(2) arrest. Yeast. 1999;15:1097–1109. doi: 10.1002/(SICI)1097-0061(199908)15:11<1097::AID-YEA437>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 46.Wrensch M, Jenkins RB, Chang JS, et al. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet. 2009;41:905–908. doi: 10.1038/ng.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.