

Abstract
In this paper, we present the results of proof-of-concept experiments using a novel photocleavable and mass spectrometry identifiable cross-linker pcPIR (photocleavable Protein Interaction Reporter). pcPIR can be dissociated under UV irradiation either off- or on-line before the introduction to the mass spectrometers. Photo dissociation of cross-linkers is different from either the gas phase or the chemical cleavage of cross-linkers. Different types of cross-links can be identified using the pcPIR mass relationships, where the mass of cross-linked precursor equals the sum of the masses of the released products and reporter. Since pcPIR is cleaved prior to the entrance to the mass spectrometer, the released peptides are available to be sequenced with routine CID MS/MS experiments and database search algorithms. In this report, the pcPIR strategy of identifying the cross-linked peptides with on- and off-line photocleavage coupled with novel targeted data dependent LC-MS/MS is demonstrated with the use of standard peptides, BSA and human hemoglobin tetramer protein complex.
Keywords: photocleavable protein interaction reporter (pcPIR), cross-linker, protein interaction
INTRODUCTION
Chemical cross-linking coupled with mass spectrometry is a commonly used technique to characterize proteins, interactions and complex topologies1-6. Cross-linking is a process that covalently links two residues/proteins in close proximity and thus the maximum length of the cross-linker has been used to provide approximate distance measurements between the two cross-linked sites. By stabilizing protein complexes with new covalent bonds, cross-linking strategies have unique capabilities for mapping protein interactions, especially transient and weak interactions in native biological systems. However, there are few successful applications on complicated biological systems4, 7, because system complexity and product heterogeneity have generally precluded large-scale applications of cross-linking strategies. Various types of products can be produced in the cross-linking reactions, such as dead-end, intra-, and inter-peptide cross-links, multiply modified peptides, unmodified peptides, and other nonspecific products. Among all these types of products, the desired inter-cross-linked products are of low abundance, which creates significant challenges for detection and identifcation5, 6, 8. To overcome these problems, a number of approaches have been developed, which are discussed in several recent reviews9-11. For example, these strategies include the reduction of system complexity by introducing an affinity tag to the cross-linked products6, 7, 11, 12; or implementing specific signatures in the cross-linked products. Some examples use the isotopic labeled cross-linkers/proteins13-15, and some other examples involve the IR- or UV- dissociation of cross-links, which are reported by the Brodbelt’s group16, 17. In their UVPD strategy, only the conjugated peptides were dissociated under UV condition, which specially distinguished the conjugated peptides from the other non-conjugated peptides. Other than these methods, cleavable cross-linkers such as chemically cleavable18 or gas phase/mass spectrometry cleavable cross-linkers5-7, 10, 19 are also widely used.
Significant efforts have been spent on software tool development when non-cleavable cross-linkers are employed4, 21, 22. For example, recently Singh et al reported an open-modification strategy which considers one peptide in the cross-linked complex as a modification with unknown mass on the other peptide22. However, the identification of cross-linked peptides/sites is still difficult due to the huge database size. If the number of possible cross-linked peptides is n, the database size for possible cross-linked products will be n2. In contrast, cleavable cross-linking strategies have the advantage that the intact peptides can be released after dissociation of the cross-linker and thus each peptide can be identified with traditional MS/MS experiments and database search algorithms. According to the cleavage mechanisms, cleavable cross-linkers can be cataloged as chemically cleavable18, gas phase/mass spectrometry cleavable3, 7, 19 and photocleavable15 cross-linkers. Isotopically-coded chemically cleavable cross-linkers and software tool have been reported before20. Our group has developed a series of gas phase cleavable cross-linkers, embodied in Protein Interaction Reporter (PIR) technology3, 5-8, 23, which allows release of two intact peptides as well as a reporter ion after low-energy MS/MS dissociation. The released peptides can be searched against restricted database and identified with accurate mass. The identities can be further validated with tandem MS followed by prior activation experiments. An important feature of PIR strategy is that with the built-in specific mathematical relationship between the two released intact peptides, reporter and the cross-linked precursor, different types of cross-linked products can easily be distinguished3. There are few photocleavable cross-linkers reported till now. Borchers’ group reported a photocleavable cross-linker BiPS15. BiPS is partially cleavable under MALDI condition to produce unsymmetrical released products. The cross-link relationship is identified with characteristic doublet isotopically labeled peaks and the mass relationships. In this way, each peak’s intensity is reduced by half because of the isotopic label; and the unsymmetrical cleavage of the cross-linker leaves two different modification groups on the peptides, which increases the peptide search space by a factor of two. Alternatively, cleavable cross-linker technologies, though promising have their own limitations too. Chemically cleavable cross-linkers are cleaved off-line and thus the relationship between the cross-linked precursors and the released products becomes intractable during LC separation. In addition, extra sample preparation or clean up steps usually lead to more sample loss. Gas phase cleavable cross-linkers can overcome these limitations; however, gas phase dissociation of the cross-linked precursor can produce released peptides with low charge states. The total available charge is limited by the cross-linked precursor, and therefore gas phase released peptides are usually observed as 1+ or 2+ ions, which may not be optimal for MS/MS analysis.
To combine the PIR concept and the advantages of other cleavable cross-linking strategies, we developed a novel photocleavable Protein Interaction Reporter (pcPIR) cross-linking strategy. pcPIR incorporates two photocleavable bonds, a mass encoded reporter tag and an affinity group in the structure (Figure 1a). The photocleavable bond has been employed previously in other cross-linking or labeling reagents, which usually contain only one photocleavable bond to release the whole cross-linked product from support23-25. However, we applied it to the PIR concept and developed a cross-linker with two photocleavable arms that can be dissociated under UV conditions, which leads to the release of intact peptides from the cross-linked precursors. As a new member of PIR cross-linker family, pcPIR retains the advantages of PIR technology, i.e., cleavage of the PIR cross-linked product occurs on-line after LC separation, therefore the cross-linked precursor and released peptides will appear with a specific temporal relationship in the chromatogram. As a consequence, the PIR mathematical relationship is applicable to pcPIR cross-linked peptides, thus allowing identification of cross-links and types. Since the cleavage happens before the introduction to mass spectrometers, the released peptides can be identified with targeted data-dependent MS/MS experiments and traditional database search algorithms. Moreover, pcPIR offers additional advantages over previous PIR cross-linkers. The possibility of producing peptide fragments during release of the intact peptides from the cross-linked precursors is reduced with photo dissociation. In addition, photo cleavage takes place in solution phase rather than in gas phase inside the mass spectrometers; therefore the released intact peptides are usually observed more highly charged, which generally allows production of more comprehensive fragmentation spectra during MS/MS analyses. All these features will eventually enable further applications of pcPIR on native biological systems. In this paper, we present the proof-of-concept results of pcPIR strategy; describe the synthesis of the pcPIR cross-linker, photocleavage characterization, affinity purification strategy and applications using standard peptides, BSA and human hemoglobin tetramer protein complex.
Figure 1.


pcPIR structure
(a) Structure of pcPIR
(b) Cleavage of inter-peptide-cross-link
EXPERIMENTAL
Materials
Angiotensin II (A), Bradykinin (B), bovine serum albumin (BSA), human hemoglobin (Hb) and Tergitol solution (70% NP40 in water) were purchased from Sigma-Aldrich (St. Louis, MO) and used without purification. Fmoc-Arg-PMC, Fmoc-Lys-Biotin, Fmoc-Lys-Fmoc-OH and the hydroxymethyl photolinker (4-[4-Hydroxymethyl-2-methoxy-5-nitrophenoxy)butanoic acid) were purchased from Novabiochem (San Diego, CA). Monomeric Avidin Ultralink Resin and Mass Spectrometry-Grade Trypsin Endoproteinase were purchased from Pierce (Rockford, IL), the Amicon Ultra −0.5mL 10K Centrifugal Filters were from Millipore (Billerica, MA), and Phenylmethylsulfonyl Fluoride (PMSF) was from GBionsciences (Maryland Heights, MO).
Synthesis of pcPIR cross-linker
The cross-linker was synthesized by using a 431A Peptide Synthesizer (Applied Biosystems, Foster City, CA). The procedure was slightly modified from that used for the synthesis of previous PIR compounds3. Fmoc-Arg-PMC was coupled to HMPB-MBHA resin using DCC, and then Fmoc-Lys-Biotin and Fmoc-Lys-Fmoc-OH as a branching point, were linked in sequence. On each arm, a hydroxymethyl photolinker was coupled as a cleavable point and a carboxyl group was introduced by coupling with succinyl anhydride, which was then activated by forming the esters with N-hydroxysuccinimide (NHS). The final product (Figure 1a) was deprotected and cleaved from the super acidic sensitive resin with 95% TFA in water or 1% TFA in Dichloromethane and purified with HPLC. The yield of the final product was about 30% and the steps of coupling Fmoc-Lys-Fmoc-OH and succinyl ahbydride are the yield limiting steps. The purity of the final compound was more than 95% pure.
Cross-linking of standard peptides and their analysis
1 μL of 10mM Angiotensin II (10 nmol) was mixed with 1 μL of 10mM Bradykinin (10 nmol) in water and dried by speed vacuum overnight. 1 μL of 10 mM pcPIR in DMSO (10 nmol) was added, and the reaction mixture was incubated at room temperature in dark with shaking overnight.
On-line photocleavage was accomplished with a VSL-337 nitrogen UV laser instrument (Spectra-physics, Mountain View, CA). UV laser was focused through a focusing lens (f = 11.5 cm) orthogonally onto an ESI tip, which had been made by etching a piece of fused silica capillary (360 μm × 30 μm). The instrument used in this experiment was a 7-Tesla ESI-FTICR-MS (Apex-Q Bruker Daltonics, Billerica, MA). The released intact peptides from UV laser irradiation were further fragmented with CID in the hexapole by decreasing the collision cell voltage to around −18 V. Samples generated for off-line UV cleavage experiments were analyzed with a 4800 MALDI-TOF/TOF mass spectrometer (Applied Biosystems, Foster City, CA). The cross-linked sample was spotted onto a MALDI plate, and a “UV off” MS spectrum was collected. Then the MALDI plate was irradiated under a UV lamp with the wavelength maximum of 365 nm, and a “UV on” MS spectrum was collected. Then the released peptides were fragmented with CID MS/MS.
Mascot generic format (mgf) files were obtained for each MS/MS spectrum of the released peptides. The database used in this study was E. coli whole genome database (downloaded from NCBI in 2008) spiked with Angiotensin II and Bradykinin sequences. The search was accomplished with software Mascot and the parameters were as follows: enzyme was set as none; 3 possible missed cleavages; and the remaining tag from cross-linker was considered as a variable modification on Lysine or N-termini.
Cross-linking of protein/protein complex and analyses
A cross-linked BSA sample was prepared by following the protocol described by Rinner and Aebersold et al4, except that the cross-linker applied in current experiment was pcPIR. Briefly, 100 μL of 15 μM (1 mg/mL) BSA in water and 1 μL of 100 mM pcPIR in DMSO were mixed and incubated at 35 °C in dark with shaking for 30 min. The sample was then reduced and alkylated with dithiothreitol (DTT) and iodoacetamide (IAA), and digested with 0.5% w/w trypsin at 37 °C for 16 h (overnight). The protein digest was purified with C18 Sep-Pak® cartridges (Waters, Milford, MA) and fractioned with Macro Spin Column SCX (The Nest Group, Inc., Southborough, MA). The enriched cross-linked products were further fractioned with HPLC (nanoAcquity Waters, Milford, MA) and detected with LTQ-FT MS (Thermo Fisher Scientific Inc., Waltham, MA). A 30-cm long C18 column was made in house by packing fused silica capillary (360 μm × 75 μm) with MAGIC C18AQ 100A 5U beads (Michrom Bioresources, Inc., Auburn, CA). A 2-cm long trap column was prepared similarly by packing fused silica capillary (360 μm × 100 μm) with MAGIC C18AQ 200A 5U beads. A two-step-LC-MS-identification strategy was developed. Two LC experiments with exactly the same gradient were run in sequence. The first LC-MS was run with the laser turned on at the intensity to allow detection of both the cross-linked precursor and its released peptides. The instrument resolution was set at 100K to obtain high mass accuracy data. X-links8 searches were used to identify the pcPIR relationships with mass error tolerance of 10 ppm. The m/z values of the identified released peptides and LC elution time were then used to generate a mass-and-time targeted inclusion list, which was used in the second data-dependent LC-MS/MS run. The second LC-MS/MS experimental set up was as follows: one MS with 60K resolution was followed by three MS/MS experiments, precursor masses were obtained from mass-and-time targeted list, dynamic exclusion repeat and exclusion duration were both 15 sec. LC-MS/MS data were then searched against yeast and reversed yeast database spiked with BSA sequence with both Mascot and SEQUEST software to identify the sequences of the released peptides. 3 possible missed cleavages were allowed, the precursor error tolerance was 15 ppm and the fragmentation error tolerance was 0.6 Da. The remaining tag from pcPIR was treated as a variable modification on Lysine or protein N-termini. The following LC gradient was employed in both LC runs: 0-60 min 5%-60% buffer B, 60-85 min flushing with 85% buffer B and 85-120 min equilibrating with 5% buffer B (buffer A: 0.1% formic acid in DI water; buffer B: 95% acetonitrile and 0.1% formic acid in DI water).
Cross-linking and analyses of human hemoglobin (Hb) were performed with the same procedures. 100 μL of 15 μM (1 mg/mL) Hb in water solution was cross-linked with 1 μL of 100 mM pcPIR in DMSO. The remaining steps followed those used in the BSA cross-linking experiment. In this case, a database of yeast and reversed yeast spiked with the sequences of human hemoglobin α and β subunits was used for database search.
Avidin-biotin affinity purification
5 nmol pcPIR cross-linked Angiotensin-Bradykinin sample was mixed with about 5 nmol BSA digest (0.33 mg protein) in 100 mM NH4HCO3 solution. Half of the sample was saved as control and the other half was mixed with 20 uL monomeric avidin slurry and incubated at room temperature in dark for 1.5 hrs. The beads were then washed with 100mM NH4HCO3 with 0.1% NP40 by 3 times, and 100 NH4HCO3 solution by 3 times. Finally the beads were eluted with 75% acetone nitril and 0.5% trifloricacid in water solution by 4 times, each time using 100 uL eluting solution.
100 μL of 15 μM BSA was reacted with 1 μL of 100 mM pcPIR at 35 °C in dark for 0.5 hr. Then the cross-linked protein sample was cleaned with 100mM NH4HCO3 in Amicon filters for 3 times. The cross-linked protein sample was then reduced, alkylated and digested as described previously. Then half of the sample was saved as control and the other half was further enriched with avidin affinity purification. PMSF was added to the experimental half to a final concentration of 1 mM to inhibit trypsin acitivity. 40 uL monomeric avidin slurry was incubated with the mixture at room temperature in dark for 2 hrs, and the beads were washed and eluted as described before, with 200 uL eluting solution each time.
RESULTS and DISCUSSIONS
pcPIR concept and strategy
The difficulty in unambiguously identifying the cross-linking relationships as well as the cross-linked peptide sequences requires new cross-linkers and software tools, which can enable efficient detection of the cross-linked products and the analyses of the obtained mass spectrometric data. The goal of pcPIR cross-linker development is to specifically detect the cross-linking relationships and eventually identify the cross-linked sites on proteins, which is achieved by fragmentation of the released intact peptides and database search algorithms. The structure of pcPIR is shown in Figure 1a, which has two reactive groups, two photocleavable groups (highlighted between red []) and a mass encoded reporter containing a biotin affinity tag. Under UV irradiation, the cross-linker is cleaved into the released peptides and the reporter as shown in Figure 1b.
The general experimental workflow for pcPIR strategy is shown in Figure 2a. The cross-linked products are obtained with cross-linking reactions of proteins, followed by digestion and enrichment of the desired products. The ultimate goal of identification of the cross-linked peptide/protein with the cross-linked sites can be achieved in a two-step-LC-MS-identification strategy. The first accurate-mass-based LC-MS experiment is used to identify the cross-linking relationships and the second targeted LC-MS/MS experiment allows identification of the released peptide sequences/cross-linked sites. For the first step, LC-MS is performed with UV laser to promote the cleavage of pcPIR labeled precursors. The identification of cross-linking relationships and types are determined with previously developed software X-links8, the principle of which is based on the specific mathematical relationships between the cross-linked precursors and the released peptides. The relationship is only determined based on whether the detected peaks can fit in one of the three cross-links’ equations (Figure 2b). As discussed previously8, higher mass accuracy is required to greatly reduce the false discovery rate. For the second step, traditional shotgun LC-MS/MS and database search methods are applied to identify the released peptides from the detected relationships. An inclusion list with m/z values and retention time of these released peptides are generated from the previous X-links search results. The exact same gradient was used for both LC runs. In addition, the laser can be set at higher power to ensure optimal release of the intact peptides for better signal. Ideally, only one LC run with 3 scan types will be needed to complete all the analyses. Scan 1 detects the MS of the cross-linked parents; scan 2 detects the photocleavage released peptides; and scan 3 performs data-dependent MS/MS on the released peptides, based on the observed cross-linking relationships. In the future, software will be developed to analyze the cross-linking relationships with scans 1 and 2 on-line, and to control the mass spectrometer to fragment the released peptides in scan 3.
Figure 2.


Scheme and relationship identification mechanism
(a) Scheme of pcPIR strategy
(b) Mathematical relationships between cross-linked precursors and released peptides: inter-cross-linked precursor = reporter + released peptide A + released peptide B intra-cross-linked precursor = reporter + doubly modified released peptide dead-end = reporter + released peptide + OH
On-line/off-line photocleavage and identification of the cross-linked peptides
An on-line photocleavage method was set up using ESI ion source mass spectrometers (Supplemental Figure 1a). With this method, the cross-linked products can be cleaved after LC separation but before MS detection. As shown in Figure 3a, a pair of spectra, with UV off and on, was collected during direct infusion of the sample. In this experiment, standard peptides Angiotensin II (A) and Bradykinin (B) were cross-linked. Neither of the peptides has Lysine in the sequence and thus the highest reactive sites are the two N-termini. Therefore inter-cross-linked (X-pcPIR-X) or dead-end (DE-X) products were expected. By comparing the MS spectra with UV off (upper) and UV on (lower), it is clearly observed that all the cross-linked precursors, A-pcPIR-A, A-pcPIR-B, B-pcPIR-B and DE-A, DE-B, have a decreased signal when the UV laser is switched on; the released intact peptide ions, released Angiotensin II (RA2+) and released Bradykinin (RB2+), only appear in the spectrum with UV on; and finally the peak intensities for unmodified peptides Angiotensin II (A2+) and Bradykinin (B2+) remain unchanged. Figure 3b shows the complementary extracted ion chromatograms (EICs) of the cross-linked parent and the cleavage product (released Bradykinin). When UV laser is turned on, the parent intensity decreases and the released product increases. Within 2.4 sec, the cleavage reaches maximum. When laser is turned off, the parent drops and the released product increases. With such special “flicker” peaks, the cross-linked products can be easily distinguished from the non-cross-linked products. The differential comparison can be used to quickly distinguish the unmodified peptides and allows identification of the cross-linking relationships, which is based on a mass relationship for all the inter-cross-linked products: the cross-linked precursor equals the sum of neutral mass of released peptide A (RA), released peptide B (RB) and the reporter. Here for A-pcPIR-B, the equation is: A-pcPIR-B (experimental neutral mass 3459.5450) = RA (1145.5246) + RB (1159.5496) + reporter (1154.5067), and the error is 10 ppm. Similarly, intra-peptide-cross-link = released peptide + reporter, and dead-end = released peptide + reporter + OH. All the mathematical relationships of the 3 cross-link types are listed in Figure 2b. For more complicated samples, previously developed software X-links using the same concept can automatically assign cross-linking relationships and types8. In short, software X-links searches through the whole LC-MS to determine whether there are masses that can fit one of the three equations from each neighboring spectrum. Once found, the MS peaks are reported as cross-link candidates. In addition, the cross-link types are assigned in the same process, because different types should fit in different equations. X-links has been proven to be able to identify cross-links and distinguish different types from our previous publications6, 7. Although it was originally designed for PIR cross-linkers, it can be adjusted for pcPIR calculations, with changes in the cross-linker parameters, e.g. neutral masses of cross-linker, reporter and the modification group left on the released peptides due to cleavage. With the present pcPIR cross-linker, the reporter ion was not always detected in the spectra, however the calculated mass of the reporter can be included by default in the software. This identification method is advantageous over isotopically-coded cross-linking strategies because the cross-linked or labeled peaks are not split due to the different isotopic labels; and therefore the sensitivity of the experiment is increased. As mentioned before, the released peptides can be further identified with CID fragmentation as shown in Supplemental Figure 2a. The peaks marked with “M” are the intact released peptides with the remaining tag modification (mass = 100.0160). This tag is symbolized with a short black line. Fragment ions with the tag modification are marked as “a, b, y” ions while those without modifications are marked as “a°, b°, y°” ions. The * symbol denotes noise peaks. In addition, MALDI MS methods are usually considered complimentary to ESI MS methods. Different peptides can often be identified from these methods, and by combining the results together, better coverage is expected. pcPIR cross-linker is therefore also coupled to MALDI mass spectrometry, using an off-line UV lamp cleavage method as shown in Supplemental Figure 1b. At least 1 hr is needed for cleavage, and it produces the same released Angiotensin (RA) and released Bradykinin (RB) as the on-line cleavage (S. Figure 3a), which are fragmented with CID MS/MS (S. Figure 3b). With the same released peptides, the ESI method and the MALDI method can be used as a confirmation to each other. Interestingly, RA+ and RB+ peaks can also be observed in the “UV off” spectrum, but the ratios of their intensities to Angiotensin or Bradykinin peaks are much lower than that in the “UV on” spectrum. This can be explained by the possible photocleavage with the MALDI laser. The wavelength of MALDI laser is 355 nm, which is close to the central wavelength (365 nm) of the UV lamp used for photocleavage. In addition, the complete cleavage with extended UV lamp irradiation can further confirm the identified relationships. Finally, we evaluated the peptide/protein identification with routine database search algorithm by searching the database of E. coli whole genome spiked with Angiotensin II and Bradykinin. As shown in Supplemental Table 1, Angiotensin II and Bradykinin with pcPIR modifications on N-termini were identified as the only two significant hits.
Figure 3.


On-line photocleavage of pcPIR cross-linked Angiotensin II (A) and Bradykinin (B).
(a) Upper: MS spectrum before UV irradiation; Lower: MS spectrum after UV irradiation. Insets: upper, cross-linked precursors; lower, released products
(b) Complementary extracted ion chromatograms (EICs) of the cross-linked parent A-pcPIR-B and released Bradykinin (RB). UV cleavage reaches the maximum within 2.4 sec.
Characterization of on-line UV laser cleavage
The sample flow rate towards the MS inlet through the ESI spray tip is a critical factor of the photocleavage efficiency. With a lower flow rate, one would predict that the available time for the photocleavable bonds to absorb photons and dissociate is longer and thus the cleavage efficiency is higher. A photocleavable peptide (S. Figure 4) was used for the flow rate optimization. The laser pulse frequency was set at 20 Hz for all the experiments. Photocleavage efficiency was studied under different conditions by changing only the sample flow rate. The observed relationship between flow rate and photocleavage efficiency is shown in S. Figure 5. Every data point was collected from a triplicate experiment. As predicted, photocleavage efficiency increases as flow rate decreases. At 0.07 μL/min, the cleavage of the precursor ion is near completion. One-arm-cleaved product reaches a plateau when the flow rate increased up to 0.3 μL/min, because it is an intermediate product between the un-cleaved intact precursor and the completely cleaved product (reporter). On the other hand, the flow rate also affects the mass spectrometry signal. With a flow rate lower than 0.3 μL/min, the peak intensities in the spectrum were observed to decrease. The optimal mass spectrometry signals were obtained when the flow rate was higher than 0.3 μL/min (data not shown). Since it is necessary to detect both the precursor and product peaks for pcPIR relationship calculation, a balance was chosen to be 0.3 μL/min to 0.5 μL/min, which resulted in both good cleavage efficiency and good peak intensities.
Affinity purification of the cross-linked/labeled products
With biotin incorporated in the structure of pcPIR, the cross-linked or labeled products can be affinity purified with avidin-biotin interaction. pcPIR cross-linked Angiotensin II and Brandykinin (A-pcPIR-B) was spiked into BSA tryptic digestion and used to demonstrate this strategy. S. Figure 6a and 6b show the TICs (total ion chromatogram) and EICs (extracted ion chromatogram) of A-pcPIR-B without and with avidin affinity purification. Without purification, A-pcPIR-B EIC peak was below the noise level and its MS peak was not observable. However after purification, the A-pcPIR-B chromatographic peak became one of the most abundant peaks in the TIC, and the MS peak can be clearly seen (inset spectrum). We further tested the purification method using the pcPIR cross-linked BSA sample. BSA tryptic peptide ion 582.32 2+ is one of the most abundant ions without purification. It is also observed with a greatly reduced intensity after purification, due to non-specific binding. Dead-end precursor 730.68 3+ is also observed both before and after purification. So the ratios of dead-end over non-labeled peptide in both cases are used to calculate the enrichment factor of the labeled products. As listed in S. Table 2,_without avidin purification the dead-end peak height is about 7% of the tryptic peptide peak height. However after avidin purification, the labeled peak is about 4 times higher than that of the peptide peak. Therefore the avidin affinity purification gives about 60 times enrichment of the labeled product. Furthermore, in some cases the purification is even more important, as shown in S. Figure 6c. Inter-cross-linked precursor ion 798.65 4+ appears after avidin purification (c1); however its EIC peak was below the noise level before purification and its MS peak was not observed. The two experiments with or without avidin capture were analyzed with exactly the same LC gradient, therefore the same component should elute at the same retention time (±1 min). c2 shows that in −1 min window, a similar m/z of tryptic peptide 797.75 5+ was observed, which may suppress the cross-link’s signal. c3 and c4 are the same retention time and +1 min windows, in which no desired product was observed. The labeled/cross-linked products were observed in this case due to the improved dynamic range gained from affinity purification.
pcPIR application on BSA
BSA was employed as a model protein to demonstrate the pcPIR strategy in the initial studies. Strong cation exchange (SCX) chromatography is another way to enrich the cross-linked products. The cross-linked products, with two N-termini, should carry more charges than unmodified peptides4. Figure 4 shows an example of the identified inter-cross-linked pair and the released peptides. Several other BSA tryptic peptide peaks can also be observed, most of which have charge states equal or higher than 4+. The highly charged cross-linked precursor and the two released peptides are shown in the inset spectra. Due to the presence of other BSA tryptic peptides, these peaks are still not among the highest intensity peaks in the spectrum. Therefore it is likely that these peaks would not be selected for fragmentation with traditional peak-intensity-dependant MS/MS analyses. However, the pcPIR relationship was first identified with X-links and MS/MS was triggered with the mass-and-time targeted inclusion list. These released peptides are specifically selected, fragmented and identified, despite their relatively low peak intensities (Figure 4b). Combining the results from SCX and avidin purification experiments, the peptides identified with Mascot and SEQUEST database searches are listed in Table 1. The modified Lysines with remaining tag from pcPIR are underlined in the sequences. Finally, the inter-cross-linked peptide pairs and the dead-end labeled peptides were identified with the following criteria: 1) The cross-linked precursor and released peptides exhibited a temporal relationship in the LC chromatogram such that they appear to “co-elute”. 2) Their pcPIR mass relationships were accurately obeyed (within 10 ppm). 3) The sequences of the released peptides were identified as statistically significant hits in the database search (Expect score < 0.05, precursor delta mass < 10 ppm). 4) Two LC runs with the same elution profile were employed in this experiment. So the retention time of the same released peptide should be similar in the two LC runs (within a ± 1.5 min window). The pcPIR cross-linked peptide pairs are confirmed with all these criteria met (Table 2). The pairs marked with * were reported previously by others4.
Figure 4.


An example of inter-cross-linked pair from cross-linking BSA experiment
(a) Insets: MS spectra of two released peptides and the inter-cross-linked precursor
(b) MS/MS spectra of the two released peptides
Table 1.
Identified released peptides from pcPIR cross-linking BSA experiment
| Observed m/z | Observed mass | Calculated mass | Delta mass (ppm) | Expect | Peptide sequence |
|---|---|---|---|---|---|
| 459.2583 | 916.5021 | 916.4978 | 4.72 | 0.0017 | R. SLGKVGTR. C |
| 474.2653 | 946.5161 | 946.5124 | 3.94 | 0.0020 | R. LSQKFPK. A |
| 545.7903 | 1089.5661 | 1089.5666 | −0.40 | 0.0016 | R. EKVLTSSAR. Q |
| 551.3061 | 1100.5976 | 1100.5978 | −0.16 | 0.00013 | R. ALKAWSVAR. L |
| 431.8784 | 1292.6132 | 1292.6109 | 1.81 | 0.0021 | R. DTHKSEIAHR. F |
| 648.3063 | 1294.5980 | 1294.5976 | 0.33 | 7.5 e-5 | R. CASIQKFGER. A |
| 450.5507 | 1348.6301 | 1348.6299 | 0.17 | 0.00013 | R. FKDLGEEHFK. G |
| 820.4227 | 1638.8309 | 1638.8287 | 1.35 | 5.9 e-05 | R. LCVLHEKTPVSEK. V |
| 870.4800 | 1738.9454 | 1738.9465 | −0.62 | 0.00025 | R. KVPQVSTPTLVEVSR. S |
| 549.6454 | 1645.9143 | 1645.9039 | 6.35 | 0.0041 | K. LKHLVDEPQNLIK. Q |
| 682.6900 | 2045.0482 | 2045.0252 | 11.3 | 0.022 | K. SLHTLFGDELCKVASLR. E |
| 554.2300 | 2212.8909 | 2212.8936 | −1.22 | 0.004 | K. VHKECCHGDLLECADDR. A |
Table 2.
Identified inter-cross-linked pairs and dead-ends from pcPIR cross-linking BSA experiment
| Cross-linked precursor mass |
Released peptide A mass |
Released peptide B mass |
Error (ppm) |
Cross-linking/labeling | Distance (Å) |
|---|---|---|---|---|---|
| 3017.5340 | 946.5133 | 916.4970 | 5.65 | R. LSQKFPK. A -pcPIR- R. SLGKVGTR. C | 25.6 |
| 3190.5913* | 1089.5678 | 946.5133 | 1.11 | R. EKVLTSSAR. Q -pcPIR- R. LSQKFPK. A | 21.9* |
| 3201.6261* | 1100.5989 | 946.5133 | 2.26 | R. ALKAWSVAR. L -pcPIR- R. LSQKFPK. A | 14.9* |
| 3365.6129 | 1294.5982 | 916.4970 | 3.28 | R. CASIQKFGER. A -pcPIR- R. SLGKVGTR. C | 29.6 |
| 3709.8572 | 1638.8293 | 916.4970 | 6.54 | R. LCVLHEKTPVSEK. V -pcPIR- R. SLGKVGTR. C | 26.5 |
| 2189.0297 | 916.4970 | −0.25 | R. SLGKVGTR. C -pcPIR- OH | ||
| 2219.0444 | 946.5133 | −0.97 | R. LSQKFPK. A -pcPIR- OH | ||
| 2362.1022 | 1089.5678 | 0.49 | R. EKVLTSSAR. Q -pcPIR- OH | ||
| 2373.1384 | 1100.5989 | 2.63 | R. ALKAWSVAR. L -pcPIR- OH | ||
| 2565.1405 | 1292.6083 | −0.41 | R. DTHKSEIAHR. F -pcPIR- OH | ||
| 2567.1374 | 1294.6032 | 0.37 | R. CASIQKFGER. A -pcPIR- OH | ||
| 2621.1634 | 1348.6297 | 0.17 | R. FKDLGEEHFK. G -pcPIR- OH | ||
| 2911.3661 | 1638.8293 | 1.22 | R. LCVLHEKTPVSEK. V -pcPIR- OH | ||
| 2918.4322 | 1645.9046 | −1.94 | K. LKHLVDEPQNLIK. Q -pcPIR- OH | ||
| 3011.4816 | 1738.9470 | 0.45 | R. KVPQVSTPTLVEVSR. S -pcPIR- OH | ||
| 3317.5640 | 2045.0219 | 2.67 | K. SLHTLFGDELCKVASLR. E -pcPIR- OH | ||
| 3485.4323 | 2212.8964 | 0.76 | K. VHKECCHGDLLECADDR. A -pcPIR- OH |
pcPIR application on Hemoglobin tetramer protein complex
Human hemoglobin tetramer complex (Hb) was employed for the initial proof-of-concept experiment of pcPIR application on studying the topology of protein complexes or protein-protein interactions. The tetramer Hb complex is formed by two α subunits and two β subunits. Figure 5 shows an example of an inter-subunit-cross-linked pair: the cross-linked precursor and photocleavage released peptide are shown in the inset spectra, and the MS/MS fragmentation of the released peptide is shown in Figure 5b. The sequence of the released peptide was identified with Mascot search algorithm, with a precursor peak error of 0.45 ppm and an Expect score of 0.0023. In this case, both released peptides have the same sequence, which indicates a homo-dimer inter-cross-link between the two β subunits of Hb. The crystal structure of the Hb four subunits is shown in Figure 6, where the cross-link is visualized as a red line. Both the cross-linked residues are exposed to solvent and are in close proximity (approximately 26.9 Å). The same residue was also identified as a dead-end labeled product, suggesting its high reactivity (data not shown). The distance between two lysine residues is an important factor that impacts which sites on protein(s) are cross-linked. Table 2 lists such distances from results of pcPIR cross-linking BSA experiments. The identified inter-cross-linked pairs with closest distances were also identified in previous DSS cross-linking experiments4. Compared to DSS (11.4 Å in length), pcPIR cross-linker has a longer spacer arm length (maximum of 40 Å); however the distance between the cross-linked sites may be less than 40 Å, due to the flexibility of cross-linker’s structure. This statement is supported by the fact that the distances between cross-linked sites from BSA are observed to range from about 15 Å to about 30 Å (S. Table 3). It should be noted that shorter cross-linkers, for example DSS, are commonly used in providing strict distance constraints of the cross-linked sites. In protein structure studies, this is the primary desirable information. However in protein-interaction studies, the identities of the cross-linked peptides are most useful. Therefore, longer and more flexible cross-linkers, such as pcPIR, are more beneficial. These structures allow cross-linking of proteins or protein subunits with a wider range of distances, as is shown in the Hb experiments.
Figure 5.


An example of inter-cross-linked pair from cross-linking Hb experiment
(a) Insets: MS spectra of the released peptide and the inter-cross-linked precursor
(b) MS/MS spectrum of the released peptide
Figure 6.
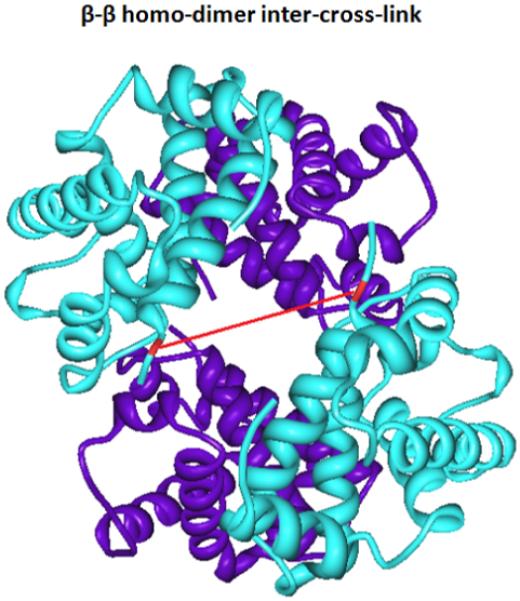
The crystal structure of Hemoglobin four subunits and the identified inter-subunit-cross-linked pair
Evaluation of pcPIR
One major advantage of PIR strategies, including pcPIR, is that only a small known residual mass from the cross-linker (mass = 100.0160 Da) is treated as a modification group when performing database search7, thus the database size increases linearly with increasing numbers of peptide sequences. On the other hand, in the experiments with non-cleavable cross-linkers, the MS/MS database search space increases by square with increasing numbers of candidate peptide sequences. In addition, the released peptides can be treated as normal tryptic peptides with modifications of known mass. Therefore the database search algorithms can be greatly simplified. Furthermore, pcPIR has other advantages. 1) The cleavage mechanism of pcPIR is orthogonal to that of the collision activated cleavage; therefore the cleavage is more specific, only targeting the photocleavable bonds in the cross-linker. Such a specific cleavage on cross-linker eliminates the possibility of detecting any fragments from peptides during the step of releasing intact peptides from the cross-linked precursors. 2) The ESI and MALDI methods are complementary to each other, and the best coverage should be obtained by combining the results from both methods. A special advantage in the ESI method is that the released peptides are generated with higher charge states, so they can produce more comprehensive fragmentation during MS/MS analyses. 3) On-line photocleavage can be coupled with LC separation. This is the first time that a photocleavable PIR cross-linker is reported, which can be cleaved on-line after LC-separation and the photo dissociation degree is tunable by controlling the laser intensity or the sample flow rate. On-line cleavage takes place after LC separation; therefore LC elution information of different products can be retained. One problem in chemically cleavable cross-linking strategies is that the cleavage happens off-line and thus the LC elution time of the cleavage products are no longer relevant to that of their precursors. However, in the pcPIR strategy the cross-linked precursor and cleavage products appear under the same LC chromatogram peak. This “co-elution” of precursor and cleavage products strongly supports the cross-linking relationship identifications. pcPIR can also be cleaved off-line and analyzed in situ with MALDI ion source instruments, in which way the LC “co-elution” feature is also preserved. 4) Finally, pcPIR can be used more conveniently than chemically cleavable cross-linkers, since no post-cleavage cleaning step is needed.
CONCLUSIONS
A novel photocleavable and mass spectrometry identifiable cross-linker, pcPIR, was designed, synthesized and demonstrated with standard peptides, BSA and hemoglobin tetramer protein complex. The photo dissociation of cross-linker between LC separation and MS detection preserves the intact released peptides and the mass relationships between the precursor and the released peptides. With accurate masses and LC information, cross-linking relationships and thus cross-link types can be identified. The subsequent LC-MS/MS run with mass-and-time targeted inclusion list allows the identification of the released peptide sequences and cross-linked sites on proteins using traditional database search algorithms. In such a way, database search is simplified, because the size of the database increases only linearly with the increase of the possible cross-linked candidates. The biotin group in the structure also enables avidin affinity purification of the cross-linked products. Such features of pcPIR make it promising for the studies of the more complicated biological systems in the future.
Supplementary Material
ACKNOWLEDGEMENT
This research was supported by NIH grant 5R01RR023334, NSF grant DBI-0352451 and UW Proteomics Resource (#UWPR95794).
REFERENCES
- (1).Sinz A. Mass Spectrom Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- (2).Jin Lee Y. Mol Biosyst. 2008;4:816–823. doi: 10.1039/b801810c. [DOI] [PubMed] [Google Scholar]
- (3).Tang X, Munske GR, Siems WF, Bruce JE. Anal Chem. 2005;77:311–318. doi: 10.1021/ac0488762. [DOI] [PubMed] [Google Scholar]
- (4).Rinner O, Seebacher J, Walzthoeni T, Mueller LN, Beck M, Schmidt A, Mueller M, Aebersold R. Nat Methods. 2008;5:315–318. doi: 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tang X, Yi W, Munske GR, Adhikari DP, Zakharova NL, Bruce JE. J Proteome Res. 2007;6:724–734. doi: 10.1021/pr060480e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhang H, Tang X, Munske GR, Zakharova N, Yang L, Zheng C, Wolff MA, Tolic N, Anderson GA, Shi L, Marshall MJ, Fredrickson JK, Bruce JE. J Proteome Res. 2008;7:1712–1720. doi: 10.1021/pr7007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang H, Tang X, Munske GR, Tolic N, Anderson GA, Bruce JE. Mol Cell Proteomics. 2009;8:409–420. doi: 10.1074/mcp.M800232-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Anderson GA, Tolic N, Tang X, Zheng C, Bruce JE. J Proteome Res. 2007;6:3412–3421. doi: 10.1021/pr070035z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gingras AC, Gstaiger M, Raught B, Aebersold R. Nat Rev Mol Cell Biol. 2007;8:645–654. doi: 10.1038/nrm2208. [DOI] [PubMed] [Google Scholar]
- (10).Back JW, de Jong L, Muijsers AO, de Koster CG. J Mol Biol. 2003;331:303–313. doi: 10.1016/s0022-2836(03)00721-6. [DOI] [PubMed] [Google Scholar]
- (11).Trakselis MA, Alley SC, Ishmael FT. Bioconjug Chem. 2005;16:741–750. doi: 10.1021/bc050043a. [DOI] [PubMed] [Google Scholar]
- (12).Itoh Y, Cai K, Khorana HG. Proc Natl Acad Sci U S A. 2001;98:4883–4887. doi: 10.1073/pnas.051632998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Muller DR, Schindler P, Towbin H, Wirth U, Voshol H, Hoving S, Steinmetz MO. Anal Chem. 2001;73:1927–1934. doi: 10.1021/ac001379a. [DOI] [PubMed] [Google Scholar]
- (14).Taverner T, Hall NE, O’Hair RA, Simpson RJ. J Biol Chem. 2002;277:46487–46492. doi: 10.1074/jbc.M207370200. [DOI] [PubMed] [Google Scholar]
- (15).Petrotchenko EV, Xiao K, Cable J, Chen Y, Dokholyan NV, Borchers CH. Mol Cell Proteomics. 2009;8:273–286. doi: 10.1074/mcp.M800265-MCP200. [DOI] [PubMed] [Google Scholar]
- (16).Gardner MW, Vasicek LA, Shabbir S, Anslyn EV, Brodbelt JS. Anal Chem. 2008;80:4807–4819. doi: 10.1021/ac800625x. [DOI] [PubMed] [Google Scholar]
- (17).Gardner MW, Brodbelt JS. Anal Chem. 2009;81:4864–4872. doi: 10.1021/ac9005233. [DOI] [PubMed] [Google Scholar]
- (18).Bennett KL, Kussmann M, Bjork P, Godzwon M, Mikkelsen M, Sorensen P, Roepstorff P. Protein Sci. 2000;9:1503–1518. doi: 10.1110/ps.9.8.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Soderblom EJ, Goshe MB. Anal Chem. 2006;78:8059–8068. doi: 10.1021/ac0613840. [DOI] [PubMed] [Google Scholar]
- (20).Petrotchenko EV, Olkhovik VK, Borchers CH. Mol Cell Proteomics. 2005;4:1167–1179. doi: 10.1074/mcp.T400016-MCP200. [DOI] [PubMed] [Google Scholar]
- (21).Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, Dollinger G. Proc Natl Acad Sci U S A. 2000;97:5802–5806. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Singh P, Shaffer SA, Scherl A, Holman C, Pfuetzner RA, Larson Freeman TJ, Miller SI, Hernandez P, Appel RD, Goodlett DR. Anal Chem. 2008;80:8799–8806. doi: 10.1021/ac801646f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chowdhury SM, Munske GR, Tang X, Bruce JE. Anal Chem. 2006;78:8183–8193. doi: 10.1021/ac060789h. [DOI] [PubMed] [Google Scholar]
- (24).Hammond N, Koumi P, Langley GJ, Lowe A, Brown T. Org Biomol Chem. 2007;5:1878–1885. doi: 10.1039/b704587e. [DOI] [PubMed] [Google Scholar]
- (25).Lemaire R, Stauber J, Wisztorski M, Van Camp C, Desmons A, Deschamps M, Proess G, Rudlof I, Woods AS, Day R, Salzet M, Fournier I. J Proteome Res. 2007;6:2057–2067. doi: 10.1021/pr0700044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.