

Abstract
Arsenic is a well known environmental toxicant but the mechanism by which it causes cytotoxicity is poorly understood. Arsenite induces apoptosis in glutathione (GSH)-deficient GCS-2 cells by causing cell cycle dysfunction and down-regulating critical signaling pathways. This study was designed to examine the effect of arsenite on redox-sensitive phosphatidylinositol 3-kinase (PI3K)/Akt, a signaling pathway involved in cell survival and growth, and transcription factor, activating protein-1 (AP-1). Arsenite significantly diminished Akt and c-Fos levels and caused accelerated degradation of these proteins by ubiquitnation. Arsenite also induced cell cycle arrest and apoptosis. The cell cycle arrest involved the down-regulation of cyclin A2, cyclin D1, cyclin E, cyclin dependent kinases (CDK) 2, CDK4, and CDK6. Apoptosis involved down-regulation of anti-apoptotic proteins Bcl-2, Bcl-xL, survivin, and inhibitor of apoptosis protein (IAP) and up-regulation of pro-apoptotic protein Bax. Taken together, our results suggest that a possible mechanism of arsenite-induced toxicity under low/no GSH conditions, is to negatively regulate GCS-2 cell proliferation by attenuating Akt and AP-1 by ubiquitination and causing cell cycle dysfunction and apoptosis.
Keywords: Glutathione, Akt, AP-1, Arsenic, ubiquitination, cell cycle, apoptosis, redox, signal transduction
Introduction
Arsenic, a well-documented cytotoxin and a human carcinogen, has been linked to hyperkeratosis, cancer of the bladder, skin, and lung, and cardiovascular disorders [Kligerman et al., 2007; IARC., 2003; Kitchin., 2001]. Arsenicals act as environmental carcinogens and as therapeutic agents in the treatment of cancer [Kitchin., 2001; Thomas., 2007; Verstovsek et al., 2006]. How chronic exposure to arsenic, causes cancer, cell cycle arrest, cell injury and cell death, and the molecular mechanisms involved in arsenic toxicity remain incompletely understood.
Thiol-containing compounds play a vital role in many biologically significant reactions. Of these, GSH, a principal non-protein intracellular thiol, is present in millimolar concentrations in many organs [Meister., 1988; Kosower et al., 1978]. GSH acts as an antioxidant and is believed to protect cells against toxins and xenobiotics through the formation of GSH conjugates [Lieberman et al., 1995; Ballatori et al., 1998; Kala et al., 2004]. GSH also plays an important role in signaling pathways involving reactive oxygen species [Dickinson et al., 2002; Allen et al., 2000].
PI3K/Akt signaling pathway has been regarded as one of the vital pro-survival pathways in the cell [Alessi et al., 1997; Maddika et al., 2008]. Many types of stimuli including toxic insults activate Akt and that in turn regulates basic cellular functions such as growth, proliferation, transcription, translation, cell cycle, and apoptosis [Alessi et al., 1997; Maddika et al., 2008; Yuan et al., 2008; Ruggero et al., 2005; Nimbalkar et al., 2003]. Although many studies support the role of PI3K-Akt pathway in cell proliferation and cell survival, PI3K pathway has also been implicated in the promotion of cell death [Duronio., 2008; Marone et al., 2008]. Other than cell survival, Akt pathway also regulates cell cycle progression at G1-S and G2-M phases by either phosphorylating directly or regulating the expression indirectly of various substrates including cyclin D family [Liang et al., 2003; Chang et al., 2003]. CDKs (CDK2, CDK4, CDK6), members of the serine/threonine family of cyclin dependent kinases are key regulators of cell cycle progression [Malumbres et al., 2005]. By binding to partners cyclin A or cyclin E and the subsequent phosphorylation, they get activated during G1 and S phases of the cell cycle.
Studies have shown that As (III) modulates the expression and DNA binding activities of various transcription factors associated with cell proliferation and cell death including tumor suppressor 53 (p53), nuclear factor-kB (NF-κB), a Y-box specific transcription factor-1 (YB-1), activator protein-1 (AP-1), and a GC-box specific transcription factor 1(SP-1) [Salazar et al., 1997; Barchowsky et al., 1996; Kaltreider et al., 1999; Simenova et al., 2000]. Available data suggest that As (III) may modulate AP-1 dependent gene transcription and contribute to either induction of cell proliferation or acceleration of cell death [Simenova et al., 2000; Trouba et al., 2000].
Various intracellular and extracellular stress signals can result in the activation of pro-apoptotic proteins such as Bcl2-associated X protein (Bax) and inactivation of anti-apoptotic Bcl2 family members such as Bcl2 or Bcl-XL [Maddika et al., 2007]. PI3k/Akt and MAP kinases have been shown to play a role in arsenic-induced mitochondrial cell death in tumor cells [Tabellini et al., 2005; Sanchez et al., 2008].
Using GSH-deficient GCS-2 cells, we have previously demonstrated that arsenite induces ubiquitination and apoptotic cell death [cytochrome C leakage and DNA fragmentation; Habib et al., 2007] and that p53 regulates Hsp90β during arsenite-induced cytotoxicity [Habib et al., 2009]. In the current study, we show that arsenite down-regulates Akt and c-Fos by ubiquitination, decreases AP-1 activity and causes cell cycle dysregulation and apoptotic cell death. Our results support the concept that arsenite down-modulates PI3k/Akt and AP-1 pathways under GSH-deficient conditions and the cytotoxicity of arsenite culminates in cell cycle dysregulation and apoptotic cell death.
Experimental Procedures
Reagents
Polyclonal antibodies to Akt, c-Fos, cell cycle and apoptosis proteins were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) or Cell Signal Technologies (Beverly, MA). Anti-ubiquitin antibodies were obtained from StressGen Biotechnologies (Victoria, British Columbia, Canada). Secondary horseradish peroxidase-conjugated antibodies were from Bio-Rad Laboratories (Richmond, CA). [γ-32P]ATP was purchased from Perkin-Elmer Life Sciences (Boston, MA).
Cell Culture
BDC-1 and GCS-2 cells are epithelial cells derived from 3.5-day postcoitus (dpc) embryos from both wild-type and Glutamate cysteine ligase catalytic subunit (Gclc)-deficient mice [Shi et al., 2000]. They have been passaged hundreds of times and they are morphologically stable. All cell culture studies used M15 complete medium and cultures were done essentially as described earlier [Habib et al., 2007; Shi et al., 2000]. All GSH-dependent GCS-2 cells (mutant cells) were maintained in medium containing 2.5 mM GSH and NAC-dependent GCS-2 cell lines were cultured in medium supplemented with 2 mM NAC. BDC-1 cells (wild type controls) were maintained in medium without GSH or NAC.
Treatment of Cells
Sodium arsenite was purchased from Sigma (St. Louis, MO). A 100 μM stock solution was prepared in PBS, pH 7.4 and was used at final concentrations ranging from 0–2 μM. Briefly, the GCS-2 cells were seeded at 1.5 × 106 cells per 10 cm dish, allowed to grow for 48 h either in the presence of 2.5 mM GSH or 2mM NAC, depleted of GSH or NAC for 24 h, and treated with various concentrations of arsenite for up to 21 h. BDC-1 cells were treated under similar conditions as GCS-2 cells except that they were grown in the absence of GSH or NAC.
Measurement of Thiols
Thiols were measured using the high-performance liquid chromatography-electrochemical detection method of Kleinman and Richie [Kleinman et al., 1995] with minor modifications.
Immunoprecipitation
Cells were washed twice with ice-cold PBS, and harvested in cell lysis buffer ( 50 mM Tris-HCL, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1 mM disodium EDTA, 1% sodium deoxycholate) containing the protease inhibitors (Roche Biochemicals, Indianapolis, IN). Cells were lysed by sonication and the protein supernatants were clarified by centrifugation at 25,000 × g for 10 min at 4 °C. Protein concentration was determined by the BCA protein assay reagent kit (Pierce, Rockford, IL). Akt and c-Fos from clarified lysates were immunoprecipitated with 1μg of specific polyclonal antibody against Akt or c-Fos (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and 20 μl of protein A/G plus agarose (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for 2h at 4°C. Immunoprecipitated proteins were washed under stringent conditions, resolved on 7.5% SDS-polyacrylamide gels, transferred on to nitrocellulose membranes (Amersham Pharmacia Biotech, Piscataway, NJ), and analyzed by western blotting.
Western Blotting
After various treatments, cells were collected by centrifugation and resuspended in lysis buffer [50 mM Tris-HCl, 150 mM NaCl, 1% Triton-X-100, 0.1% SDS, 1 mM EDTA, 1% sodium deoxycholate, 20 mM NaF, 1 mM Na3VO4, 50 μM Na2MoO4, 10 mM Sodium pyrophosphate, supplemented with protease inhibitor mixture (Roche Applied Science, Indianapolis, IN)]. 100 μg of total protein was routinely used for Western blots. Protein was separated on 10% SDS-polyacrylamide gels, transferred to nitrocellulose membranes, incubated with the indicated antibodies and developed by ECL technology (Amersham Biosciences Corp, Piscataway, NJ) according to manufacturer’s instructions. The blots were stripped and reprobed with mouse actin antibody. The band intensities on the films were quantified using a GS-800 calibrated scanning densitometer (Bio-Rad, Hercules, CA). Quantitative expression of specific proteins was normalized to the expression of actin.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA assays were performed using the Gel Shift Assay System (Promega) according to the manufacturer’s protocol. The double-stranded oligonucleotides containing the putative AP-1 binding sites (underlined) were as follows: 5′-CGCTTGATGAGTCAGCCGGAA-3′. In the binding reactions, γ32P-labeled DNA probes were incubated with 10 μg of nuclear extract. Binding reactions were performed at room temperature for 20 min and the DNA-protein complexes were resolved by electrophoresis on 5% nondenaturing Tris-Borate-EDTA polyacrylamide gels and visualized by autoradiography. Signals were quantified using PhosphorImager and ImageQuant™ software. The AP-1 binding activity was further confirmed by coincubation with unlabeled AP-1 oligonucleotides (cold probe competition) or with antibodies to c-Fos (supershift assay).
Statistical Analysis
Data are expressed as mean ± S.D. Statistical evaluation of data was performed using Student’s t test, considering p value of ≤ 0.05 as significant.
Results
Arsenite is a negative regulator of Akt under low/no GSH conditions
Previous studies from our laboratory have demonstrated that low/no GSH levels predispose GCS-2 cells to arsenite-induced apoptosis [Habib et al., 2007]. To define the mechanisms contributing to arsenite-induced toxicity in GCS-2 cells, in the current study, we determined if low GSH levels and arsenite are involved in Akt-mediated signal transduction. Preliminary dose response and time course studies indicated that arsenite caused a time- and dose-dependent decrease in Akt levels in GCS-2 cells with maximal effect observed at 0.5 μM at 21 h (data not shown). Using antibodies specific for Akt, we found that arsenite significantly decreased Akt levels (2.8 ± 0.3 fold, n=3; p ≤ 0.05 compared to untreated GCS-2 cells) in GCS-2 cells (Fig. 1A, top panel). Akt expression remained comparable in untreated GCS-2 cells compared to untreated or arsenite treated BDC-1 cells. Arsenite had no effect on Akt expression in BDC-1 cells.
Fig. 1.
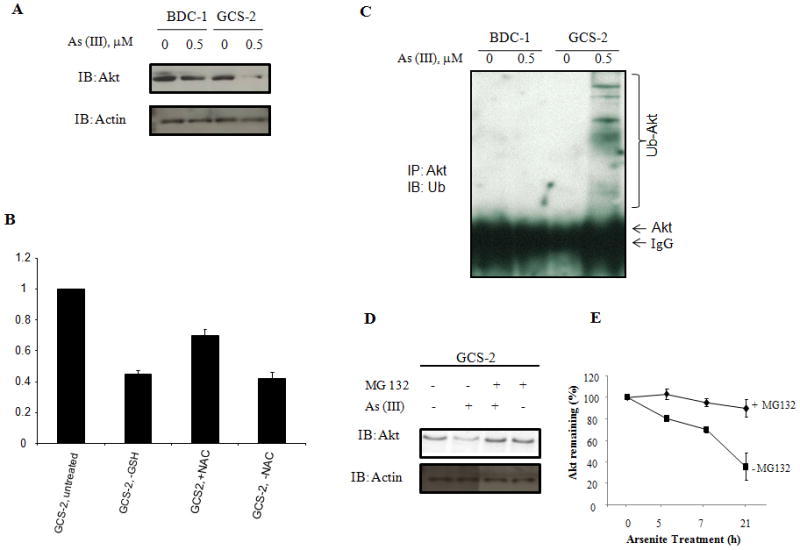
Effect of arsenite on Akt levels and stability in GCS-2 cells. (A) Western blot analysis of Akt protein levels. Culture and treatment of cells is done as described in Ref. 31. BDC-1 and GCS-2 cells were incubated with 0.5 μM arsenite (indicated along the top) for 21 h, cell extracts separated by SDS-PAGE and analyzed by immune-blotting with anti-Akt antibody. Equivalent loading was verified with anti-actin antibody (bottom). (B) Quantification of Akt expression by densitometry. Effect of arsenite on Akt levels was determined in GSH- and NAC-dependent cells. Culture and treatment of cells is done as described in Ref. 31. Cells were treated and extracts were separated on SDS-PAGE as in (A) and quantified by densitometry. The results are expressed as mean ± SD of triplicate determinations. (C) Effect of arsenite on Akt ubiquitination in GCS-2 cells. Akt was immunoprecipitated with anti-Akt, separated by SDS-PAGE, and analyzed by immunoblot with anti-ubiquitin antibody. (D) Effect of proteasome inhibitor MG132 on arsenite-induced degradation of Akt. Cells were treated with and without 0.5 μM arsenite and with without 2.5 μM MG132. Cell extracts were prepared and analyzed as described above and immunoblotted with anti-Akt antibody. (E) Quantification of Akt expression by densitometry. Effect of 0.5 μM arsenite on Akt levels was determined in GSH-dependent GCS-2 cells with and without MG-132 treatment. Cells were treated and extracts were separated on SDS-PAGE as in (D) and quantified by densitometry. The results are expressed as mean ± SD of triplicate determinations.
Thiol antioxidant NAC partially restores Akt levels
We have previously reported that NAC does not complex with arsenite [Habib et al., 2007]. To determine if other thiol antioxidants such as NAC could restore Akt to its original levels in GCS-2 cells, we exposed GCS-2 cells previously rescued with NAC to arsenite. NAC partially reversed the levels of Akt in GCS-2 cells treated with arsenite in the presence of NAC if it is present continuously during arsenite exposure while we found that in the absence of NAC, Akt levels diminished to the same extent as in the case of GCS-2 cells treated with arsenite in the absence of GSH (Fig. 1B)
Arsenite targets Akt to proteasomal degradation
Polyubiquitination often serves as a signal for proteasomal degradation. We examined the possibility that Akt is proteolytically degraded upon arsenite treatment via the ubiquitin-proteasomal system. We immunoprecipitated Akt from total cellular extracts with anti-Akt antibody and analyzed by immunoblot with anti-ubiquitin antibody. In GCS-2 cells treated with arsenite, we found bands in the high molecular weight region (>55 kDa) ranging in size from 100 to 250 kDa (Fig. 1C). These data suggest that Akt is polyubiquitinated in the GCS-2 cells after arsenite treatment. We also confirmed the above results by immunoprecipitating Akt from total cellular extracts with anti-ubiquitin antibody and immunoblotting with anti-Akt antibody (data not shown). There was no evidence of Akt ubiquitination in untreated GCS-2 cells or untreated BDC-1 cells or arsenite treated BDC-1 cells.
To confirm whether Akt degradation is mediated through proteolysis, we exposed the GCS-2 cells to arsenite in the presence or absence of MG-132, a 26S proteasome inhibitor before the cells were harvested for immunoblotting. Co-exposure with MG 132 restored Akt to its original levels (Fig. 1D). Lactacystin, another proteasomal inhibitor also significantly abolished the arsenite-induced Akt degradation (data not shown). These results suggest that arsenite induces Akt ubiquitination and degradation mainly through the 26S proteasome pathway and the proteasomal inhibitors could block the down-regulation of Akt. We quantified the time-dependent change in Akt levels upon arsenite exposure in the presence or absence of MG-132 treatment and the results are shown in Fig. 1E.
Arsenite treatment attenuates c-Fos levels and targets c-Fos for ubiquitination
We next determined the status of AP-1 constituent c-Fos by immmuno blot analysis of total cellular extracts from untreated and arsenite treated BDC-1 and GCS-2 cells. This analysis was focused on c-Fos as this is a major component of the AP-1-DNA binding complex. We found that arsenite treatment under low/no GSH conditions decreased c-Fos levels in GCS-2 cells by 3.1 fold (3.1 ± 0.2, n=3; p ≤ 0.05; Fig. 2A).
Fig. 2.
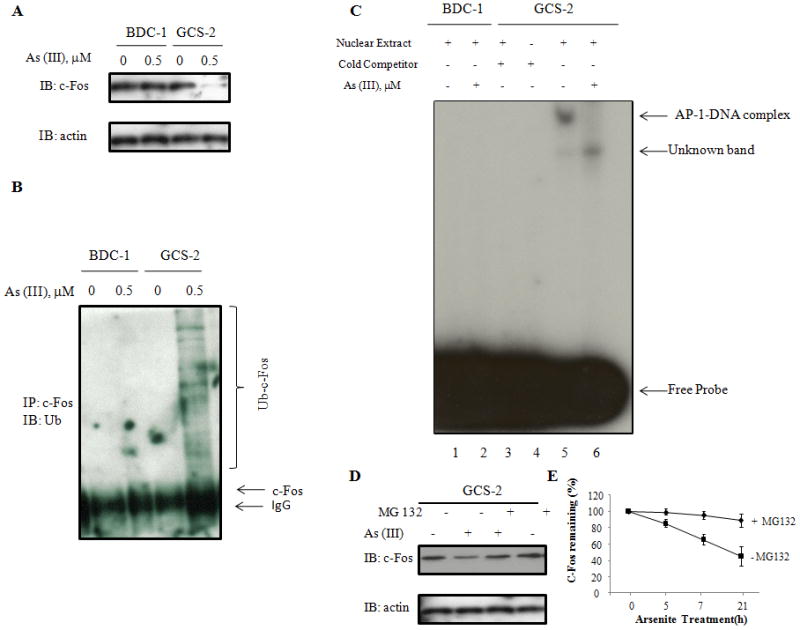
Effect of arsenite on c-Fos levels, degradation and AP-1binding activity in GCS-2 cells. (A) Western blot analysis of c-Fos protein levels. Arsenite treated and untreated cell extracts were resolved by SDS-PAGE and immunoblotted with antibodies against c-Fos. Equal loading was verified with anti-actin antibody (bottom panel). The blot presented is a representative of three independent experiments. (B) Effect of arsenite on c-Fos ubiquitination in GCS-2 cells. C-Fos was immunoprecipitated with anti-c-Fos, separated by SDS-PAGE, and analyzed by immunoblot with anti-ubiquitin antibody. (C) Effect of arsenite on AP-1 binding in GCS-2 cells. Gel mobility shift assay with nuclear extracts (BDC-1 and GCS-2) and 32P-labeled oligonucleotide containing TGAGTCAG without excess cold competitor and no arsenite (lanes 1 and 5 respectively); BDC-1 and GCS-2 nuclear extracts treated with 32P-labeled oligonucleotide without excess cold competitor and 0.5 μM arsenite treatment (lanes 2 and 6 respectively); GCS-2 nuclear extracts treated with 32P-labeled oligonucleotide and excess cold competitor but without arsenite treatment (lane 3); 32P-labeled oligonucleotide TGAGTCAG with no nuclear extract and no arsenite treatment but with excess cold competitor (lane 4). For experimental conditions, see “Experimental Procedures”. The data presented is a representative of three independent experiments. (D) Effect of proteasome inhibitor MG132 on arsenite-induced degradation of c-Fos. Cells were treated with and without 0.5 μM arsenite and with without 2.5μM MG132. Cell extracts were prepared and analyzed as described above and immunoblotted with anti-c-Fos antibody. (E) Quantification of c-Fos expression by densitometry. Effect of 0.5μM arsenite on c-Fos levels was determined in GSH-dependent GCS-2 cells with and without MG-132 treatment. Cells were treated and extracts were separated on SDS-PAGE as in (D) and quantified by densitometry. The results are expressed as mean ± SD of triplicate determinations.
We further investigated the possibility that ubiquitination is the reason for the marked diminution in c-Fos levels in GCS-2 cells by arsenite. c-Fos was immunoprecipitated from cellular extracts with anti-c-Fos and analyzed by western blotting with an anti-ubiquitin antibody. In GCS-2 cells treated with arsenite, we observed a wide-array of bands in the high molecular weight region (>40 kDa) indicating that c-Fos is ubiquinated in these cells (Fig. 2B). We verified the above results by immunoprecipitating c-Fos from cellular extracts with an anti-ubiquitin antibody and blotting with an anti-c-Fos antibody (data not shown). We did not see any evidence of ubiquitination in either untreated or arsenite treated BDC-1 cells or untreated GCS-2 cells.
Arsenite treatment decreases AP-1 binding activity
The second step of our study was to determine if arsenic treatment and the decrease in c-Fos levels had affected AP-1 binding activity in GCS-2 cells. We end labeled AP-1 consensus sequence 5′-CGCTTGATGAGTCAGCCGGAA-3′DNA fragment [Hu et al., 2002] with [γ-32p] ATP and used it as a probe or as specific competitor without labeling in EMSA. Nuclear extracts from either untreated or arsenite treated BDC-1 or GCS-2 cells were incubated with the probe in vitro. Nuclear extracts from untreated and arsenite treated BDC-1 cells were unable to bind the probe (Fig. 2C, lanes 1 and 2). While extracts from untreated GCS-2 cells exhibited a high AP-1 binding activity, arsenite inhibited binding by more than 90% (Fig. 2A, compare lane 5 Vs lane 6). In addition, the specific binding could be competed out by excess cold competitor or by omitting nuclear extract from the reaction (lanes 3 and 4 respectively). Moreover, c-Fos specific antibody was able to supershift the AP-1 bound DNA band confirming the specificity of the binding whereas the unknown band (seen on lane 6) was unaffected by the antibody treatment (data not shown).
Proteasomal inhibitors block arsenite-induced down-regulation of c-Fos
We further explored if the rapid degradation of c-Fos was mediated via proteolysis. We exposed the GCS-2 cells to arsenite both in the presence or absence of MG-132, a 26S proteasome inhibitor before we collected and analyzed the cells by immunoblotting. Co-exposure of the cells with both arsenite and MG-132 restored the arsenite-induced c-Fos degradation (Fig. 2D). Another proteasomal inhibitor lactacystin also significantly attenuated arsenited-induced c-Fos proteolysis (data not shown). These data indicate that arsenite-induced degradation of c-Fos occurs via the 26S proteasomal pathway and the proteasomal inhibitors can block c-Fos proteolysis. Quantification of the time-dependent change in c-Fos levels upon arsenite exposure in the presence or absence of MG-132 treatment is shown in Fig. 2E.
Effect of arsenite on the expression of cell cycle regulators in GCS-2 cells
PI3K/Akt pathway has been implicated in the regulation of either cell growth or apoptosis [Tabellini et al., 2005]. Thus, it is important to determine if there is a link between arsenite exposure and cell cycle regulation. We hypothesized that the down-regulation and degradation of both Akt and c-Fos upon arsenite treatment might result in direct regulation of the cell cycle genes. Western blot analysis was performed to determine the levels of various cell cycle related regulators. The levels of cyclins A2, D1, E and the kinases CDK2, CDK4, and CDK6 were all down-regulated in GCS-2 cells treated with arsenite compared to untreated cells (Fig. 3).
Fig. 3.
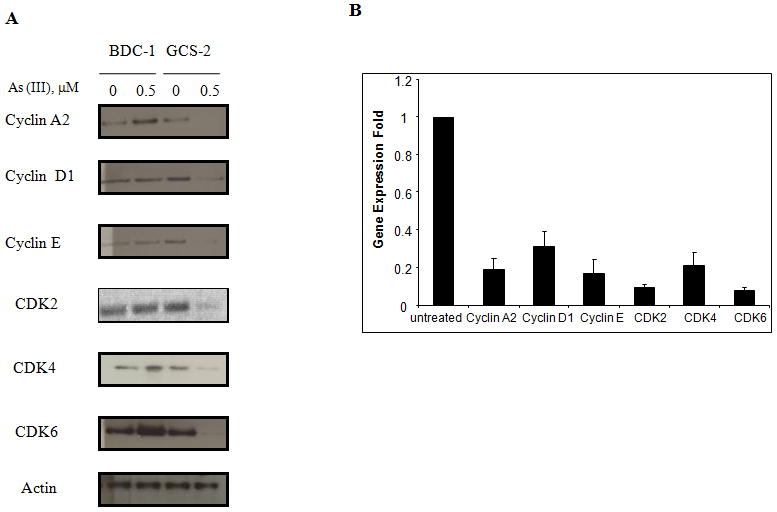
Arsenite-induced down-regulation of cell cycle regulators in GCS-2 cells. (A) Effect of arsenite on cyclin A2, cyclin D1, cyclin E, CDK-2, CDK-4, and CDK-6 expression in GCS-2 cells. Cell extracts were immuno-blotted with respective antibodies. Equal protein loading was verified with anti-actin antibody. (B) Quantification of cell cycle regulator expression by densitometry. Cells were treated and extracts were separated on SDS-PAGE as in (A) and quantified by densitometry. The results are expressed as mean ± SD of triplicate determinations. Expression of each cell cycle regulator was compared against its own untreated GCS-2 control cells and the expression of GCS-2 control cells corresponding to each cell cycle regulator was arbitrarily represented as one fold in the figure for comparison purposes.
Arsenite exposure decreases the levels of anti-apoptotic proteins and increases the levels of Bax
We have previously demonstrated that arsenite induces apoptosis in GCS-2 cells [Habib et al., 2007]. We examined the status of apoptosis regulatory factors by immunoblotting and found that low GSH levels and arsenite treatment cooperatively reduced anti-apoptotic proteins such as Bcl-2, Bcl-xL, survivin, and IAP to varying levels in GCS-2 cells. On the other hand, level of pro-apoptotic protein Bax increased by 1.8 fold (Fig. 4). These observations corroborate our earlier findings on arsenite-induced apoptotic cell death in GCS-2 cells.
Fig. 4.
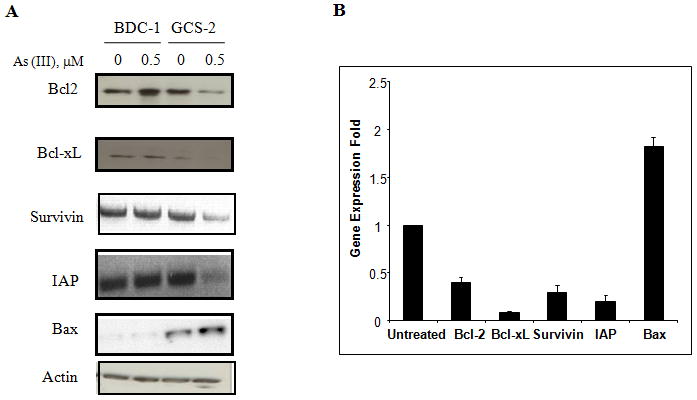
Effect of arsenite on anti-apoptotic and pro-apoptotic protein levels in GCS-2 cells. (A) Effect of arsenite on anti-apoptotic proteins Bcl-2, Bcl-xL, survivin, IAP and pro-apototic prtein Bax expression in GCS-2 cells. Cell extracts were immuno-blotted with respective antibodies. Equal protein loading was verified with anti-actin antibody. (B) Quantification of apoptotic protein expression by densitometry. Cells were treated and extracts were separated on SDS-PAGE as in (A) and quantified by densitometry. The results are expressed as mean ± SD of triplicate determinations. Expression of each anti-apoptotic and pro-apoptotic protein expression was compared against its own untreated GCS-2 control cells and the expression of GCS-2 control cells corresponding to each apoptotic protein was arbitrarily represented as one fold in the figure for comparison purposes.
Discussion
Several modes of action have been proposed for arsenic toxicity including its ability to act as a co-carcinogen, induce DNA damage, inhibit DNA repair, alter DNA methylation patterns, and induce cellular proliferation and cell death [Kligerman et al., 2007]. It is quite possible that more than one mode of action is operational at any given time and none of these are mutually exclusive in nature. Arsenic is also employed as a therapeutic agent in the treatment of cancer and some of the mechanisms that are implicated in the anti-tumor cytotoxicity of arsenic include its ability to induce apoptotic cell death by modulating the redox homeostasis in the cell [Habib et al., 2007; Davison et al., 2004].
As (III) has a high avidity for interaction with sulfhydryl groups. GSH being the major non-protein thiol plays an important role in arsenic detoxification and its depletion enhances cellular sensitivity to arsenic cytotoxicity and arsenic-induced apoptosis while increased GSH levels promote resistance to arsenic-induced apoptosis [Miller et al., 2002; Thompson et al., 2009]. Previous studies from our laboratory have shown that exposure of GSH-depleted GCS-2 cells to low concentrations of arsenite leads to apoptotic cell death [Habib et al., 2007]. The current study was undertaken to identify some of the molecular mechanisms that can explain the cytotoxic activities of arsenic under very low/absent levels of GSH in GCS-2 cells.
We have examined the effects of very low (submicromolar) concentrations of arsenic on gene expression in GSH deficient cells. These concentrations more closely mimic the conditions generally associated with arsenic exposure in polluted environments [Miller et al., 2002]. Arsenic is thought to exert a growth stimulatory effect at lower concentrations while higher concentrations cause apoptosis [Bode et al., 2002]. These opposing paradoxical effects of arsenic can apparently explain its actions as both a carcinogen and a chemotherapeutic agent.
There are several reports that suggest that arsenic can initiate many signaling cascades within a cell but identifying those pathways is crucial to our understanding the toxicity of arsenic. PI3k/Akt pathway plays a vital role in cell survival by regulating caspase-mediated apoptosis and inhibition of this signaling could lead to apoptosis. Several laboratories have examined a role for Akt in arsenic-induced toxicity but the data are confusing [Ouyang et al., 2006; Mann et al., 2008]. It has been shown that sensitivity of leukemic cell lines toward arsenic-induced apoptosis is determined by the level of activation of PI3K/Akt pathway [Tabellini et al., 2005]. Tsou et al have demonstrated that arsenite treatment enhanced proteasomal degradation of Akt in vascular endothelial cells while Mann et al have shown that arsenite decreased not only Akt protein levels but also Akt activity [Mann et al., 2008; Tsou et al., 2005]. Our results show that arsenite treatment resulted in a rapid decrease in the Akt level. Moreover, arsenite also promoted the ubiquitination of Akt, thereby contributing to the down-regulation of Akt (Fig. 1). These observations suggest that the inhibitory effect of arsenite on Akt is mediated, at least in part, by the ubiquitin-proteasome degradation, thereby contributing to apoptosis in GCS-2 cells. In contrast, Ouyang et al have shown in mouse epidermal cells that arsenite treatment induced Akt activity leading to an enhanced cyclin D1 level [Ouyang et al., 2006].
To further elucidate the molecular mechanisms of arsenic modulated gene expression, we studied the DNA binding activities of AP-1, a redox sensitive transcription factor (Fig. 2C). It is known that AP-1 is modulated by a variety of physiological stimuli including changes in the redox status of the cell [Cavigelli et al., 1996]. This transcription factor in turn regulates a number of physiological processes including cell growth, proliferation, differentiation, and cell death [Hu et al., 2002; Drobná et al., 2003]. It is possible that inhibition of their activation may shift the control from cell survival to cell death. Under our experimental conditions, AP-1 DNA binding activity is suppressed and c-Fos level decreases in the GCS-2 cells (Fig. 2B). Although previous studies have demonstrated that inorganic arsenite enhanced AP-1 DNA binding activity and c-Fos phosphorylation, most of these studies employed acute exposure of cells to very high concentrations of arsenic (ranging from 50 μM to 400 μM; ref. 42, 43). Hu et al studied human GM487 fibroblasts after both short-term exposure (up to 5 μM and 24 h) and long-term semi-continuous exposure (up to 0.5 μM and 10 weeks) to arsenic. They concluded that the short-term exposure up-regulated AP-1 binding activity and c-Fos expression while long-term semi-continuous exposure decreased both AP-1 DNA binding activity and c-Fos protein levels [Hu et al., 2002]. Direct comparison between our observations and the above-mentioned studies is difficult because of the inherent differences in arsenic exposure conditions and the absence of documentation of actual GSH levels in these cells after exposure to arsenic. Other studies have demonstrated that c-Fos shows a somewhat biphasic response with a slight up-regulation at lower doses (10 μM) and down-regulation at higher doses (40 μM) of arsenic exposure [Yedjou et al., 2009].
In mammalian cells, cyclins A, D and E are key regulators involved in G1 to S phase transition. Several investigators have examined the effects of arsenic on cell cycle but the reported results are conflicting and confusing [Yedjou et al., 2009; Hwang et al., 2006]. Our observations on the down-regulation in the protein levels of key cell cycle components (Fig. 3) such as cyclin A, cyclin D, cyclin E and the associated kinases concur with the results reported by Woo et al in HUVEC suggesting a mitotic cell cycle arrest thus resulting in the prevention of progression of the cell cycle from G1 to S phase [Woo et al., 2005]. On the other hand, Yedjou et al [Yedjou et al., 2009] have shown that exposure of HL-60 cells to 30 μM arsenic had no significant effect on the levels of cyclin D1 but enhanced the expression of cyclin A which is required for both the S and G2 phases of the cell cycle. Arsenic concentrations used in the above experiments are about 60 fold higher than what we had used in our studies. Besides, as the GCS-2 cells lack GSH and the ability to methylate arsenicals, there is a greater tendency for the intracellular accumulation of inorganic arsenic thus accentuating the cytotoxicity of arsenic.
GSH synthesis inhibitor, L-buthionine sulfoximine (BSO), can enhance arsenic-induced cytotoxicity in tumor cells [Davison et al., 2004; Kito et al., 2002]. Moreover, it has been shown that thiol antioxidant NAC can inhibit arsenic-induced apoptosis [Santra et al., 2007]. Our earlier studies have demonstrated that GSH depletion and exposure to arsenic induced apoptotic cell death in GCS-2 cells [Habib et al., 2007]. Mitochondria play a central role in both extrinsic and intrinsic apoptotic pathways [Baysan et al., 2007]. Bcl-2 family of proteins regulates the release of cytochrome c and AIF culminating in the activation of caspase cascade. Studies have reported that the down-regulation of Bcl-2 played a pivotal role in the apoptosis of cancer cells [Akao et al., 1998]. Bcl-2, a suppressor of programmed cell death is known to homodimerize with itself and forms heterodimers with Bax [Oltvai et al., 1993]. Bax, a promoter of cell death is functionally neutralized by heterodimerization with Bcl-2. The ratio of Bcl/Bax protein could ultimately tip the balance toward cell survival or cell death. The interactions among Bcl family of proteins (Bax, Bcl-2, Bcl-xL, etc.) stimulate the release of cytochrome c to induce apoptosis [Gross et al., 1999]. Our findings on the gene expression of cell death proteins (Fig. 4) are in general agreement with studies by Tun-Kyi et al. and Baysan et al [Baysan et al., 2007; Tun-Kyi et al., 2008].
In summary, our present study reveals that under conditions of reduced GSH availability, arsenite down-regulated Akt and c-Fos via the ubiquitin-prteasome-mediated degradation. Together with our previous report that arsenite induces apoptosis through the accumulation of ubiquitin-protein conjugates, we have demonstrated that arsenite induces apoptosis by affecting the cell cycle and the apoptotic machinery.
Acknowledgments
This work was supported by NIH Grant ES-08668. I am grateful to Dr. Michael W Lieberman, The Methodist Hospital Research Institute, for his support during this investigation and Dr. Mohamed Habib for critically reading the article.
Abbreviations used
- AP-1
activating protein-1
- CDK
cyclin dependent kinase
- GSH
glutathione
- NAC
N-acetyl cysteine
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
References
- 1.Akao Y, Mizoguchi H, Kojima S, Naoe T, Ohishi N, Yagi K. Arsenic induces apoptosis in B-cell leukaemic cell lines in vitro: activation of caspases and down-regulation of Bcl-2 protein. Br J Haematol. 1998;102:1055–1060. doi: 10.1046/j.1365-2141.1998.00869.x. [DOI] [PubMed] [Google Scholar]
- 2.Alessi DR, Deak M, Casamayor A, Caudwell F, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol. 1997;7:776–789. doi: 10.1016/s0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- 3.Allen RG, Tresini M. Oxidative stress and gene regulation. Free Rad Biol Med. 2000;28:463–499. doi: 10.1016/s0891-5849(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 4.Ballatori N, Wang W, Lieberman MW. Accelerated methyl mercury elimination in gamma-glutamyl transpeptidase-deficient mice. Am J Pathol. 1998;152:1049–1055. [PMC free article] [PubMed] [Google Scholar]
- 5.Barchowsky A, Dudek EJ, Treadwell MD, Wetterhahn KE. Arsenic induces oxidant stress and NF-κ B activation in cultured aortic endothelial cells. Free Radic Biol Med. 1996;21:783–790. doi: 10.1016/0891-5849(96)00174-8. [DOI] [PubMed] [Google Scholar]
- 6.Baysan A, Yel L, Gollapudi S, Su H, Gupta S. Arsenic trioxide induces apoptosis via the mitochondrial pathway by upregulating the expression of Bax and Bim in human B cells. Int J Oncol. 2007;30:313–318. [PubMed] [Google Scholar]
- 7.Bode AM, Dong Z. The paradox of arsenic: molecular mechanisms of cell transformation and chemotherapeutic effects. Crit Rev Oncol Hematol. 2002;42:5–24. doi: 10.1016/s1040-8428(01)00215-3. [DOI] [PubMed] [Google Scholar]
- 8.Cavigelli M, Li WW, Lin A, Su B, Yoshioka K, Karin M. The tumor promoter arsenite stimulates AP-1 activity by inhibiting a JNK phosphatase. EMBO J. 1996;15:6269–6279. [PMC free article] [PubMed] [Google Scholar]
- 9.Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCubrey JA. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer therapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- 10.Davison K, Mann KK, Waxman S, Miller WH., Jr JNK activation is a mediator of arsenic trioxide-induced apoptosis in acute promyelocytic leukemia cells. Blood. 2004;103:3496–3502. doi: 10.1182/blood-2003-05-1412. [DOI] [PubMed] [Google Scholar]
- 11.Dickinson DA, Forman HJ. Cellular glutathione and thiols metabolism. Biochem Pharmacol. 2002;64:1019–1026. doi: 10.1016/s0006-2952(02)01172-3. [DOI] [PubMed] [Google Scholar]
- 12.Drobná Z, Jaspers I, Thomas DJ, Styblo M. Differential activation of AP-1 in human bladder epithelial cells by inorganic and methylated arsenicals. FASEB J. 2003;17:67–69. doi: 10.1096/fj.02-0287fje. [DOI] [PubMed] [Google Scholar]
- 13.Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008;415:333–344. doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- 14.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 15.Habib GM, Shi ZZ, Lieberman MW. Glutathione protects cells against arsenite-induced toxicity. Free Radic Biol Med. 2007;42:191–201. doi: 10.1016/j.freeradbiomed.2006.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Habib GM, Shi ZZ, Lieberman MW. p53 regulates Hsp90β during arsenite-induced cytotoxicity in glutathione-deficient cells. Arch Biochem Biophys. 2009;481:101–109. doi: 10.1016/j.abb.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Y, Jin X, Snow ET. Effect of arsenic on transcription factor AP-1 and NF-κB DNA binding activity and related gene expression. Toxicol Lett. 2002;133:33–45. doi: 10.1016/s0378-4274(02)00083-8. [DOI] [PubMed] [Google Scholar]
- 18.Hwang BJ, Utti C, Steinberg M. Induction of cyclin D1 by submicromolar concentrations of arsenite in human epidermal keratinocytes. Toxicol Appl Pharmacol. 2006;217:161–167. doi: 10.1016/j.taap.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 19.IARC. Some drinking water disinfectants and contaminants, including arsenic. International agency for research on cancer; Lyon, France: 2003. Monographs on the evaluation of carcinogenic risks to humans. [PMC free article] [PubMed] [Google Scholar]
- 20.Kala SV, Kala G, Prater CI, Sartorelli AC, Lieberman MW. Formation and urinary excretion of arsenic triglutathione and methylarsenic diglutathione. Chem Res Toxicol. 2004;17:243–249. doi: 10.1021/tx0342060. [DOI] [PubMed] [Google Scholar]
- 21.Kaltreider RC, Pesce CA, Ihnat MA, Lariviere JP, Hamilton JW. Differential effects of arsenic (III) and chromium (VI) on nuclear transcription factor binding. Mol Carcinog. 1999;25:219–229. [PubMed] [Google Scholar]
- 22.Kitchin AT. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicol Appl Phrmacol. 2001;172:249–261. doi: 10.1006/taap.2001.9157. [DOI] [PubMed] [Google Scholar]
- 23.Kito M, Akao Y, Nohishi, Yagi K, Nozawa Y. Arsenic trioxide-induced apoptosis and its enhancement by buthionine sulfoximine in hepatocellular carcinoma cell lines. Biochem Biophys Res Commun. 2002;291:861–867. doi: 10.1006/bbrc.2002.6525. [DOI] [PubMed] [Google Scholar]
- 24.Kleinman WA, Richie JP., Jr Determination of thiols and disulfides using high performance liquid chromatography with electrochemical detection. J Chromatogr B Biomed Appl. 1995;672:5101–5106. doi: 10.1016/0378-4347(94)00194-a. [DOI] [PubMed] [Google Scholar]
- 25.Kligerman AD, Tennant AH. Insights into the carcinogenic mode of action of arsenic. Toxicol Appl Pharmacol. 2007;222:281–288. doi: 10.1016/j.taap.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Kosower NS, Kosower EM. The glutathione status of cells. Int Rev Cytol. 1978;54:109–160. doi: 10.1016/s0074-7696(08)60166-7. [DOI] [PubMed] [Google Scholar]
- 27.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 28.Lieberman MW, Barrios R, Carter BZ, Habib GM, Lebovitz RM, Rajagopalan S, Sepulveda AR, Shi ZZ, Wan DF. Gamma-glutamyl transpeptidase. What does the organization and expression tell us about its function? Am J Path. 1995;147:1175–1185. [PMC free article] [PubMed] [Google Scholar]
- 29.Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, Eshraghi M, Manda KD, Wiechek E, Los M. Cell survival, cell death and cell cycle pathways are interconnected: Implications for cancer therapy. Drug Resistance Updates. 2007;10:13–29. doi: 10.1016/j.drup.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Maddika S, Ande SR, Wiechec E, Hansen LL, Wesselborg S, Los M. Akt-mediated phosphorylation of CDK2 regulates its dual role in cell cycle progression and apoptosis. J Cell Science. 2008;121:979–988. doi: 10.1242/jcs.009530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 32.Mann KK, Colombo M, Miller WH., Jr Arsenic trioxide decreases Akt protein in a caspase-dependent manner. Mol Cancer Ther. 2008;7:1680–1687. doi: 10.1158/1535-7163.MCT-07-2164. [DOI] [PubMed] [Google Scholar]
- 33.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 34.Meister A. Glutathione metabolism and its selective modification. J Biol Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- 35.Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 36.Nimbalkar D, Henry MK, Quelle FW. Cytokine activation of phosphiinositide 3-kinase sensitizes hematopoietic cells to cisplatin-induced death. Cancer Res. 2003;63:1034–1039. [PubMed] [Google Scholar]
- 37.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 38.Ouyang W, Li J, Ma Q, Huang C. Essential roles of PI-3K/Akt/NFkB pathway in cyclin D1 induction by arsenite in JB6 C141 cells. Carcinogenesis. 2006;21:864–873. doi: 10.1093/carcin/bgi321. [DOI] [PubMed] [Google Scholar]
- 39.Ruggero D, Sonenberg N. The Akt of translational control. Oncogene. 2005;24:7426–7434. doi: 10.1038/sj.onc.1209098. [DOI] [PubMed] [Google Scholar]
- 40.Salazar AM, Ostrowsky-Wegman P, Mendez D, Miranda E, Garcia-Carranca A, Rojas E. Induction of p53 protein expression by sodium arsenite. Mutat Res. 1997;381:259–265. doi: 10.1016/s0027-5107(97)00207-8. [DOI] [PubMed] [Google Scholar]
- 41.Sanchez Y, Amran D, Fernabdez C, de Blas E, Aller P. Genistein selectively potentiates arsenic trioxide-induced apoptosis in human leukemia cells via reactive oxygen species generation and activation of reactive oxygen species-inducible protein kinases (p38-MAPK, AMPK) Int J Cancer. 2008;123:1205–1214. doi: 10.1002/ijc.23639. [DOI] [PubMed] [Google Scholar]
- 42.Santra A, Chowdhury A, Ghatak S, Biswas A, Dhalil GK. Arsenic induces apoptosis in mouse liver is mitochondria dependent and is abrogated by N-acetylcysteine. Toxicol Appl Pharmacol. 2007;220:146–155. doi: 10.1016/j.taap.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 43.Shi ZZ, Osei-Frimpong J, Kala G, Kala SV, Barrios RJ, Habib GM, Lukin DJ, Danney CM, Matzyk MM, Lieberman MW. Glutathione synthesis is essential for mouse development but not for cell growth in culture. 2000;97:5101–5106. doi: 10.1073/pnas.97.10.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simenova PP, Wang S, Toriumi W, Kommineni C, Matheson J, Unimye N, Kayama F, Harki D, Ding M, Vallyathan V, Luster MI. Arsenic mediates cell proliferartion and gene expression in the bladder epithelium: association with AP-1 transactivation. Cancer Res. 2000;60:3445–3453. [PubMed] [Google Scholar]
- 45.Tabellini G, Tazzari PL, Bortul R, Evangelisti C, Billi AM, Grafone T, Martinelli G, Baccarini M, Martelli AM. Phosphoinositide 3-kinase/Akt inhibition increases arsenic trioxide-induced apoptosis of acute promyelocytic and T-cell leukaemias. Brit J Haematol. 2005;130:716–725. doi: 10.1111/j.1365-2141.2005.05679.x. [DOI] [PubMed] [Google Scholar]
- 46.Thomas DJ. Molecular processes in cellular arsenic metabolism. Toxicol Appl Pharmacol. 2007;222:365–373. doi: 10.1016/j.taap.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 47.Thompson JA, White CC, Chan JY, Kavanagh TJ, Fausto N, Franklin CC. Distinct Nrf1/2-independent mechanisms mediate As 3+-induced glutamate-cysteine ligase subunit gene expression in murine hepatocytes. Free Radic Biol Med. 2009;46:1614–1625. doi: 10.1016/j.freeradbiomed.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trouba KJ, Wauson EM, Vorce RL. Sodium arsenite induced dysregulation of proteins involved in proliferation signaling. Toxicol Appl Pharmacol. 2000;164:161–170. doi: 10.1006/taap.1999.8873. [DOI] [PubMed] [Google Scholar]
- 49.Tsou TCT, Tsai FY, Hsieh YW, Li LA, Yeh SC, Chang LW. Arsenite induces endothelial cytotoxicity by down-regulation of vascular endothelial nitric oxide synthase. Toxicol Appl Pharmacol. 2005;208:277–284. doi: 10.1016/j.taap.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 50.Tun-Kyi A, Qin J-Z, Oberholzer PA, Navarini AA, Hassel1 JC, Dummer R, Dobbeling U. Arsenic trioxide down-regulates antiapoptotic genes and induces cell death in mycosis fungoides tumors in a mouse model. Ann Oncol. 2008;19:1488–1494. doi: 10.1093/annonc/mdn056. [DOI] [PubMed] [Google Scholar]
- 51.Verstovsek S, Giles F, Quintás-Cardama A, Prez N, Ravandi-Kashani F, Beran M, Freireich E, Kantajian H. Arsenic derivatives in hematologic malignancies: a role beyond acute promyeolcytic leukemia. Hematol Oncol. 2006;24:181–188. doi: 10.1002/hon.787. [DOI] [PubMed] [Google Scholar]
- 52.Woo SH, Park MJ, An S, Lee HC, Jin HO, Lee SJ, Gwak HS, Park IC, Hong SI, Rhee CH. Diarsenic and tetraarsenic oxide inhibit cell cycle progression and bFGF- and VEGF-induced proliferation of human endothelial cells. J Cell Biochem. 2005;95:120–130. doi: 10.1002/jcb.20329. [DOI] [PubMed] [Google Scholar]
- 53.Yedjou CG, Tchounwou PB. Modulation of p53, C-fos, RARE, cyclin A, and cyclin D1 expression in human leukemia (HL-60) cells exposed to arsenic trioxide. Mol Cell Biochem. doi: 10.1007/s11010-009-0160-z. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]