

Abstract
Angiogenesis is crucial for embryogenesis, reproduction, and wound healing and is a critical determinant of tumor growth and metastasis. The multi-functional signal transducer Ras is a proto-oncogene and frequently becomes mutated in a variety of human cancers, including angiosarcomas. Regulation of Ras is important for endothelial cell function and angiogenesis. Hyperactivation of Ras is linked with oncogene-induced senescence in many cell types. Given links between vascular malformations and angiosarcoma with activated Ras signaling we sought to determine the consequence of sustained Ras activation on endothelial cell function. We find sustained Ras activation in primary endothelial cells leads to prolonged activation of pro-growth signaling, accompanied by a senescence bypass, enhanced proliferation, autonomous growth, and increased survival. Moreover, Ras severely compromises the ability of these cells to organize into vascular structures, instead promoting formation of planar endothelial sheets. This abnormal phenotype is regulated by PI-3′-kinase signaling highlighting the therapeutic potential of agents targeting this axis in dealing with vascular morphogenic disorders and vascular normalization of tumors.
Keywords: Angiogenesis, Ras, endothelial, signaling, morphogenesis
Introduction
Angiogenesis occurs during tissue remodeling, wounding and inflammation, as well as in tumors and their activated stroma. The quiescent vascular endothelium undergoes phenotypic changes including, increased proliferation, migration, and ultimately tubulogenesis. These changes are highly coordinated, requiring input from a number of signaling molecules (1). The loss of proper regulation can have devastating consequences.
During development, abnormalities in angiogenesis are lethal to the embryo. In the post-natal state, abnormal control of vascular growth and remodeling results in vascular malformations, hemangiomas, and angiosarcoma. Abnormalities in vascular morphogenesis and repair may be more discreetly involved in vascular diseases such as stroke, aneurysms, and atherosclerosis (2). Emerging data suggests that the abnormal vasculature found in tumors can directly impact metastasis (3). Thus, an understanding of the signaling that controls proper vascular morphogenesis, has the potential to impact a wide range of conditions, including tumor progression.
The small GTPase, Ras is a potentially critical regulator of proper endothelial cell growth and differentiation. In humans, loss of function mutations in RASA1, which normally downregulates Ras, has been associated with vascular malformations (4). Patients affected with the genetic disease NF1, resulting in the loss of neurofibromin, another negative regulator of Ras, suffer from a range of cardiovascular abnormalities, in addition to their enhanced predisposition to a range of tumors (5). Existing studies in animals bolster the importance of Ras regulation in the proper establishment and maintenance of a vasculature. Mice engineered without NF1 or RASA1 suffer from embryonic lethality (6, 7) primarily from their loss of function in the vasculature, as the lethal phenotype is recapitulated by endothelial specific knockout of these proteins (8). Mice haploinsufficient for NF1 show enhanced angiogenesis in a corneal and retinal neovascularization assay (9). The importance of normal Ras regulation in the growth control of endothelial cells is underscored by the finding that devastating tumors of endothelial cell origin, angiosarcomas, both sporadic and following exposure to vinyl chlorides, are associated with Ras mutations (10, 11). Collectively these data suggest that unregulated Ras can compromise normal vascular growth control, morphogenesis, and function.
Despite evidence at the organism level for Ras as a critical regulator of endothelial function, limited data is available on the consequences of Ras activation at the cellular level. In other cell types, unopposed signaling from Ras is often accompanied by a cell cycle arrest or senescent phenotype, which safeguards against uncontrolled growth. This “oncogene-induced senescence”, has been demonstrated both in vivo and in vitro in an array of cell types including melanocytes (12), fibroblasts (13), lymphocytes (14), and mammary epithelium (15). To understand the effects of sustained Ras signaling in primary endothelial cells we used sustained and inducible expression of activated H-Ras. Surprisingly our data suggest that human endothelial cells bypass Ras-induced senescence, with resulting autonomous growth, enhanced survival, as well as a markedly compromised ability to organize properly into vascular structures.
Materials and Methods
Cell culture
HUVECs (Human Umbilical Vein Endothelial Cells) were from Cascade Biologics (Portland, Oregon, USA) or VEC Technologies (Troy, NY, USA). Cells were cultured as previously described (16). Cells were made quiescent in serum free MCDB-131 supplemented with 1 % penicillin/streptomycin and 2 mM L-glutamine (SF), where noted. Stimulation was performed with complete growth media (GM). Primary fibroblasts were isolated from human foreskins and grown in DMEM containing 10% FBS and 1% penicillin/streptomycin. Normal human epidermal melanocytes were generously provided by Dr. Andrew Aplin (Thomas Jefferson University) and cultured in MGM-4 media purchased from Lonza. The endothelial and fibroblast co-culture assay was conducted in EGM-2 media from Lonza. HDMVECs (Human Dermal Microvascular Endothelial Cells) were isolated from human foreskins following trypsinization, disassociation, and CD31 affinity purification with magnetic beads (DYNAL). Isolated cells were cultured in EGM-2 MV from Lonza. Except as noted, all experiments were done with HUVECs.
Western blotting
Western blotting used the following antibodies: mouse anti-pERK, rabbit anti-ERK2, mouse anti-p16, mouse anti-p53 (Santa Cruz Biotechnology), rabbit anti-pAkt(S473), rabbit anti-Akt, mouse anti-cyclin D1, mouse anti-p21, rabbit anti-Cleaved caspase 3, rabbit anti-Total caspase 3 (Cell Signaling Technology), mouse anti-HA (Covance), mouse anti-Pan Ras (Oncogene Research, Calbiochem), mouse anti-flag (Sigma). All antibodies were used at a dilution of 1:1000 overnight at 4°C. Other conditions were the same as in (16) except exposures were captured on a Kodak 4000 MM imager. All figures are representative of at least three independent experiments.
Plasmid construction, virus production, and generation of stable cell lines
Cells were infected with either tetracycline inducible lentivirus or retrovirus as indicated. Details are provided in Supplementary material.
Measurement of DNA synthesis and growth assays
Endothelial cells were serum starved for 24 h, after which complete growth media (GM) was added as a mitogenic stimulus for 16 h. Measurements of BrdU incorporation were performed as previously described (17). Growth assays were conducted as described previously (13). Population doublings were calculated using the formula: Population Doublings = Log(Final cell number/Initial cell number)/Log2. Cumulative population doublings represent the sum of population doublings from all previous passages.
Co-culture assay
This assay was performed as previously described (18) with the following modification. Endothelial cells were mixed with fibroblasts (1:20) and seeded onto a gelatin coated 12-well plate in EGM-2 media for 12–28 days (media replaced after every 2–3 days). Cells were stained live with a FITC-tagged UEA-1 lectin (Sigma-Aldrich) or fixed in 3.7% formaldehyde and visualized with either UEA-1 lectin or anti-CD31 (Clone JC70A, DAKO, 1:50).
Results
Stable expression of activated Ras in primary endothelial cells
To generate endothelial cells expressing activated Ras we cloned an HA-tagged H-RasV12 mutant into two different viral vectors. One construct, a retroviral vector, permitted expression levels of activated Ras at levels close to the endogenous Ras (Supplementary Fig. S1B) and Ras activation was readily detected in a pull-down assay under quiescent conditions (Supplementary Fig. S1C). Recent results suggest that oncogene-induced senescence may be a function of oncogene expression levels (15). In order to regulate expression, (and expand cells in the event Ras induced senescence) we also engineered an inducible lentivirus where expression of Ras was regulated by the stronger Tet-Op7-CMV promoter (Fig. 1A). This permits over-expression at levels at least 10-fold higher than endogenous (Fig. 1D). Importantly, this provided excellent regulation of the transgene, with no detectable expression of the HA-epitope in the absence of induction (Fig. 1C). Induction with doxycycline ranging from 0.05–1 μg/mL allowed robust Ras expression in a dose-dependent fashion (Fig. 1C). We used a dose of 50 ng/mL of doxycycline in subsequent experiments.
Figure 1. Inducible expression of activated Ras in primary endothelial cells.

(A) pSLIK-RasV12-Venus lentiviral vectors were used to obtain HUVECs stably expressing activated Ras (B) infection efficiencies are shown by the histograms for Venus expression. (C) Cells were induced with doxycycline and Ras expression was measured. (D) Serum starved HUVECs were induced with 50 ng/ml doxycycline and whole cell lysates were probed as indicated. Data are representative of three independent infections on independent lots of endothelial cells.
Signaling pathways activated in primary endothelial cells in response to Ras activation
We next determined the signaling downstream of Ras in these cells. Both ERK and Akt are known to regulate cell responses critical to angiogenesis including growth and survival (16, 19, 20) and are often activated in a Ras-dependent manner (16). However, chronic Ras signaling has been described to down regulate these signals in some cell types (21). We found induction of activated Ras was sufficient to promote ERK and Akt activation that was maintained over at least a 3 day period (Fig. 1D). We observed enhanced Ras-induced signaling events even after periods of prolonged cell culture, including cells infected with low-copy number retroviruses (data not shown; Supplementary Fig. S1B). These data suggest that negative feedback of Ras signaling, implicated in inducing cellular senescence in some cell types (21), does not occur in endothelial cells. Reports also suggest that Ras activation in some primary cells activates p38 MAPK, resulting in senescence (22–24). However, no sustained Ras-induced p38 activation in primary endothelial cells was observed (data not shown). Thus, several signals responsible for senescence in other cell types do not appear to occur in endothelial cells.
Senescence is bypassed in primary endothelial cells upon Ras activation
As expression of active Ras showed no down-regulation of growth signaling, we reasoned that a senescence checkpoint might not be activated in primary endothelial cells following Ras activation. To directly test this, we infected primary melanocytes (a cell type well-described to undergo Ras induced senescence) (12) with an inducible Ras expressing lentivirus, and compared them to Ras-expressing primary endothelial cells. We monitored population doubling and found that induction of active Ras expression resulted in slowed growth of the melanocytes within 7 days of culture and near complete senescence by day 21 (Fig. 2A), as reported (12). This was accompanied by increases in the cyclin-dependent kinase inhibitor p16 (Fig. 2B). In contrast, melanocytes left uninduced continued to grow with similar population doubling to endothelial cells and minimal induction of p16 (Fig. 2A and B). Induction of activated Ras in human endothelial cells resulted in no appreciable change in the population doubling and no accumulation of p16. In addition, no significant increase in p21CIP/WAF or p27kip were detected (data not shown), proteins often associated with Ras/Raf-mediated cell cycle arrest. In addition, we have also investigated chronic exposure to lower Ras levels, using retrovirus infections (see Supplementary Fig. S2). Lastly, similar results were observed in human dermal microvascular endothelial cells (Supplementary Fig. S6). These data argue that sustained Ras signaling does not induce proliferative senescence in human endothelial cells. Importantly, Ras expression was accompanied by increased β-galactosidase staining and elevated levels of PAI-1 (Supplementary Fig. S3 and data not shown), two commonly used cell senescence markers (13, 25). These results suggest these markers, while perhaps markers of cellular stress or metabolic status, are not reliable markers of replicative senescence.
Figure 2. Activated Ras induces growth arrest in primary melanocytes but not in endothelial cells.

(A) Control (uninduced) and Ras expressing melanocytes and endothelial cells were grown in complete growth media and population doublings monitored. This is representative data from one experiment. Similar results were obtained in at least three additional experiments. (B) In parallel, lysates were probed for p16, Pan-Ras, and ERK2 after 7 days. (C) Control and Ras expressing endothelial cells were serum starved for 24 h after which BrdU incorporation was measured. The data is from one representative experiment and the error bars represent standard error (**P< 0.01) of triplicate determinations. Similar results were obtained in at least three additional experiments. (D) Cell lysates were made simultaneously to the experiments in (C) and probed for Cyclin D1, Pan-Ras, and Akt.
Given the linkage between Ras mutations and endothelial cell tumors, we sought to determine if Ras was sufficient to induce the proliferation of primary endothelial cells. Endothelial cell proliferation was measured at the G1→S transition using a BrdU incorporation assay. We found in the absence of added mitogens, Ras activation was sufficient to drive cells into S phase (Fig. 2C, Supplementary Fig. S2A). This was accompanied by increased cyclin D1 levels (Fig. 2D). These data suggest that activation of Ras might make endothelial cells growth-factor independent, a trait often associated with cell transformation.
To determine if Ras expressing cells were actually progressing through the cell cycle, we performed growth assays where cells were plated in growth-factor free media and counted at various times after plating. As shown in Fig. 3A and B, cells induced to express active Ras increase in number. In contrast, un-induced cells quickly begin to decline. Importantly, the expression of Ras does not render these cells completely growth-factor independent, as beyond 3 days even the Ras expressing cells begin to succumb (data not shown). Furthermore, Ras-expressing endothelial cells maintained normal contact inhibition, did not grow in soft agar, nor did they form tumors in nude mice (data not shown), suggesting other growth regulatory check points are still intact.
Figure 3. Activated Ras confers a survival advantage to primary endothelial cells.

(A) Control (uninduced) and Ras expressing endothelial cells were cultured in minimal media and photographed at 0 and 72 h. (B) Cells at indicated times of serum-free culture were stained with crystal violet and extracted dye was quantified by measuring absorbance at 590 nm. Error bars represent standard error (**P< 0.01) of triplicate determinations from a single experiment. Similar results were obtained in three additional experiments. (C) Apoptosis was induced in endothelial cells by treating with serum-free M199 after which lysates were probed for cleaved caspase-3, HA-tag, and total caspase-3. (D) Normalized cleaved caspase-3 values obtained from at least three different experiments. The error bars represent standard error (**P< 0.01).
Activated Ras enhances cell survival
Activated Ras appeared to enhance survival of endothelial cells, allowing them to survive and grow autonomously in basal media while control cells failed to thrive (Fig. 3A and B, Supplementary Fig. S2B). To determine if Ras activation promoted cell survival, we utilized culture conditions known to be pro-apoptotic and directly measured the cleavage of caspase-3. The uninduced cells accumulated cleaved caspase-3 when cultured in M199 medium lacking serum. In contrast, Ras expressing cells showed little induction of cleaved caspase-3 over the same time period (Fig. 3C and D).
The role of Ras effectors in endothelial cell growth and survival
Previous studies have implicated the Ras-regulated signaling pathways, PI-3′-kinase and ERK, as important contributors to the growth and survival of human endothelial cells (16, 17, 26, 27). To better assess the roles these signals play in our observed response to Ras, we utilized two well characterized chemical inhibitors, U0126 (an inhibitor of MEK) and LY294002 (an inhibitor of PI-3′-Kinase). As shown in Supplementary Fig. S4A, these inhibitors show inhibition of Ras-induced signaling events, specific for the targeted signal. We found that proliferation induced by Ras was strongly reduced following ERK inhibition, in contrast to PI-3′-kinase inhibition (Supplementary Fig. S4B), where the effect was less pronounced. The suppression of caspase activation by Ras was attenuated by both inhibitors to similar levels, suggesting that both ERK and PI-3′-kinase control signals promoting cell survival (Supplementary Fig. S4C).
Recently Serban et al. (28) utilized adenoviral delivery of Ras effector mutants with selective signaling properties to induce angiogenesis in vivo. To genetically probe the contribution of effector pathways, we engineered endothelial cells to stably express RasV12S35 (Raf activation) and RasV12C40 (PI-3′-kinase activation). Interestingly, we found that neither of these mutants was sufficient to drive endothelial cell proliferation. However, we noted that the activation of downstream effectors in these cells was quite weak compared to RasV12 (data not shown). To insure optimal signal strength and to directly test the contributions of the Raf and PI-3′-kinase pathway to the Ras induced changes in endothelial cells, we also engineered inducible lentiviruses to express either activated Raf (ΔN-Raf1) or an activated form of Akt (myrAkt). As shown in Fig. 4, these showed excellent expression when induced with doxycycline, with a corresponding change in the associated cellular signaling comparable to changes seen upon stimulation with growth media, except for the chronic nature of the signal. As shown in Fig. 4B, neither activation of Raf nor Akt, is sufficient to induce G1 to S transition, consistent with results obtained with the effector mutants. Importantly, there was no cell-cycle arrest response, as the BrdU incorporation induced by growth medium was normal. These data suggest that ERK and PI-3′-Kinase signaling likely work in combination to drive cellular proliferation. Moreover these data suggest that an imbalance in these signals does not result in a proliferative arrest in endothelial cells, as reported in other cell types (29, 30).
Figure 4. Effect of activation of Raf and Akt on endothelial cell proliferation and survival.
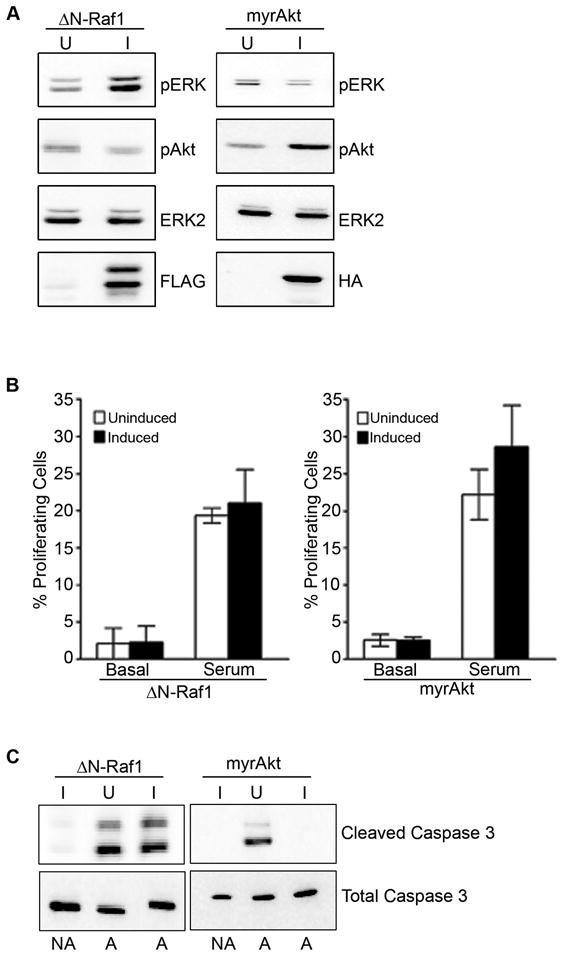
(A) Cells were serum deprived in the absence (U) or presence (I) of doxycycline to induce protein expression. Cell lysates were probed as indicated. (B) Cells treated as above were stimulated with complete growth medium (GM) as indicated and pulsed with BrdU. Data represents the % BrdU positive cells compared to total cell number. The error bars represent standard error of triplicate determinations in one experiment. Similar results were obtained in two additional experiments. (C) Apoptosis was induced in endothelial cells by treating with serum free M199, and probing lysates for cleaved and total caspase 3.
We also investigated whether the isolated signals were sufficient to alter the survival responses of the endothelial cells under apoptotic conditions. We found expression of activated Raf-1 failed to prevent cleavage of caspase-3 under conditions of serum-deprivation. In contrast, caspase-3 cleavage was completely prevented by the induction of an activated Akt, consistent with the established role of this protein in endothelial survival (31).
Sustained Ras activation alters vascular morphogenesis
The formation of blood vessels requires integrated growth, spatial orientation and vascular pruning (32). As Ras-expressing cells showed abnormal growth and survival characteristics, we hypothesized their ability to properly regulate the formation of vascular-like structures might be altered. To test this, we utilized a co-culture assay (18). This long-term assay, results in vascular structures with morphological features nearly indistinguishable from in vivo blood vessels including the formation of lumens (See Supplementary Fig. S7), tight junctions, and expression of basement membranes proteins such as collagen IV (33). While uninduced cells formed interconnected, branching and looping structures, Ras-expressing endothelial cells failed to form vascular structures. Rather these cells formed extensive planar sheets with no evidence of branching, spatial reorganization, or pruning (Fig. 5A and Supplementary Fig. S2C). This process was reversible and dependent upon Ras signaling, as turning off Ras expression in the same co-cultures, causes the sheets to regress and tubes to form. Similarly, addition of doxycycline to induce Ras expression results in formation of sheets by cells that previously had formed vascular structures (Fig. 5B). Thus, sustained Ras signaling appears to inhibit normal vascular differentiation and induction of Ras signaling in fully differentiated structures is sufficient to induce a loss of normal morphogenesis and drive the establishment of planar sheet-like structures. Recent experiments demonstrated that adenoviral H-RasV12 induced vascular permeability in vivo(28), suggesting there may be effects of Ras activation on endothelial cell junctions. The alteration of junctional proteins has been linked to abnormal vasculogenesis. To test whether stable expression of activated Ras results in changes in endothelial cell junctional integrity, we measured changes in electrical resistance by electric cell-substrate impedance sensor (ECIS, Applied BioPhysics, Inc.)(34). Our data suggest that there is no defect or enhancement in junctional integrity. Similarly, we found that VE-cadherin was expressed at cell-cell junctions, though an increased level of disorganization in Ras expressing endothelial cells was evident (Supplementary Fig. S5).
Figure 5. Activated Ras prevents normal vascular morphogenesis.

Uninduced and Ras expressing endothelial cells co-cultured with fibroblasts for 14 days and visualized by staining live endothelial cells with a FITC-tagged UEA-1 lectin. Ras expression was turned on in the uninduced cells by adding doxycycline and shut down in the induced cells by withdrawing doxycycline and the cells were again stained after an additional 14 days. The pictures (at 200X) are representative of data obtained from at least three independent experiments.
To gain insight into mechanisms underlying altered morphogenesis in the co-culture assay we used inhibitors of ERK (U0126) and PI-3′-kinase (LY294002) signaling. As this phenotype was reversible, we added inhibitors to the mature structures that formed following Ras induction, to investigate whether they would revert to a tubular type phenotype. Both inhibitors visibly reduced the overall number of endothelial cells. However, we found that the addition of the PI-3′-kinase inhibitor induced a phenotype shift whereby vascular structures appeared (Fig. 6A). In contrast, while the ERK inhibitor reduced the number of endothelial cells, those cells remained as highly planar structures (Fig. 6A). We next tested this with endothelial cells stably expressing Ras effector mutants; H-RasV12S35 known to selectively activate ERK and H-RasV12C40 known to selectively activate PI-3′-kinase signaling (28). Consistent with the inhibitor studies, expression of H-RasV12S35 mutant formed relatively normal, branching structures whereas cells expressing H-RasV12C40 formed planar sheets (Fig. 6B). These data suggest that sustained PI-3′-kinase signaling may be a principle contributor to the failed morphogenesis following sustained Ras signaling. We evaluated this further using inducible expression of either activated Raf or activated Akt. As shown in Fig. 6C, we find that expression of activated Raf results in normal morphogenic responses. In contrast, induction of activated Akt results in severely compromised morphogenesis. The expression of myr-Akt results in an increase in areas of planar sheet like structures with a small number of interspersed elongated structures. While a few areas appear to undergo some degree of normal morphogenesis, the normal elongation and branching seems to be severely compromised, with an appearance of many multi-polar cells (Fig. 6D). Collectively this data argues that proper regulation of PI-3′-kinase and Akt signaling is an essential component of vascular morphogenesis.
Figure 6. PI-3′-Kinase and Akt activation are principle contributors to the failed morphogenesis following Ras activation.

(A) Uninduced and Ras expressing endothelial cells were co-cultured for 14 days after which the cells were treated with 5 μM U0126 or 5 μM LY294002 and cultured for an additional 7 more days. Cells were fixed and stained with CD31. The pictures (at 200X) are representative of data obtained from at least three independent experiments. (B) Endothelial cells expressing Ras effector mutants or GFP alone (control) were co-cultured with fibroblasts for 14 days prior to staining with CD31. The pictures (at 200X) are representative of data obtained from at least two independent experiments. (C) HUVECS infected with inducible lentiviruses coding for either active Raf1 or active Akt were plated with fibroblasts in the absence (uninduced) or presence (induced) of doxycycline. Visualization of ΔN-Raf1 vascular structures was done at 100X by staining with FITC- UEA-1 lectin. The myr-Akt infected cultures were visualized at 200X with FITC-UEA-1. (D) Lower power (50X) visualization of myrAkt expressing co-cultures compared to uninduced.
Discussion
Oncogene-induced senescence was first demonstrated in primary human fibroblasts (13) and subsequently multiple reports describing the occurrence of this phenomenon both in vitro and in vivo (12, 14, 21, 35, 36) have appeared. Contrasting evidence showing oncogene induced transformation and increased proliferation of primary cells also exists (37, 38). These variations can be due to differences in the inherent abilities of particular cell types to respond to oncogene activation, however few studies have compared two cell types simultaneously. An alternative is that the extent and duration of oncogene activation dictates a cell’s decision to proliferate or prematurely growth arrest. This has been observed in vitro in NIH3T3 fibroblasts (39) and in vivo in the mammary epithelium where low levels of activated oncogene cause proliferation and high levels induce growth arrest (15). In this study, we show that stable and sustained Ras activation, independent of expression levels, leads to senescence bypass in primary endothelial cells, while simultaneously inducing growth arrest in primary melanocytes. These data strongly argue that the induction of senescence by Ras, is likely to be dependent upon specific factors relevant to each individual cell type.
Senescence response in melanocytes has been primarily attributed to the accumulation of the tumor suppressor p16INK4a levels (12). We found no induction of p16 in endothelial cells whereas the senescent melanocytes readily accumulated p16. In addition to p16, Ras induced senescence in primary fibroblasts is also accompanied by the accumulation of tumor suppressor p53 (13). However, we find that p53 is not induced upon Ras activation (data not shown) nor is its target gene p21CIP/WAF, suggesting p53 regulation may be differentially regulated among cell types. We also have not observed several other causative mechanisms described to induce premature senescence, e.g., the attenuation of Ras signaling events (21), or activation of p38 (24). As vascular heterogeneity is well established, we considered that cells from different vascular beds might show heterogeneous responses. However, we found that all of the key findings were also observed in primary cultures of HDMVECs, a cell type relevant to formation of hemangioma and angiosarcoma (Supplementary Fig. S6). These results highlight a unique and conserved response of endothelial cells to Ras activation. Importantly, while the inherent response of the endothelial cell in isolation may be the same, the varying microenvironment of the tissue may influence the predisposition of the endothelium to manifest an altered phenotype.
Cellular senescence acts as an initial barrier preventing a benign tumor from progressing to a malignant state (40). Interestingly, senescent cells are only detected in benign but not metastatic tumors (12, 41) supporting the notion that senescence functions as a mechanism of tumor suppression. Our findings suggest this may not be an effective checkpoint in endothelial cells and could explain the prevalence of Ras mutations in endothelial tumors (42, 43). Endothelial tumors are characterized by rapidly proliferating endothelial cells (44) that have an atypical morphology and may appear as cellular sheets (45). Our findings demonstrate that sustained Ras expression in primary endothelial cells results in enhanced proliferation, autonomous growth, and enhanced endothelial cell survival. Moreover, the ability to organize into properly formed vascular structures is compromised, with cells forming sheet-like structure like those seen in some endothelial tumors (45). Our data do not support the notion that acquisition of Ras mutations would be sufficient to induce cellular transformation, as several additional growth checkpoints were intact.
Abnormal vessels are prevalent in disorders linked to increased Ras activity. Mutations in RASA1 and NF1 genes, which down-regulate Ras, have been linked to familial hemangiomas, aneurysms, cerebral vascular malformations and an increased risk of stroke (4, 5, 46). Our data suggest that pro-proliferative signaling, defective apoptosis, as well abnormal morphogenic programming could contribute to disease conditions. Moreover, Ras mutations, even without gross manifestations, might contribute to an abnormal and unstable capillary vasculature. Recent data has demonstrated that short term adenoviral infection with activated Ras constructs results in enhanced angiogenesis in a mouse ear model. The adenoviral infection in these experiments targets both the endothelial and stromal compartments and the time of expression is limited (28); this is an important distinction since we find the morphogenic phenotype reversible. Thus microenvironmental cues, expression levels, and duration of signaling may all contribute to a failure to form normal vascular structures.
We find that Ras is sufficient to drive proliferation in the absence of mitogens and this requires ERK signaling. However, our data clearly show that activation of Raf/ERK signaling through either Ras effector mutants or activated Raf is not sufficient to drive proliferation or support survival signaling. However, these sustained signals do not interfere with normal vessel morphogenesis. In contrast, chronic activation of PI-3′-kinase signaling appears to alter morphogenesis. This finding is consistent with results from the transgenic expression of activated Akt in mice (26). These mice have altered vascular density, morphogenesis, and permeability. Inhibition of PI-3′-kinase activity is associated with a restoration of a normalized vasculature in these mice. Pro-survival signaling alone may not be the cause of the morphogenic defects following chronic Ras signaling; rather it appears that a dynamically regulated change in cellular programming is occurring. This is based on several observations: 1) treatment with U0126 inhibits Ras induced survival and decreases the cell number in the morphogenesis assay with no effect on the defect in morphogenesis induced by Ras; 2) activation of Akt completely protects cells from apoptotic stimuli, however partial morphogenic responses are seen in these cells, albeit severely compromised; and 3) the H-RasV12C40 mutant induces a defect similar to H-RasV12, though the levels of Akt activation are much lower. Thus, PI-3′-kinase related signals, in addition to Akt, may be involved.
Aberrantly formed blood vessels are known to occur in tumors and have been implicated in metastasis (3). It seems feasible that sustained and high concentrations of angiogenic growth factors could result in sustained activation of Ras signaling. The pro-mutagenic microenvironment of the tumor bed undergoing therapeutic interventions (rapid proliferation in the presence of mutagens and often radiation) could also result in de novo acquisition of endothelial Ras mutations. Indeed cytogenetic abnormalities in tumor endothelial have been detected (47) and suggested as a potential barrier to therapy. Thus, mitigating or minimizing Ras signaling in the tumor vasculature may be an effective and important adjuvant therapeutic approach to vascular normalization.
Supplementary Material
Acknowledgments
These studies were supported by the PHS (CA81419 to KP, HL077870 to PV), the AHA (0825888D to AB) and the David E. Bryant Trust. We would like to thank C. Michael DiPersio for critical reading of the manuscript. We would also like to thank Joseph E. Mazurkiewicz for his help with acquiring confocal images.
References
- 1.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–95. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438(7070):932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 3.Mazzone M, Dettori D, Leite de Oliveira R, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136(5):839–51. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73(6):1240–9. doi: 10.1086/379793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman JM, Arbiser J, Epstein JA, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet Med. 2002;4(3):105–11. doi: 10.1097/00125817-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Brannan CI, Perkins AS, Vogel KS, et al. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994;8(9):1019–29. doi: 10.1101/gad.8.9.1019. [DOI] [PubMed] [Google Scholar]
- 7.Henkemeyer M, Rossi DJ, Holmyard DP, et al. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995;377(6551):695–701. doi: 10.1038/377695a0. [DOI] [PubMed] [Google Scholar]
- 8.Gitler AD, Zhu Y, Ismat FA, et al. Nf1 has an essential role in endothelial cells. Nat Genet. 2003;33(1):75–9. doi: 10.1038/ng1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu M, Wallace MR, Muir D. Nf1 haploinsufficiency augments angiogenesis. Oncogene. 2006;25(16):2297–303. doi: 10.1038/sj.onc.1209264. [DOI] [PubMed] [Google Scholar]
- 10.Przygodzki RM, Finkelstein SD, Keohavong P, et al. Sporadic and Thorotrast-induced angiosarcomas of the liver manifest frequent and multiple point mutations in K-ras-2. Lab Invest. 1997;76(1):153–9. [PubMed] [Google Scholar]
- 11.Weihrauch M, Bader M, Lehnert G, et al. Mutation analysis of K-ras-2 in liver angiosarcoma and adjacent nonneoplastic liver tissue from patients occupationally exposed to vinyl chloride. Environ Mol Mutagen. 2002;40(1):36–40. doi: 10.1002/em.10084. [DOI] [PubMed] [Google Scholar]
- 12.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436(7051):720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 13.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 14.Braig M, Lee S, Loddenkemper C, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436(7051):660–5. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 15.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9(5):493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 16.Meadows KN, Bryant P, Pumiglia K. Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. J Biol Chem. 2001;276(52):49289–98. doi: 10.1074/jbc.M108069200. [DOI] [PubMed] [Google Scholar]
- 17.Meadows KN, Bryant P, Vincent PA, Pumiglia KM. Activated Ras induces a proangiogenic phenotype in primary endothelial cells. Oncogene. 2004;23(1):192–200. doi: 10.1038/sj.onc.1206921. [DOI] [PubMed] [Google Scholar]
- 18.Bishop ET, Bell GT, Bloor S, Broom IJ, Hendry NF, Wheatley DN. An in vitro model of angiogenesis: basic features. Angiogenesis. 1999;3(4):335–44. doi: 10.1023/a:1026546219962. [DOI] [PubMed] [Google Scholar]
- 19.Thakker GD, Hajjar DP, Muller WA, Rosengart TK. The role of phosphatidylinositol 3-kinase in vascular endothelial growth factor signaling. J Biol Chem. 1999;274(15):10002–7. doi: 10.1074/jbc.274.15.10002. [DOI] [PubMed] [Google Scholar]
- 20.Yu Y, Sato JD. MAP kinases, phosphatidylinositol 3-kinase, and p70 S6 kinase mediate the mitogenic response of human endothelial cells to vascular endothelial growth factor. J Cell Physiol. 1999;178(2):235–46. doi: 10.1002/(SICI)1097-4652(199902)178:2<235::AID-JCP13>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 21.Courtois-Cox S, Genther Williams SM, Reczek EE, et al. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10(6):459–72. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells. 2003;8(2):131–44. doi: 10.1046/j.1365-2443.2003.00620.x. [DOI] [PubMed] [Google Scholar]
- 23.Sun P, Yoshizuka N, New L, et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 2007;128(2):295–308. doi: 10.1016/j.cell.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 24.Wang W, Chen JX, Liao R, et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol. 2002;22(10):3389–403. doi: 10.1128/MCB.22.10.3389-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phung TL, Ziv K, Dabydeen D, et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell. 2006;10(2):159–70. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng H, Zhao D, Mukhopadhyay D. Flt-1-mediated down-regulation of endothelial cell proliferation through pertussis toxin-sensitive G proteins, beta gamma subunits, small GTPase CDC42, and partly by Rac-1. J Biol Chem. 2002;277(6):4003–9. doi: 10.1074/jbc.M110842200. [DOI] [PubMed] [Google Scholar]
- 28.Serban D, Leng J, Cheresh D. H-ras regulates angiogenesis and vascular permeability by activation of distinct downstream effectors. Circ Res. 2008;102(11):1350–8. doi: 10.1161/CIRCRESAHA.107.169664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998;12(19):2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pumiglia KM, Decker SJ. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A. 1997;94(2):448–52. doi: 10.1073/pnas.94.2.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90(12):1243–50. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 32.Benjamin LE. The controls of microvascular survival. Cancer Metastasis Rev. 2000;19(1–2):75–81. doi: 10.1023/a:1026552415576. [DOI] [PubMed] [Google Scholar]
- 33.Donovan D, Brown NJ, Bishop ET, Lewis CE. Comparison of three in vitro human ‘angiogenesis’ assays with capillaries formed in vivo. Angiogenesis. 2001;4(2):113–21. doi: 10.1023/a:1012218401036. [DOI] [PubMed] [Google Scholar]
- 34.Giaever I, Keese CR. Micromotion of mammalian cells measured electrically. Proc Natl Acad Sci U S A. 1991;88(17):7896–900. doi: 10.1073/pnas.88.17.7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collado M, Gil J, Efeyan A, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436(7051):642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 37.Lemoine NR, Staddon S, Bond J, Wyllie FS, Shaw JJ, Wynford-Thomas D. Partial transformation of human thyroid epithelial cells by mutant Ha-ras oncogene. Oncogene. 1990;5(12):1833–7. [PubMed] [Google Scholar]
- 38.Tuveson DA, Shaw AT, Willis NA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5(4):375–87. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 39.Sewing A, Wiseman B, Lloyd AC, Land H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol Cell Biol. 1997;17(9):5588–97. doi: 10.1128/mcb.17.9.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campisi J. Aging, tumor suppression and cancer: high wire-act! Mech Ageing Dev. 2005;126(1):51–8. doi: 10.1016/j.mad.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 41.Collado M, Serrano M. The senescent side of tumor suppression. Cell Cycle. 2005;4(12):1722–4. doi: 10.4161/cc.4.12.2260. [DOI] [PubMed] [Google Scholar]
- 42.Boivin-Angele S, Lefrancois L, Froment O, et al. Ras gene mutations in vinyl chloride-induced liver tumours are carcinogen-specific but vary with cell type and species. Int J Cancer. 2000;85(2):223–7. doi: 10.1002/(sici)1097-0215(20000115)85:2<223::aid-ijc12>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 43.Froment O, Boivin S, Barbin A, Bancel B, Trepo C, Marion MJ. Mutagenesis of ras proto-oncogenes in rat liver tumors induced by vinyl chloride. Cancer Res. 1994;54(20):5340–5. [PubMed] [Google Scholar]
- 44.Mulliken JB, Zetter BR, Folkman J. In vitro characteristics of endothelium from hemangiomas and vascular malformations. Surgery. 1982;92(2):348–53. [PubMed] [Google Scholar]
- 45.Koch M, Nielsen GP, Yoon SS. Malignant tumors of blood vessels: angiosarcomas, hemangioendotheliomas, and hemangioperictyomas. J Surg Oncol. 2008;97(4):321–9. doi: 10.1002/jso.20973. [DOI] [PubMed] [Google Scholar]
- 46.Lin AE, Birch PH, Korf BR, et al. Cardiovascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am J Med Genet. 2000;95(2):108–17. doi: 10.1002/1096-8628(20001113)95:2<108::aid-ajmg4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 47.Hida K, Hida Y, Amin DN, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004;64(22):8249–55. doi: 10.1158/0008-5472.CAN-04-1567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.