

Abstract
Rationale
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is caused by a single point mutation in a well-defined region of the cardiac type-2 ryanodine receptor (RyR2). However, the underlying mechanism by which a single mutation in such a large molecule produces drastic effects on channel function remains unresolved.
Objective
Using a knock-in (KI) mouse model with a human CPVT-associated RyR2 mutation (R2474S), we investigated the molecular mechanism by which CPVT is induced by a single point mutation within the RyR2.
Methods and Results
The R2474S/+ KI mice showed no apparent structural or histological abnormalities in the heart, but they showed clear indications of other abnormalities. Bidirectional or polymorphic VT was induced after exercise on a treadmill. The interaction between the N-terminal (aa 1–600) and central (aa 2000–2500) domains of the RyR2 (an intrinsic mechanism to close Ca2+ channels) was weakened (domain unzipping). Upon protein kinase A (PKA)-mediated phosphorylation of the RyR2, this domain unzipping further increased, resulting in a significant increase in the frequency of spontaneous Ca2+ transients. cAMP-induced aberrant Ca2+ release events (Ca2+ sparks/waves) occurred at much lower sarcoplasmic reticulum (SR) Ca2+ content as compared to the wild-type (WT). Addition of a domain-unzipping peptide, DPc10 (aa 2460–2495), to the WT reproduced the aforementioned abnormalities that are characteristic of the R2474S/+ KI mice. Addition of DPc10 to the (cAMP-treated) KI cardiomyocytes produced no further effect.
Conclusions
A single point mutation within the RyR2 sensitizes the channel to agonists and reduces the threshold of luminal [Ca2+] for activation, primarily mediated by defective inter-domain interaction within the RyR2.
Keywords: ryanodine receptor, calcium, ventricular tachycardia, sarcoplasmic reticulum
Introduction
To date, more than 70 cardiac ryanodine receptor (RyR2) missense mutations have been identified that are linked with 2 inherited forms of sudden cardiac death—catecholaminergic polymorphic ventricular tachycardia (CPVT)1 and arrhythmogenic right ventricular cardiomyopathy (ARVC) type 2.1 These mutations cluster in 3 well-defined regions of the RyR2 that correspond to malignant hyperthermia (MH) or the central core disease (CCD) mutable regions, designated as the N-terminal domain (aa 1–600), central domain (aa 2000–2500), and the C-terminal transmembrane channel domain of the skeletal muscle-type ryanodine receptor (RyR1).1 This suggests that the RyR2 shares a common domain-mediated channel regulation mechanism with RyR1. Mutations at different positions in each of these domains result in the nearly identical phenotype of channel dysfunctions such as hyperactivation of the Ca2+ channel and hypersensitization to agonists. To account for these phenomena, Ikemoto et al2, 3 proposed the so-called “domain switch hypothesis” and stated that in the resting or non-activated state, the N-terminal domain and the central domain make close contact at several sub-domains (domain zipping). Then, on physiological or pharmacological stimulation these critical inter-domain contacts are weakened, resulting in the loss of conformational constraints (domain unzipping), thus lowering the energy barrier for Ca2+ channel opening. Consistent with this hypothesis, single particle analysis of the three-dimensional structure of the RyR2 molecule revealed that the N-terminal and central domains (located in domains 5 and 6 of the so-called clamp region, respectively) are in a close apposition to each other. 4, 5
Recent reports deal with 3 types of knock-in (KI) mice with human CPVT/ARVC-associated RyR2 mutations: R4496C,6 R176Q,7 and R2474S.8 Injection of caffeine plus epinephrine or exercise induces ventricular tachycardia in these mice, indicating that these point mutations can cause lethal arrhythmias. However, the underlying mechanism by which a single mutation causes lethal arrhythmia remains unresolved. We recently reported that in failing hearts, defective inter-domain interaction within the RyR2 (aberrant unzipping of the N-terminal/central domain pair and channel activation in an otherwise resting state) causes diastolic Ca2+ leakage and contractile dysfunction.9 As shown in our previous report,9 pathological conditions (diastolic Ca2+ leakage and contractile dysfunction) are reproduced in the otherwise normal system by adding DPc10, a central domain peptide (Gly2460–Pro2495) of the RyR2 that interferes with the interaction between the N-terminal and central domains of the RyR2 and causes defective domain unzipping. George et al10 showed that functional coupling between the cytoplasmic and transmembrane domains of the RyR2 is mediated by the 3722–4610 residue region, called the I-domain and that sudden cardiac death (SCD)-linked mutations occurring in the I-domain (N4104K and R4496C) caused channel instability and Ca2+ release dysfunction owing to defective inter-domain interactions between the I-domain and the channel domain. These findings suggest that the weakened inter-domain interaction at least in these 2 regions of the RyR2 (N-terminal domain/central domain and I-domain/channel domain) is the key mechanism underlying the pathogenesis of CPVT/ARVC and heart failure.
Although the domain peptide approach provided important information regarding the underlying mechanism of the RyR2 abnormalities during heart failure and lethal arrhythmia, further in vivo studies with the mutation-linked disease model are required for a straightforward test of the inter-domain interaction hypothesis. In the present study, using the KI mouse model with a human CPVT-associated RyR2 mutation, R2474S, we investigated the molecular mechanism by which CPVT is induced by a single point mutation within the RyR2. The data presented here suggested that the introduced mutation, in fact, causes defective inter-domain interaction in the RyR2, reduces the threshold of luminal Ca2+-dependance for channel activation, sensitizes RyR2 to protein kinase A (PKA)-dependent phosphorylation, and in turn leads to CPVT.
Methods
For a detailed description, see the expanded Materials and Methods sections and Online Figures I and II in the online data supplement.
Animals
This study conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The care of the animals and the protocols used were in accordance with guidelines laid down by the Animal Ethics Committee of Yamaguchi University School of Medicine.
Statistics
Paired or unpaired t-tests were used for statistical comparisons of data obtained during the 2 different situations, while ANOVA with a post hoc Scheffe’s test was used for statistical comparison of concentration-dependent data. All data are expressed as mean ± standard error (SE). A p value less than 0.05 was considered statistically significant.
Results
There was no appreciable change in structural or functional characteristics of R2474S/+ KI mice during the resting (non-activated) state
In the absence of activation, there was no appreciable difference in the structural or functional features of the hearts between WT and KI mice. Thus, the cross-sectional view showed the identical features (Figure 1A). Echocardiography revealed no functional difference between WT and KI mice (Figure 1B). There was no appreciable change in the KI mice regarding the expression or phosphorylation levels of any of the sarcoplasmic reticulum (SR) proteins examined (Online Figure III).
Figure 1.
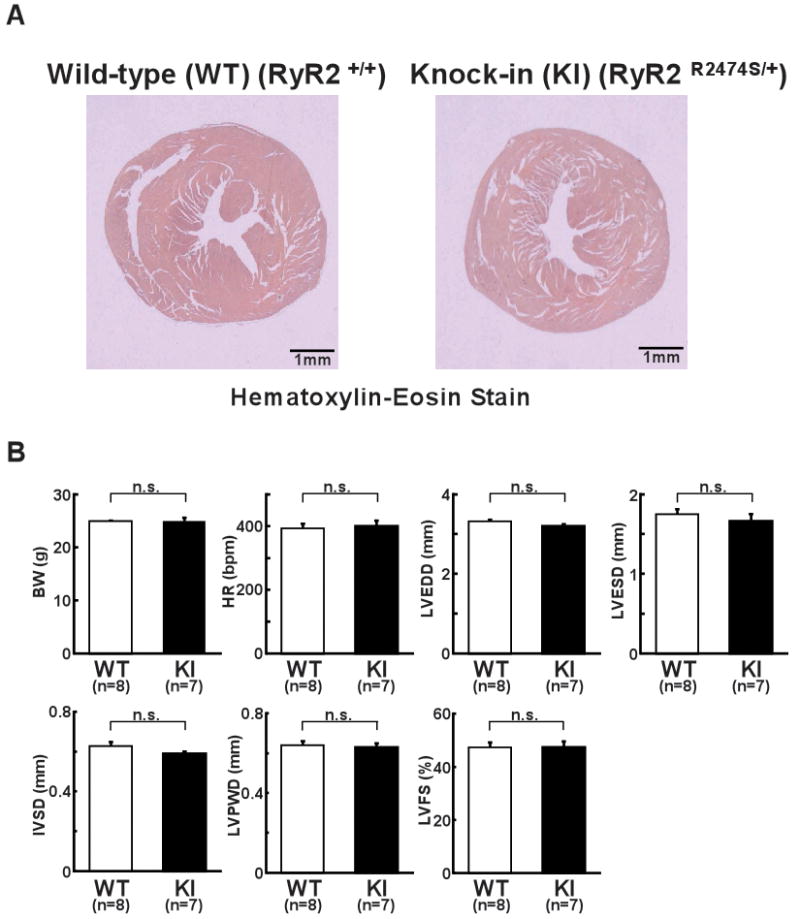
Structural and functional characterization of hearts from WT and R2474S/+ KI mice. A, Representative images of Hematoxylin-Eosin-stained hearts from WT and KI mice. B, Summarized data of body weight (BW), heart rate (HR), left ventricular end-diastolic diameter (LVEDD), left ventricular end-systolic diameter (LVESD), intra-ventricular septum diastolic thickness (IVSD), left ventricular posterior wall diastolic thickness (LVPWD), left ventricular fractional shortening ((LVEDD−LVESD)/LVEDD×100). N: the number of mice examined.
Exercise or drug (epinephrine and caffeine) administration induced VT in R2474S/+ KI mice
In the resting conscious condition, we frequently observed polymorphic ventricular premature contractions in KI mice in response to even very weak stimuli, like light or sound, but not in WT mice (data not shown). Injection of caffeine plus epinephrine (i.p.) or exercise on a treadmill induced bidirectional or polymorphic VT in KI mice, but not in WT mice (Figure 2A). The duration of VT in most KI mice was less than 30 sec (Figure 2B).
Figure 2.

Effect of epinephrine plus caffeine or exercise on ventricular arrhythmia. A, Representative ECG recordings in WT and R2474S/+ KI mice. In all KI mice examined, bidirectional or polymorphic ventricular tachycardia (VT) was induced by epinephrine (2 mg/kg of body weight (BW) i.p.) and caffeine (120 mg/kg of body weight i.p.) (n=10) (top) or exercise with treadmill (n=6) (bottom). In WT mice, neither epinephrine plus caffeine (n=8) nor exercise (n=6) produced VT. B, Durations of VT and numbers of VT episodes observed during the 5 minutes period in WT and KI mice. VT was induced by only epinephrine (2 mg/kg of body weight i.p.) without caffeine. N: the number of mice examined.
Seizures were not observed in R2474S/+ KI mice
To determine whether seizures occurred in KI mice, as previously reported,8 we monitored the behavior of mice by video recording for 1 week and assessed their susceptibility to seizures by pharmacologic induction (see expanded Materials and Methods in the online data supplement). Thus far, we have not observed spontaneous seizures in KI or WT mice. Furthermore, a pharmacological provocation test with 4-aminopyridine and caffeine showed no difference in the latency to the development of generalized tonic-clonic seizures (Online Figure IV).
Relaxation phase of cell shortening and Ca2+ transient was prolonged in the isoproterenol-activated R2474S/+ KI mice
There was no statistically significant difference in the contour or kinetic parameters of Ca2+ transient and cell shortening at baseline between the WT and KI mice (Figure 3A, Online Table I). In response to isoproterenol (ISO), however, the time from the peak to 80% decline in cell shortening or Ca2+ transient was prolonged in KI cardiomyocytes, suggesting a delay in the inactivation of Ca2+ release and/or spontaneous Ca2+ release events. Moreover, the SR Ca2+ content, determined by caffeine application, was significantly lower in KI cardiomyocytes than in WT cardiomyocytes, both before and after the addition of isoproterenol (Figure 3B). SERCA2 activity could contribute to the slowed decay kinetics of the Ca2+ transient during heart failure. Thus, we next measured the SERCA2-mediated Ca2+ uptake by monitoring the time-dependent change in the intra-SR [Ca2+] (Online Figure V). As shown, there was no appreciable change in the time course of SR Ca2+ uptake.
Figure 3.
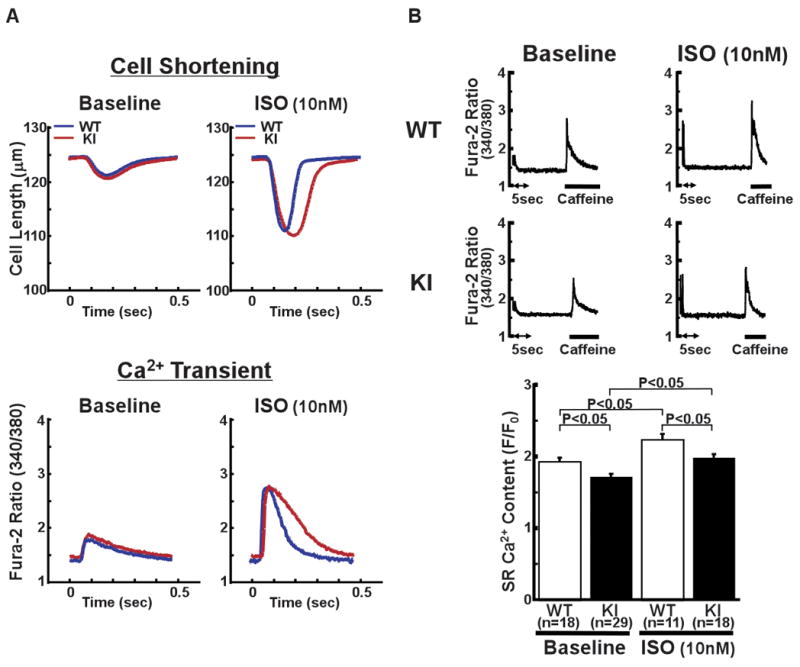
Cell shortening and intracellular Ca2+ transient in intact cardiomyocytes. A, Representative recordings of cell length and fura-2 fluorescence signal, at a pacing rate of 2 Hz, with isoproterenol (ISO) (10 nmol/L) activation (ISO) and without it (Baseline). B, Representative recording of fura-2 fluorescence signal after addition of caffeine (top figure), which is a measure of the SR Ca2+ content, and the summarized data of SR Ca2+ content (bottom). Caffeine-induced Ca2+ transient was measured by first applying a stimulation train at 2 Hz, and then by rapidly switching the super fusing solution to a solution containing 20 mmol/L caffeine for 5 to 6 seconds. N: the number of cells from 3–9 hearts.
RyR2 Ca2+ channels of R2474S/+ KI cardiomyocytes were hypersensitive to channel activation by isoproterenol, PKA phosphorylation, and luminal calcium
In KI cardiomyocytes, Ca2+ spark frequency (SpF) was higher than that in WT cardiomyocytes, both before and after the addition of isoproterenol (Figure 4A, Online Figure VIA for 3-dimensional images). In the presence of a higher concentration of isoproterenol (100 nmol/L), the frequency of spontaneous Ca2+ waves was much higher in KI cardiomyocytes than in WT cardiomyocytes and the duration of local Ca2+ release provoked by the transmission of the Ca2+ waves was markedly prolonged. (Online Figure VII). In both KI and WT cardiomyocytes, isoproterenol (10 nmol/L) increased the peak amplitude, full width at half maximum (FWHM), and full duration at half maximum (FDHM) (Online Table II). In KI cardiomyocytes, however, FDHM increased approximately twice as much as in WT cardiomyocytes (Online Table II). In response to isoproterenol (10 nmol/L), the level of PKA-dependent phosphorylation at Ser2808 of the RyR2 showed a rather modest increase that was slightly less than half of maximum phosphorylation (see Online Figure II). But in this case, the extent of the increase was nearly the same in the KI and WT cardiomyocytes (Figure 4B). These results suggest that the sensitivity of the channel to both SR content (luminal [Ca2+]) and PKA phosphorylation is markedly increased in KI channels.
Figure 4.

Local Ca2+ release events in intact cardiomyocytes and Ser2808 phosphorylation in the RyR2. A, Spontaneous Ca2+ sparks. Top: Recordings of line-scan images of fluo-4 AM fluorescence with or without isoproterenol (10 nmol/L) in intact WT and R2474S/+ KI cardiomyocytes. Bottom: Summarized data of Ca2+ spark frequency. N: the number of cells from 6–8 hearts. B, Comparison of protein kinase A (PKA)-mediated phosphorylation level at Ser2808 of the RyR2 (P-Ser2808-RyR2) in intact KI and WT cardiomyocytes. In the presence of isoproterenol (10 nmol/L), KI and WT cardiomyocytes were incubated in the lysis buffer, and centrifuged. Then, the supernatant fraction containing crude homogenate was used for the phosphorylation assay. C, Comparison of P-Ser2808-RyR2 in KI and WT cardiomyocytes. In the presence of cAMP (1 μmol/L), okadaic acid (1 μmol/L), and CaMKII inhibitor KN-93 (1 μmol/L), KI and WT cardiomyocytes were incubated in the lysis buffer, and centrifuged. Then, the supernatant containing crude homogenate was used for phosphorylation assay. N: the number of cell lysate from 3–5 heats. D, Effect of cAMP on the RyR2-FKBP12.6 association in WT and KI cardiac homogenates. (Top) Representative Western blots of the RyR2 and FKBP12.6 after immunoprecipitation (cf. online supplemental methods). (Bottom) Summarized data of the ratio of FKBP12.6 to the RyR2, which was normalized to control condition. Although FK506 dissociated FKBP12.6 from the WT RyR2, shown as positive control, cAMP (1 μmol/L) did not dissociate the RyR2-bound FKBP12.6 in WT and KI cardiac homogenates. N: the number of SR preparations from 7–14 hearts.
For further analysis of the increased sensitivity of KI channels to PKA phosphorylation and the SR Ca2+ content, we measured the PKA phosphorylation level at Ser2808 of RyR2 after addition of cAMP under the same conditions as the Ca2+ spark assay ([Ca2+] = 30 nmol/L, buffered by 0.5 mmol/L ethylene glycol tetraacetic acid (EGTA)). For this purpose, we added the Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitor KN-93 (1 μmol/L) to inhibit the effect of intrinsic CaMKII on the phosphorylation of the RyR2. We then measured both SpF and SR Ca2+ content in the absence and in the presence of 1 μmol/L cAMP in the saponin-permeabilized cardiomyocytes. There was no significant difference in the PKA phosphorylation level at Ser2808 between the KI and WT cardiomyocytes, both in the absence and the presence of cAMP (Figure 4C). Because there is marked variation in specificity among antibodies generated against Ser2808, 11 we compared the phosphorylation level at Ser2808 of RyR2 by using another different antibody against Ser2808 (Badrilla, UK). There was no significant difference in the PKA phosphorylation level at Ser2808 between KI and WT cardiomyocytes (Online Figure VIII).
To test the postulated possibility that de-stabilization of the RyR2 caused by the dissociation of FK506 binding protein 12.6 (FKBP12.6) from the RyR2 is a common pathogenic mechanism underlying heart failure and lethal arrhythmia,12, 13 we determined the RyR2-bound FKBP12.6 by using a pull-down assay. As shown in Figure 4D, between the WT and KI mice, there was no significant difference in the RyR2-bound FKBP12.6 in the absence or the presence of cAMP (1 μmol/L).
Figure 5A shows the representative traces of Ca2+ sparks in the presence of various concentrations of cAMP (0.1–1 μmol/L) (see also Online Figure VIB for 3-dimensional images of Ca2+ sparks). The dependence of SpF on the SR Ca2+ content is plotted in Figure 5B. To obtain the point at lower SR Ca2+ content, thapsigargin was added to the cardiomyocytes. Then, both Ca2+ sparks and SR Ca2+ content were measured 3, 5, and 10 minutes after the addition of thapsigargin. SpF was higher in KI than in WT cardiomyocytes, although there was a considerable reduction in the SR Ca2+ content in KI cardiomyocytes. As a result, there was a considerable left-shift of the SpF versus SR Ca2+ content plot in the case of KI cardiomyocytes (Figure 5B). The effect of cAMP on Ca2+ spark characteristics are summarized in Online Table III. Compared to the WT cardiomyocytes, both the peak amplitude and FWHM decreased whereas FDHM increased in KI cardiomyocytes, suggesting a delay in the inactivation of the RyR2. These results suggest that the threshold of luminal [Ca2+] for channel opening decreased considerably owing to the single R2474S CPVT mutation of the RyR2. To assess the maximum capacity of Ca2+ loading of SR, we added tetracaine (1 mmol/L) to inhibit Ca2+ release and then evaluated SR Ca2+ content by caffeine application. After treatment with the Ca2+ release blocker tetracaine (arrows, Online Figure IX), the SR Ca2+ content increased and reached nearly the same level (approximately 4 F/F0) in both WT and KI cardiomyocytes. This suggests that the reduced SR Ca2+ content in the KI cardiomyocytes was not due to reduced capacity of Ca2+ loading, but due to increased activation of the Ca2+ release flux relative to the influx.
Figure 5.

Decreased threshold of SR Ca2+ content to induce spontaneous Ca2+ sparks in saponin-permeabilized cardiomyocytes. A, Representative line-scan images of cardiomyocytes after addition of cAMP (0.1–1 μmol/L), at 30 nmol/L [Ca2+] buffered by 0.5 mmol/L EGTA. B, Relationship between Ca2+ spark frequency (SpF) and SR Ca2+ content. SR Ca2+ content was measured by addition of 10 mmol/L caffeine. To obtain the point at lower SR Ca2+ content, thapsigargin was added to the cardiomyocytes. Then, both Ca2+ sparks and SR Ca2+ content were measured 3, 5, and 10 minutes after the addition of thapsigargin [WT (n=6–12 hearts): (−) thapsigargin:107–441 cells for SpF and 10–84 cells for SR Ca2+ content; (+) thapsigargin: 4 cells for SpF and 4 cells for SR Ca2+ content, R2474S/+ KI (n=6–9 hearts): (−) thapsigargin: 91–361 cells for SpF and 14–46 cells for SR Ca2+ content; (+) thapsigargin: 4 cells for SpF and 4 cells for SR Ca2+ content]. C, Effect of protein kinase A (PKA) phosphorylation on Ca2+ spark frequency at comparable SR [Ca2+] content in WT and R2474S/+ KI saponin-permeabilized cardiomyocytes. For PKA phosphorylation of the RyR2, both cAMP (1 μmol/L) and okadaic acid (1 μmol/L) were added to the cardiomyocytes at 30 nmol/L [Ca2+] buffered by 0.5 mmol/L EGTA. Two minutes after the addition of cAMP, the cAMP was washed away to prevent further PKA phosphorylation. Then, thapsigargin (Thap) (0.3 μmol/L) was added. One minute later, line-scanned images were obtained. Arrow indicates the change in the data point produced by the addition of thapsigargin, Note that addition of cAMP increased SpF at a comparable SR Ca2+ content only in the KI cardiomyocytes.
D, E, Top: Representative line-scan images of cardiomyocytes after addition of DPc10 (50 μmol/L) and/or dantrolene (1 μmol/L), at 30 nmol/L [Ca2+] buffered by 0.5 mmol/L EGTA. Bottom: The dependence of SpF on the SR Ca2+ content after addition of DPc10 in the absence or in the presence of dantrolene. Arrow indicates the shift of the data points by the addition of DPc10 in the absence or in the presence of dantrolene [WT (n=5–12 hearts): {(−) DPc10: 441 cells for SpF and 84 cells for SR Ca2+ content; (+) DPc10: 13 cells for SpF and 7 cells for SR Ca2+ content; (+)DPc10, (+)dantrolene: 47 cells for SpF and 29 cells for SR Ca2+ content}, KI (n=3–9 hearts): {(−) cAMP, (−) DPc10, (−) dantrolene: 91 cells for SpF and 14 cells for SR Ca2+ content; (+) cAMP (1 μmol/L), (+) DPc10: 15 cells for SpF and 10 cells for SR Ca2+ content; (+) cAMP (1 μmol/L), (+) dantrolene (1 μmol/L): 37 cells for SpF and 25 cells for SR Ca2+ content}].
PKA-phosphorylation modulates not only RyR2 function but also SR Ca2+ load-dependence of channel activation. Thus, to clarify the direct effect of PKA-phosphorylation on RyR2 function, we measured the SpF and SR Ca2+ content in the presence of 0.3 μmol/L thapsigargin (inhibitor of SERCA2a activity) (Figure 5C). At comparable SR Ca2+ content, PKA phosphorylation increased SpF in KI cardiomyocytes, but not in WT cardiomyocytes. This suggests that PKA-dependent phosphorylation sensitized diastolic SR Ca2+ release, but only in CPVT, and not WT RyR2 channels.
Domain unzipping peptide DPc10 mimics the phenotype of KI channels in WT cardiomyocytes
As shown previously,9 DPc10 (a peptide corresponding to the 2460–2495 region of the central domain) mimics the channel disorder in the CPVT mutant (R2474S) by interfering with the channel-stabilizing inter-domain interactions between the N-terminal and central domains (i.e., domain unzipping). To assess the effect of domain unzipping on the dependence of SpF on the SR Ca2+ content, we added DPc10 to the WT saponin-permeabilized cardiomyocytes. Successful incorporation of DPc10 was confirmed by the intracellular fluorescence signal of Alexa fluor 488-labeled DPc10 (Online Figure X). DPc10 (50 μmol/L, applied externally) induced Ca2+ sparks in WT cardiomyocytes (Figure 5D, top). Importantly, addition of DPc10 caused a left-shift of the SpF/SR Ca2+ content relationship in WT cardiomyocytes (Figure 5D, bottom), resulting in the nearly identical SpF/SR Ca2+ profile as that of KI cardiomyocytes (Figure 5B). Similar to cAMP-treated KI cardiomyocytes, addition of DPc10 to WT cardiomyocytes prolonged FDHM (21.32 ± 0.31 ms (n = 27 cells) to 23.82 ± 0.54 ms (n = 48 cells); p < 0.01), but decreased the peak (1.74 ± 0.03 to 1.69 ± 0.01; p = 0.068) and FWHM (2.05 ± 0.03 μm to 2.00 ± 0.02 μm; p < 0.01). Addition of DPc10 to the (cAMP-treated) KI cardiomyocytes produced no further effect (Figure 5E). To assess whether suppression of domain unzipping reversed the left-shift of the SpF/SR Ca2+ content relationship in KI cardiomyocytes, we added dantrolene, which has corrected aberrant domain unzipping and prevented the development of heart failure14. Interestingly, dantrolene shifted back the SpF/SR Ca2+ content relationship to the right, almost towards the point corresponding to that of WT cardiomyocytes (Figure 5E). Dantrolene also prevented the DPc10-induced left-shift of the SpF/SR Ca2+ content relationship in WT cardiomyocytes (Figure 5D). Collectively, these findings indicate that the defective inter-domain interaction within the RyR2 caused by either point mutation (R2474S) or domain unzipping peptide (DPc10) reduced the threshold of luminal [Ca2+] for channel activation, leading to the phenotype of CPVT (hyper-activation of the RyR2 channel). This suggests that as in the case of MH, correction of the defective inter-domain interaction by dantrolene may provide an effective method to treat CPVT.
Luminal [Ca2+] of SR in saponin-permeabilized WT and KI cardiomyocytes
To confirm the above notion that the level of SR Ca2+ load was decreased in KI cardiomyocytes, we monitored luminal [Ca2+] in both cardiomyocytes using fluo-5N as a luminal Ca2+ probe. The average level of free diastolic SR Ca2+ decreased in KI cardiomyocytes, and upon caffeine-induced discharge of luminal Ca2+, the luminal Ca2+ reached the same basal level in both WT and KI cardiomyocytes (Figure 6).
Figure 6.
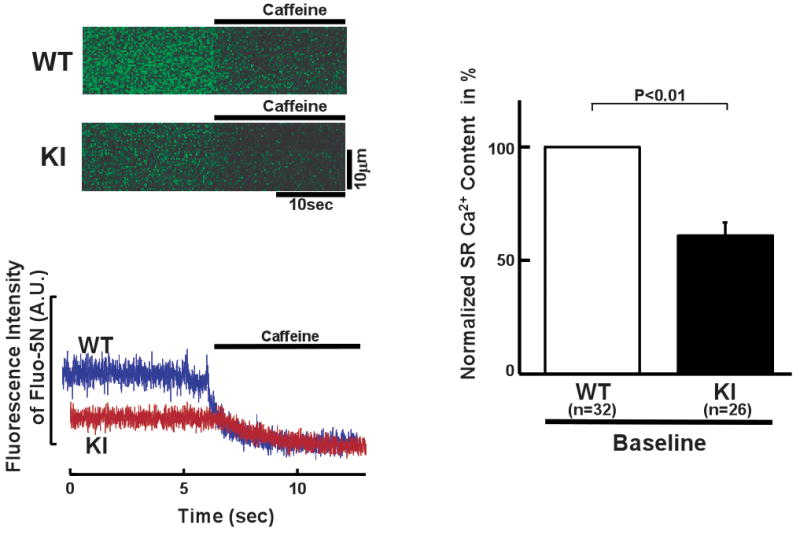
Spectroscopic determination of luminal [Ca2+] of the SR in saponin-permeabilized WT and R2474S/+ KI cardiomyocytes (see “Expanded Materials and Methods” in online data supplements for measurement of intra-SR [Ca2+]). (Left) Representative fluo-5N images before and after addition of caffeine (20 mmol/L). (Right) Summarized data of the luminal [Ca2+] normalized to control condition of WT cardiomyocytes. N: the number of cells from 3–4 hearts.
Spectroscopic evidence that the inter-domain interaction is defective in R2474S/+ KI cardiomyocytes
To investigate whether the inter-domain interaction was defective in KI mice, we used the fluorescence quench technique that permits spectroscopic monitoring of the state of the inter-domain interaction (zipped or unzipped).15 The methylcoumarin acetamido (MCA) probe that has been attached to the critical domain would be inaccessible to a bulky fluorescence quencher (QSY®-BSA conjugate) in a zipped configuration of the interacting domains, while it would become accessible to the quencher upon unzipping. As in our previous study with DPc10,9 site-specific intense fluorescence labeling of the RyR2 band was achieved using DPc10 as a carrier (Figure 7A, left lane), but there was no MCA labeling when DPc10-mut was used as a carrier (middle lane). As shown in the “cold-chase” experiment (right lane), excess unlabeled DPc10 (10 mmol/L) prevented DPc10-mediated MCA labeling. DPc10 increased the slope of the Stern-Volmer plot (KQ), a measure of the extent of unzipping between the N-terminal (aa 1–600) and central (aa 2000–2500) domains in WT SR (Figure 7B, left). Addition of cAMP (1 μmol/L) had no effect (no unzipping) in WT SR (Figure 7B, left). In contrast, KI SR showed a high KQ value (Figure 7B, right). Addition of cAMP (1 μmol/L) further increased the KQ, which was comparable with the value of WT SR with added DPc10 (Figure 7B, right). Addition of DPc10 on the top of cAMP (1 μmol/L) produced no further increase in KQ. Interestingly, dantrolene (1 μmol/L) partially reversed the KQ that had been increased by cAMP (Figure 7B, right). Dantrolene also prevented the DPc10-induced unzipping in WT SR (Figure 7B, left). These KQ values are summarized in Figure 7C.
Figure 7.

Spectroscopic monitoring of domain-domain interaction between the N-terminal (aa 1–600) and central (aa 2000–2500) domains in the RyR2. A, Site-directed fluorescence labeling of the RyR2 with MCA. MCA fluorescence labeling took place only when DPc10-SAED was used to mediate site-specific labeling (left lane). No MCA fluorescence was seen when DPc10-mut-SAED was used (middle lane) or when an excess concentration of DPc10 (10 mmol/L) was added during the labeling (‘cold chase’, right lane). B, Left: Stern-Volmer plots of the MCA fluorescence quenching data with QSY®7-BSA of WT mice. Right: Comparison of the Stern-Volmer plots of the MCA fluorescence quenching data of R2474S/+ KI mice with those of WT mice. Note that the KQ increased in the SR vesicles from KI to a similar level as that of DPc10-added WT SR vesicles. The values of KQ are means±SE of 5–9 experiments using 2–3 SR preparations from 20–30 mice heart. C, Summarized data of KQ shown in Figures 7B.
Arrhythmogenic characteristics of the membrane potential in R2474S/+ KI cardiomyocytes
Because altered membrane potential events represent an important landmark in arrhythmogenesis,16 we measured membrane potentials of WT and KI cardiomyocytes by recording the di-8-ANEPPS fluorescence while pacing in the presence of 30 nmol/L isoproterenol (Online Figure XI). WT cardiomyocytes showed no spontaneous after potential (Figure 8A) and no spontaneous Ca2+ transient (Figure 8B). However, KI cardiomyocytes showed spontaneous Ca2+ transients and spontaneous after potentials in response to isoproterenol (30 nmol/L) when we increased the pacing rate from 1 to 5 Hz (Figures 8A and 8B). Interestingly, the spontaneous after potentials and Ca2+ transients disappeared in the presence of dantrolene (1 μmol/L), which corrected the defective inter-domain interaction between the N-terminal and central domains (Figures. 8A and 8B).
Figure 8.

Provocation of triggered activity and spontaneous Ca2+ transient in R2474S/+ KI cardiomyocytes. Representative recording of di-8-ANEPPS fluorescence (A) and Ca2+ transients (B) before and after addition of isoproterenol (30 nmol/L) in intact WT and KI cardiomyocytes. Summarized data are shown at the bottom of each figure. After-potentials (indicated by arrows and shown in the insets) and spontaneous Ca2+ transients took place after pacing (5Hz) in KI cardiomyocytes. N: the number of hearts.
Discussion
Many point mutations have been found in RyR2 in patients with CPVT. Several pieces of biochemical evidence suggest that these mutations cause defective channel gating, leading to diastolic Ca2+ leakage.17 More direct evidence that a single point mutation in the RyR2 is the primary cause of channel dysfunctions in CPVT patients have been obtained by recent studies with a knock-in (KI) mouse model.6–8 A single point mutation (R4496C) introduced in the RyR2 was found to cause bidirectional or polymorphic VT in the KI mice.6 In isolated cardiomyocytes from the R4496C KI mice, both delayed after depolarization (DAD) and triggered activity were induced on stimulation with isoproterenol. 18 A more recent study using the same model 19 showed that a dramatic increase in the Ca2+ sensitivity of the RyR2 channel resulted in the increased frequency of Ca2+ sparks and Ca2+ waves, which was further amplified by either isoproterenol or high pacing rates. Further studies7,8 confirmed mutation-linked dysfunction of RyR2, namely spontaneous Ca2+ release events and DAD. These studies show a close inter-relationship between the single point mutation, increased Ca2+ sensitivity of the channel gating, and the resulting lethal arrhythmia. However, the underlying mechanism by which a single mutation in such a large molecule causes drastic effects on cardiac function has remained unclear.
Defective inter-domain interaction is the source mechanism of mutation-linked channel disorder
The most important new aspect of the present study is the finding that introduction of the R2474S CPVT mutation into the central domain of RyR2 induced a defective interaction between the central domain and the N-terminal domain, as predicted from the “domain switch hypothesis” (cf. Introduction), and this caused channel dysfunction similar to that of CPVT patients in KI mice. The three lines of evidence are consistent with this. First, DPc10 (aa 2460–2495), which contains the mutable R2474 residue and is known to interfere with normal inter-domain interaction between the N-terminal and central domains,9 reproduced the abnormal cellular Ca2+ events seen in the R2474S/+ KI mice (e.g., increased frequency of Ca2+ sparks) in an otherwise normal system (i.e., in cardiomyocytes isolated from WT mice). However, the addition of DPc10 to the (cAMP-treated) R2474S KI cardiomyocytes produced no further effect, suggesting that the defective inter-domain interaction (aberrant domain unzipping) had already taken place in the KI cardiomyocytes. Second, The R2474S mutation, introduced into the central domain of RyR2 of KI mice, did produce defective inter-domain interaction between the N-terminal and central domain (aberrant domain unzipping), as evidenced by the accessibility of the fluorescent probe MCA attached to the N-terminal domain to a high molecular weight fluorescence quencher QSY®7-BSA was considerably higher in the KI RyR2 than the WT RyR2. Finally, dantrolene, which corrects aberrant domain unzipping, did suppress aberrant phenomena characteristic of CPVT KI mice, such as reduced threshold of luminal Ca2+ for channel activation, spontaneous Ca2+ sparks, and DAD.
PKA-dependent phosphorylation of the RyR2 at Ser2808 facilitates domain unzipping only in the CPVT mutant ryanodine receptor
An interesting new finding in the present study is that the threshold of luminal [Ca2+] for activation of Ca2+ sparks was much lower in R2474S/+ KI mice than in WT mice. In other words, the sensitivity of the RyR2 channel to activation by luminal [Ca2+] was increased in R2474S/+ KI mice. More importantly, we could reproduce this sensitized channel gating to luminal [Ca2+] that is characteristic of the R2474S/+ KI mice, in WT cardiomyocytes by adding DPc10 (Figure 5D). This provides further support for the notion that the aberrant channel gating in R2474S/+ KI mice is produced by defective inter-domain interaction between the N-terminal and central domains.
This study also showed that the level of PKA-dependent phosphorylation of the RyR2 at Ser2808 was virtually indistinguishable between KI and WT RyR2s (Figure 4B, C), yet PKA phosphorylation produced a much larger effect in increasing the frequency of Ca2+ sparks (SpF) in the KI cardiomyocytes than the WT myocytes (Figures 4A and 5A). This suggests that the CPVT mutation also sensitizes the channel to PKA phosphorylation-dependent activation. In the three-dimensional image of the RyR2, Ser2808, the site of PKA phosphorylation, has been localized in the vicinity of the boundary between the N-terminal (aa 1–600) and the central domains (aa 2000–2500).20 Earlier cryo-electron microscopy single particle study of RyR1 21 also showed that domain 5 (including the N-terminal domain) and domain 6 (including the central domain) at the clamp region are indeed in close apposition to each other in the resting state, whereas these domains become separated in the activated (channel-open) state (e.g., in the presence of cAMP and activating Ca2+). Thus, it is tempting to suggest that PKA phosphorylation at Ser2808 accelerates domain unzipping in the KI channel, where domain unzipping has already progressed because of a weakened inter-domain interaction caused by the CPVT mutation.
A new molecular mechanism for CPVT
Although recent studies using KI mouse models have demonstrated that CPVT is caused by mutation-linked dysregulation of intracellular Ca2+ events and membrane potential events (DAD and triggered activity), it is still unclear how a single mutation changes the conformational state of the RyR2, leading to leaky channel. We propose a new molecular mechanism underlying CPVT (Online Figure XI). In the normal channel, domain-domain interaction between the N-terminal (aa 1–600) and central (aa 2000–2500) domains is maintained in a zipped state, and thus, the channel is stabilized, preventing Ca2+ leakage and DAD at a physiological range of SR Ca2+ contents. In the mutant channel, the stabilized inter-domain interaction is disrupted, causing aberrant domain unzipping; domain unzipping is further aggravated by the PKA-phosphorylation of Ser2808, located at the boundary between the 2 domains at the clamp region.20 In turn, the threshold of luminal [Ca2+] for channel activation is decreased (cf. ref. 17). Together, this results in SR Ca2+ leakage, DAD, and lethal arrhythmia. It should be noted, however, that DAD-triggered arrhythmia can also be induced by intracellular Ca2+ overload, for example, the toxic arrhythmogenic effects of the cardiac glycosides.22
In contrast to a previous report, 8 the RyR2-FKBP12.6 link was not disrupted in R2474S/+ KI mice. In the present study, the level of protein kinase A (PKA)-dependent phosphorylation at Ser2808 of the RyR2 showed only a modest increase in response to 1 μmol/L cAMP (slightly less than half of maximum phosphorylation; see Online Figure II). This level of phosphorylation may not have been high enough to dissociate FKBP12.6 from RyR2. Nonetheless, the fact that such a modest increase in the PKA-phosphorylation at Ser2808 induced domain unzipping and the resulting Ca2+ leakage in KI mice, suggests that the defective inter-domain interaction within the RyR2 is the source mechanism of CPVT and that FKBP dissociation is not a direct cause of defective channel gating in CPVT. As we previously reported, 9 however, it is also true that FKBP dissociation induces domain unzipping, which may account for the destabilized channel gating in heart failure. Thus, either a CPVT-type mutation or FKBP dissociation in heart failure commonly induces domain unzipping as an independent trigger, resulting in aberrant Ca2+ release in diseased hearts.
The reduction in SR Ca2+ content in heart failure may be partly due to increased NCX function and the increased SR Ca2+ leakage (in addition to reduced SERCA function), causing contractile dysfunction as well as arrhythmia. We previously demonstrated that dantrolene corrects defective inter-domain interactions within the RyR2 in failing hearts, inhibits spontaneous Ca2+ leakage, and in turn improves cardiomyocyte function in failing hearts.14 In this study, we showed that dantrolene was equally effective in the CPVT-type mutated RyR2 as in failing hearts. This indicates that channel dysfunction in CPVT and heart failure are caused by a common mechanism, that is, defective inter-domain interaction within the RyR2.
According to a recent report by Lehnart et al,8 R2474S KI mice, which harbors the same mutation as those used in the present study, exhibited spontaneous generalized tonic-clonic seizures. In our KI mice, however, we did not observe spontaneous tonic-clonic seizures. We have no explanation for this difference. It may be ascribable to differences in the background of the mouse model. In generating the RyR2 KI mice, we used ES cells derived from C57BL/6J mouse (no difference in the background between ES cell line mice and KI mice), but Lehnart et al. used those from 129 mice, followed by backcrossing with C57BL/6J mice.
In this, study, we evaluated the phosphorylation status only at Ser2808. However, because beta-adrenergic stimulation has been reported to activate CaMKII, 11, 23 a further investigation is clearly needed to assess the role of other phosphorylation sites (e.g. Ser2814 and Ser2030) on CPVT-type channel disorder.
In conclusion, a single point mutation within the RyR2 sensitizes the RyR2 channel to activation by luminal [Ca2+] (i.e., a decreased threshold of luminal [Ca2+] for channel activation), and in turn induces spontaneous Ca2+ sparks and DAD, leading to CPVT. More importantly, this aberrant channel opening is primarily mediated through defective inter-domain interaction between the N-terminal (aa 1–600) and central (aa 2000–2500) domains.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by grants-in-aid for scientific research from The Ministry of Education in Japan (grant Nos. 20390226 to MY, 20590868 to TY, 20591805 to SK, 19209030 to MM), a grant from Takeda Science Foundation (to MY), and a grant from the National Heart, Lung and Blood Institutes (HL072841 to NI). The authors declare no competing financial interests.
Non-standard Abbreviations and Acronyms
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- RyR
ryanodine receptor
- WT
wild-type
- KI
knock-in
- PKA
protein kinase A
- MH
malignant hyperthermia
- CCD
central core disease
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- SR
sarcoplasmic reticulum
- VT
ventricular tachycardia
- SpF
Ca2+ spark frequency
- FWHM
full width at half maximum
- FDHM
full duration at half maximum
- CaMKII
Ca2+/Calmodulin-dependent protein kinase II
- MCA
methylcoumarin acetamido
- DAD
delayed after depolarization
- SERCA
SR Ca2+-ATPase
- PLB
phospholamban
- CASQ
calsequestrin
- NCX
Na+/Ca2+ exchanger
Footnotes
Disclosures
None.
References
- 1.Yano M, Yamamoto T, Ikeda Y, Matsuzaki M. Mechanisms of Disease: ryanodine receptor defects in heart failure and fatal arrhythmia. Nat Clin Pract Cardiovasc Med. 2006;3:43–52. doi: 10.1038/ncpcardio0419. [DOI] [PubMed] [Google Scholar]
- 2.Ikemoto N, Yamamoto T. Regulation of calcium release by interdomain interaction within ryanodine receptors. Front Biosci. 2002;7:d671–683. doi: 10.2741/A803. [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto T, El-Hayek R, Ikemoto N. Postulated role of interdomain interaction within the ryanodine receptor in Ca2+ channel regulation. J Biol Chem. 2000;275:11618–11625. doi: 10.1074/jbc.275.16.11618. [DOI] [PubMed] [Google Scholar]
- 4.Liu Z, Wang R, Zhang J, Chen SR, Wagenknecht T. Localization of a disease-associated mutation site in the three-dimensional structure of the cardiac muscle ryanodine receptor. J Biol Chem. 2005;280:37941–37947. doi: 10.1074/jbc.M505714200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang R, Chen W, Cai S, Zhang J, Bolstad J, Wagenknecht T, Liu Z, Chen SR. Localization of an NH2-terminal disease-causing mutation hot spot to the “clamp” region in the three-dimensional structure of the cardiac ryanodine receptor. J Biol Chem. 2007;282:17785–17793. doi: 10.1074/jbc.M700660200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 7.Kannankeril PJ, Mitchell BM, Goonasekera SA, Chelu MG, Zhang W, Sood S, Kearney DL, Danila CI, De Biasi M, Wehrens XH, Pautler RG, Roden DM, Taffet GE, Dirksen RT, Anderson ME, Hamilton SL. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci U S A. 2006;103:12179–12184. doi: 10.1073/pnas.0600268103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, Lederer WJ, Kass RS, Morley G, Marks AR. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118:2230–2245. doi: 10.1172/JCI35346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oda T, Yano M, Yamamoto T, Tokuhisa T, Okuda S, Doi M, Ohkusa T, Ikeda Y, Kobayashi S, Ikemoto N, Matsuzaki M. Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation. 2005;111:3400–3410. doi: 10.1161/CIRCULATIONAHA.104.507921. [DOI] [PubMed] [Google Scholar]
- 10.George CH, Jundi H, Walters N, Thomas NL, West RR, Lai FA. Arrhythmogenic mutation-linked defects in ryanodine receptor autoregulation reveal a novel mechanism of Ca2+ release channel dysfunction. Circ Res. 2006;98:88–97. doi: 10.1161/01.RES.0000199296.70534.7c. [DOI] [PubMed] [Google Scholar]
- 11.Huke S, Bers DM. Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80–85. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 13.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi S, Yano M, Suetomi T, Ono M, Tateishi H, Mochizuki M, Xu X, Uchinoumi H, Okuda S, Yamamoto T, Koseki N, Kyushiki H, Ikemoto N, Matsuzaki M. Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing inter-domain interactions within the ryanodine receptor. J Am Coll Cardiol. 2009;53:1993–2005. doi: 10.1016/j.jacc.2009.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto T, Ikemoto N. Spectroscopic monitoring of local conformational changes during the intramolecular domain-domain interaction of the ryanodine receptor. Biochemistry. 2002;41:1492–1501. doi: 10.1021/bi015581z. [DOI] [PubMed] [Google Scholar]
- 16.Liu N, Priori SG. Disruption of calcium homeostasis and arrhythmogenesis induced by mutations in the cardiac ryanodine receptor and calsequestrin. Cardiovasc Res. 2008;77:293–301. doi: 10.1093/cvr/cvm004. [DOI] [PubMed] [Google Scholar]
- 17.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR) Proc Natl Acad Sci U S A. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2R4496C knock-in mouse model. Circ Res. 2006;99:292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- 19.Fernández-Velasco M, Rueda A, Rizzi N, Benitah JP, Colombi B, Napolitano C, Priori SG, Richard S, Gómez AM. Increased Ca2+ Sensitivity of the Ryanodine Receptor Mutant RyR2R4496C Underlies Catecholaminergic Polymorphic Ventricular Tachycardia. Circ Res. 2009;104:201–209. doi: 10.1161/CIRCRESAHA.108.177493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng X, Xiao B, Cai S, Huang X, Li F, Bolstad J, Trujillo R, Airey J, Chen SR, Wagenknecht T, Liu Z. Three-dimensional localization of serine 2808, a phosphorylation site in cardiac ryanodine receptor. J Biol Chem. 2007;282:25929–25939. doi: 10.1074/jbc.M704474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serysheva II, Schatz M, van Heel M, Chiu W, Hamilton SL. Structure of the skeletal muscle calcium release channel activated with Ca2+ and AMP-PCP. Biophys J. 1999;77:1936–1944. doi: 10.1016/S0006-3495(99)77035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hauptman PJ, Kelly RA. Digitalis. Circulation. 1999;99:1265–1270. doi: 10.1161/01.cir.99.9.1265. [DOI] [PubMed] [Google Scholar]
- 23.Curran J, Hinton MJ, Ríos E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leakage in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.