

Abstract
A new solid-state NMR-based strategy is established for the precise and efficient analysis of orientation and dynamics of transmembrane peptides in fluid bilayers. For this purpose, several dynamically averaged anisotropic constraints, including 13C and 15N chemical shift anisotropies and 13C-15N dipolar couplings, were determined from two different triple-isotope-labeled WALP23 peptides (2H, 13C, and 15N) and combined with previously published quadrupolar splittings of the same peptide. Chemical shift anisotropy tensor orientations were determined with quantum chemistry. The complete set of experimental constraints was analyzed using a generalized, four-parameter dynamic model of the peptide motion, including tilt and rotation angle and two associated order parameters. A tilt angle of 21° was determined for WALP23 in dimyristoylphosphatidylcholine, which is much larger than the tilt angle of 5.5° previously determined from 2H NMR experiments. This approach provided a realistic value for the tilt angle of WALP23 peptide in the presence of hydrophobic mismatch, and can be applied to any transmembrane helical peptide. The influence of the experimental data set on the solution space is discussed, as are potential sources of error.
Introduction
In a number of recent publications, the orientation of transmembrane peptides has been discussed, particularly in the context of hydrophobic mismatch (1,2). The standard NMR method of probing the tilt and rotation angle of a peptide in a bilayer is based on the polarization inversion with spin exchange at magic angle (PISEMA (3)) experiment, which measures the 15N chemical shift and 15N-H dipolar coupling on each amide bond. More recently, as an alternative approach, the geometric analysis of labeled alanines (GALA) was developed based on 2H NMR quadrupolar splitting (4–6). With this method, relatively small tilt angles (typically 5–10°, 5.5° for WALP23 in 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC)), were determined for WALP peptides, which are model peptides designed as mimics of α-helical transmembrane segments of membrane proteins. In a recent study, 15N PISEMA and 2H GALA NMR approaches were compared on a similar peptide (GWALP23 in 1,2-dilauroylphosphatidylcholine), yielding in both cases a fairly small tilt angle of ∼13–10° (7). Similar angle values were obtained by oriented circular dichroism (CD) (8) and attenuated total reflection (ATR) Fourier transform infrared (9) spectroscopies. These results are in apparent contradiction with recent molecular dynamics (MD) simulations, which show that these transmembrane peptides are subject to large-amplitude whole-body motions and that they adopt large tilt angles of, e.g., 31° ± 12° for WLP23 (10) or 33.5° ± 9° (11) for WALP23 in DMPC. Although the authors of these MD simulations themselves point out some limitations, such as the length required to properly sample the peptide's behavior, the simulations clearly reveal that neglecting dynamical averaging in 2H quadrupolar splitting interpretation leads to erroneous tilt angles. Therefore, an accurate analysis of the NMR data requires models that include dedicated peptide motions. It has been discussed already that dynamical averaging may be an important factor in the interpretation of PISEMA experiments (12,13). Simulations of quadrupolar splitting of deuterated alanines in a peptide helix reveal that dynamical averaging has an even more pronounced effect on the quadrupolar splittings. This is illustrated in Fig. 1.
Figure 1.
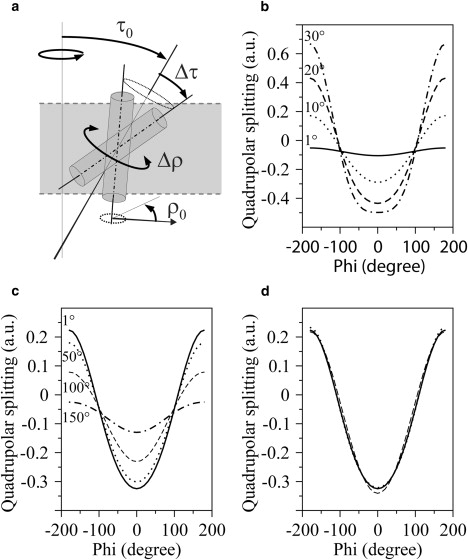
Influence of dynamical averaging on quadrupolar splitting. (a) Dynamic model describing the orientation and the dynamics of the peptide by a wobbling (Δτ) in a cone around the averaged value of the helix tilt angle (τ0) and an oscillations (Δρ) around the averaged value of helix rotation angle (ρ0). The fast axial diffusion takes place around the bilayer normal. (b–d) The three graphs represent the dependence of the quadrupolar splitting as a function of the label position around the helix (characterized by the ψ angle). (b) Variation for four tilt angles, τ0 (1°, 10°, 20°, and 30°), with no wobbling and no oscillation. (c) Variation for a tilt angle of τ0 = 12°, with oscillations of Δρ = 1°, 50°, 100°, and 150°. (d) Comparison of three dynamical models, (τ0 = 12°, Δτ = 0°, Δρ = 0°), (τ0 = 6°, Δτ = 10°, Δρ = 0°), and (τ0 = 18°, Δτ = 0°, Δρ = 80°). All plots were obtained for the same value of helix rotation angle (ρ0).
Fig. 1 b shows that using the GALA approach (no wobbling, no oscillation), a larger tilt angle leads to a stronger variation of the quadrupolar splitting for various label positions. If oscillations about the long axis of the peptide are included in the simulations, these variations of the quadrupolar splitting are dampened. This behavior is illustrated in Fig. 1 c for a peptide with a tilt angle of 12° with Δρ varying from 1° to 150°. In such a way, averaging via oscillations gives rise to an apparent tilt angle smaller than its actual value. Finally, in Fig. 1 d, wobbling-in-a-cone motions around the helix axis are also included in the simulations, and it is shown that similar curves can be achieved for different sets of parameters.
From these simulations, it is obvious that quadrupolar splittings alone are not sufficient to distinguish various dynamical models as soon as wobbling and oscillations are taken into account, and that neglecting such peptide motions can lead to an underestimation of the tilt angle (such as 12° instead of 18°). Similar behavior was described recently in a study by Strandberg et al. (13). It is therefore essential to add different experimental anisotropic interactions (such as chemical shift anisotropy and dipolar coupling) when performing a dynamical analysis. The recent work of Vostrikov et al. (7) is along this line, combining 2H NMR and PISEMA experiments. However, the data were analyzed with a semistatic, two-parameter model, with the associated artifacts illustrated above (the term semistatic is employed since a global, isotropic order parameter, scaling down the NMR interaction, is often included).
In this study, we establish an approach based on complementary anisotropic constraints to analyze the dynamics of membrane peptides, which to our knowledge is a new approach. For this purpose, two triple-isotope-labeled WALP peptides have been synthesized. Additional anisotropic NMR parameters, including two 15N amide chemical shift anisotropies (CSAs), two 13C carbonyl CSAs, and two 15N-13C dipolar couplings (DCs) between spins connected via the peptide bond were determined and combined with the six previously determined alanine methyl 2H quadrupolar splittings (6). The previous deuterium NMR data already showed that the peptide is tilted (i.e., that its most probable orientation is not the bilayer normal) and that fast axial diffusion is active along the bilayer normal but not along the α-helix axis (the bilayer normal is an axis of symmetry for the movement, the α-helix axis is not). Therefore, a simple Maier-Saupe orienting potential (14) cannot be used to describe the peptide motion. The data analysis is thus based on a helix rigid-body motion characterized by (see Fig. 1 a): 1), a fast axial diffusion of the peptide around the bilayer normal; 2), an averaged position of the peptide in the bilayer defined by a tilt angle, τ0 (between the diffusion axis and the α-helix axis), and a rotation angle, ρ0 (around the α-helix axis); 3), a wobbling amplitude, Δτ, around the averaged tilt angle, τ0; and 4), an oscillation amplitude, Δρ, around the averaged rotation angle, ρ0.
The anisotropic NMR interactions each explore a large range of orientations of the principal axis system (PAS) with respect to the helix axis, with the 15N CSA being mostly sensitive to tilt and wobbling (as in the PISEMA approach), whereas the other anisotropic constraints are sensitive to a combination of the four parameters, each in a specific way. This appears to be essential for the identification of a unique motional model capable of fitting all the experimental data.
Materials and Methods
Materials
DMPC was obtained from Avanti Polar Lipids (Alabaster, AL) and used without further purification. Two different 2H, 13C, and 15N isotopically labeled WALP23 peptides (acetyl-GW2(LA)8LW2A-NH2) were synthesized using Fmoc/tBu solid-phase synthesis, WALP23(2H3-Ala13,13C1-Ala11,15N-Leu12), referred to as WALP23#1, and WALP23(2H3-Ala7,13C1-Ala13,15N-Leu14), referred to as WALP23#2. Deuterium-labeled L-alanine (3,3,3-d3, 99%), 13C1-labeled L-alanine (1-13C, 99%), and 15N-labeled L-leucine (15N, 98%) were purchased from Cambridge Isotope Laboratories (Andover, MA). Fmoc (9-fluorenylmethyloxycarbonyl) was used to protect its amino functionality, as described by ten Kortenaar et al. (15), before being used in the peptide synthesis. The molecular mass of the synthesized peptides was verified by mass spectrometry and the purity was analyzed by high-performance liquid chromatography (HPLC) using a C4 reverse-phase HPLC column. For analytical and, if necessary, preparative HPLC, a solvent system with solvent A, composed of 95% water, 5% acetonitrile, and 0.1% TFA (trifluoroacetic acid), and solvent B, composed of 80% acetonitrile, 20% isopropanol, and 0.1% TFA, was used. The purity of the peptides used in this study was >95%.
NMR sample preparation
Static parameters of 15N and 13C anisotropic interactions were determined from lyophilyzed WALP23 peptide, and 6.5 μmol of powder of each peptide were packed into a 3.2-mm rotor. The 2H static quadrupolar coupling constant of Ala methyl was measured on 60 mg of dry WALP23#2 peptide. For WALP23 inserted into large unilamellar vesicles of DMPC, solutions of 20 mM DMPC in chloroform were prepared, and the lipid concentration was determined by the Rouser phosphorus assay. Solutions of WALP23 were prepared with a concentration of 500 μM in trifluoroethanol and the concentration of WALP23 was determined by absorption spectroscopy using an extinction coefficient of 22,400 M−1 cm−1 at 280 nm. Unoriented samples, i.e., liposomes, were used instead of oriented bilayers to allow complete control of the hydration level. Typically, 0.7 μmol of WALP23 peptides and 70 μmol DMPC were mixed in solution (1:100 peptide/lipid molar ratio). The organic solvents were evaporated under a nitrogen flow and further removed under vacuum overnight (ca. 10−2 mbar). Subsequently, the samples were hydrated with Milli-Q water and lyophilized to yield a fluffy powder, facilitating a thorough hydration at the low hydration levels needed. The unoriented samples were hydrated with deuterium-depleted water to 33% (water/(water + lipid + peptide), w/w) and incubated overnight at 37°C to equilibrate the hydration throughout the sample.
NMR experiments
Solid-state magic angle spinning (MAS) experiments were carried out on a Bruker (Billerica, MA) Avance narrow-bore spectrometer operating at 700.13 MHz for 1H. A Bruker 3.2-mm MAS triple-tuned solenoid coil was used for 13C and 15N experiments. Nonspinning 2H and 15N spectra were recorded on a Bruker Avance narrow-bore spectrometer operating at 500.13 MHz for 1H with a 5-mm simple-resonance and a 7-mm double-resonance probe, respectively, both equipped with a solenoid coil oriented at 90° with respect to the magnetic field. The sample temperature was 313 K in every case. 13C spectra were referenced to 2,2-dimethyl-2-silapentane-5-sulphonate and 15N spectra to liquid ammonia, using the 1H chemical shift of lipid's methyl resonances (0.85 ppm) as an internal reference for each nucleus.
13C and 15N static and averaged CSA parameters were determined from standard cross-polarization (CP) MAS spectra. Isotropic chemical shifts were determined at a spinning frequency of 10 kHz. CSAs were recorded by spinning side-band analyses. Typically, 4 kHz and 3 KHz, respectively, were used for the 13C and 15N static CSA determinations, whereas 0.75 kHz and 1kHz were used for the averaged CSA of WALP23 peptides into DMPC liposomes. 15N CSA of WALP in lipid bilayers was also determined from the static NMR powder spectrum. 13C-15N dipolar couplings were determined using a standard REDOR (16) experiment at a spinning frequency of 10 kHz, the ΔS/S0 REDOR curves were fitted in a standard manner (17) using Mathcad software (17–19). Deuterium NMR spectra were acquired using a standard quadrupolar echo pulse sequence (see Supporting Material for details on NMR experiments).
Determination of experimental static and dynamically averaged CSA
13C CSA parameters were obtained by the analysis of spinning side-band intensity using Dmfit freeware (http://crmht-europe.cnrs-orleans.fr/dmfit/).The static CSA and the asymmetry parameter were defined as δaniso = δ33 − δiso and η = (δ22 − δ11)/(δ33 − δiso), respectively, with |δ33 − δiso| ≥ |δ11 − δiso| ≥ |δ22 − δiso|. The 13C averaged CSA values were determined by using a Hertzfeld-Berger analysis to extract the components of the 13C dynamically averaged axially symmetric CSA, defined as CSAexp = 3/2 (δ90° − δiso), where δ90° is the chemical shift of peptide in membrane oriented at 90° with respect to the magnetic field and δiso is the isotropic chemical shift. 15N averaged axially symmetric CSAexp values were determined from δ90° and δiso, respectively, measured on a powder static 15N spectrum and on a CP-MAS spectrum at 10 kHz of spinning frequency (6).
Quantum chemistry calculation
The peptide's geometry and the 15N and 13C CSA static tensor components (more specifically, principal axis orientation with respect to the helix geometry) are required for the calculation of dynamically averaged anisotropic interactions. Both the structure and the static theoretical NMR spectroscopic data were determined using a quantum chemical approach.
All quantum chemical calculations were performed using the Gaussian 03 suite of programs (20). Molecular geometries of WALP23 were optimized starting from a perfect canonical α-helix structure (Φ = −58° and Ψ = −47°) using the ONIOM hybrid approach (21,22) that divides the system into an active and an inactive part. The active part (seven residues centered on the Ala11-Leu12 or the Ala13-Leu14 peptide bond) was calculated using the hybrid density functional theory B3LYP method (23,24) associated with a Pople-type double ξ basis set augmented by polarization functions on all atoms (namely, 6-31G(d,p)) (25). The inactive part of the system (the rest of the peptide) was calculated using a semiempirical (AM1) approach (26–28). NMR chemical shielding tensors were computed at the B3LYP/6-31G(d,p) level of theory on the whole peptide using the gauge including atomic orbital method for the numerous advantages it offers (29–33).
To allow the calculation of motionally averaged CSA parameters, the molecular frame (M) was chosen as follows: the z axis is the main inertia axis of the α-helix; the x axis is perpendicular to the z axis and contains the Leu12 α-carbon; the y axis completes the direct orthonormal frame. Thus, , , and (see Table 2 and Results and Discussion) are the Euler angles associated with the rotation matrix between the CSA tensor principal axis system PAS and the molecular frame M using the zyz convention. 15N-13C1 dipolar-coupling and methyl Ala 2H quadrupolar-coupling tensor orientations are defined by the spherical coordinates (Θ and Φ) of the 13C-15N and Cα-Cβ bond in the molecular frame. The associated numerical values were extracted from the quantum chemical optimized geometries.
Table 2.
Tensor components of the interactions used in the fitting procedure and experimental NMR parameters reflecting the peptide's dynamical behavior
| Chemical shift tensors | ||||||
|---|---|---|---|---|---|---|
| δaniso∗ (ppm) | η∗ | † (°) | † (°) | † (°) | CSAexp‡ (ppm) | |
| [13C]Ala11 | −84.2 | 0.59 | −82.0 | 97.7 | 323.3 | −16.4 ± 1.6 |
| [13C]Ala13 | −85.1 | 0.62 | −77.9 | 102.7 | 135.8 | −20.1 ± 2.0 |
| [15N]Leu12 | 104.6 | 0.20 | −163.3 | 15.1 | 20.5 | −105.6 ± 5.0 |
| [15N]Leu14 | 104.5 | 0.19 | −138.0 | 15.6 | 168.0 | −114.6 ± 5.0 |
| Dipolar interactions | ||||
|---|---|---|---|---|
| K∗ (Hz) | Θ† (°) | Φ† (°) | DCexp‡ (Hz) | |
| [13C]Ala11-[15N]Leu12 | 1010 | 68.9 | 312.4 | 313 ± 100 |
| [13C]Ala13-[15N]Leu14 | 1010 | 69.3 | 114.6 | 286 ± 100 |
| Quadrupolar interactions | ||||
|---|---|---|---|---|
| K∗ (Hz) | Θ† (°) | Φ† (°) | ΔνQ‡ (Hz) | |
| CD3-Ala7 | 37,700 | 58.7 | 85.5 | 500 ± 500 |
| CD3-Ala9 | 37,700 | 58.1 | 247.8 | 5725 ± 500 |
| CD3-Ala11 | 37,700 | 58.7 | 50.3 | 1000 ± 500 |
| CD3-Ala13 | 37,700 | 58.2 | 212.3 | 6025 ± 500 |
| CD3-Ala15 | 37,700 | 58.6 | 15.0 | 500 ± 500 |
| CD3-Ala17 | 37,700 | 58.3 | 176.9 | 6075 ± 500 |
Experimental static values measured on dry peptides.
Computed values.
Dynamically averaged values (measured on membrane-inserted peptide, at a wt % water of 33%). CSAexp = δ90° − δ0° = 3/2(δ90° − δiso) for peptide in axial diffusion around the lipid bilayer normal. δaniso = δ33 − δiso, η = (δ22 − δ11)/(δ33 − δiso), with |δ33 − δiso| ≥ |δ11 − δiso| ≥ |δ22 − δiso|, and (Θ, Φ) are the angles defining the CSA and dipolar or quadrupolar tensor orientations in the molecular frame (see Materials and Methods). Experimental uncertainties were estimated from two independent experiments and several sets of fitting parameters. They were not sensitive to line width, and the values finally retained and given in this table are very conservative (10% relative error in most cases, 5% for 15N CSAs).
Computation of the dynamically averaged anisotropic interactions and experimental fittings
Using the static anisotropic tensor information obtained as described above, dynamically averaged interactions were computed using a four-parameter model of motion involving tilt (τ0), wobbling in a cone of angle (Δτ), rotation (ρ0), and oscillation of amplitude (Δρ) (see Fig. 1 a). Internal dynamics of the peptide's helical structure, although certainly present, were assumed to be negligible based on the observation by molecular dynamics that the helical structure is well preserved during the simulations (11).
Both quadrupolar and dipolar splittings were computed from the expression
where the constant KQC/DC depends on the type of interaction, quadrupolar (QC) or dipolar coupling (DC), respectively; defines the orientation of the bilayer normal, N, relative to the principal axis frame of dipolar or quadrupolar interaction tensor (where the C-D quadrupolar tensor is quasiaxially symmetric). This equation is given for bilayers oriented with the bilayer normal parallel to the magnetic field.
The CSA effects depend on the spherical coordinates and of the bilayer normal in the principal axis frame (PAF) of the CSA tensor (where η is the asymmetry parameter):
Following the Wigner rotation matrix formalism (34), equations and were expressed as functions of the PAS orientation relative to the molecular frame (denoted M) and M orientation relative to the diffusion frame (denoted N).
Peptide motion in the submicrosecond timescale leads to time-modulated and angles, and was included by integrating and over the angles describing the movements, i.e., a variation of τ between τ0 − Δτ and τ0 + Δτ and of ρ between ρ0 − Δρ and ρ0 + Δρ, as described in the Supporting Material.
The dynamically averaged theoretical anisotropic interactions (QCs, DCs, and CSAs) were then calculated and compared to the experimental data. The sum of their squared differences was minimized to obtain the best fit. To avoid the traps of local minima typical for gradient-based approaches, we have used GOSA (35), a multivariate global optimization program based on simulated annealing (http://www.bio-log.biz). To estimate errors of the best-fit parameters, a Monte Carlo approach was employed (36). A set of 100 combinations of experimental values was randomly generated within the limits of experimental error, and the fit was performed for each of them. This gave a representation of multiple minima in the four-parameter space (see, e.g., rows F1 and F2 in Table 3), and the range of possible values for each fitted parameter. Multidimensional integration was performed with the CUBA library (37).
Table 3.
Best-fit parameters using various subsets of anisotropic NMR constraints and uniform distributions
| Number: nature of constraints | τ0 (°) tilt | Δτ (°) wobbling | ρ0 (°) rotation | Δρ (°) oscillation | |
|---|---|---|---|---|---|
| A | 12: complete ensemble (SSD = 0.36) | 20.8 ± 1.4 | 12.2 ± 4.5 | 146 ± 7 | 84 ± 21 |
| B | 10: remove 15N-13C DC (SSD = 0.33) | 21.0 ± 1.5 | 10.8 ± 6.3 | 146 ± 7 | 86 ± 19 |
| C | 14: adding two simulated 15N-H DCs∗ | 20.4 ± 1.8 | 11.3 ± 7.3 | 146 ± 6 | 82 ± 23 |
| D | 6: 2 15N CSA, 2 13C CSA, 2 CD3 ΔνQ | 21.9 ± 1.3 | 8.0 ± 6.5 | 149 ± 8 | 90 ± 20 |
| E | 8: 6 CD3 + 2 15N CSA | 21.2 ± 1.8 | 11.5 ± 5.5 | 147 ± 7 | 83 ± 28 |
| F1 | 6: CD3 ΔνQ alone (sol a: SSD = 0.3) | 2.8 ± 0.9 | 31.9 ± 1.7 | 147 ± 8 | 80 ± 21 |
| F2 | 6: CD3 ΔνQ alone (sol b: SSD = 0.6) | 21.2 ± 1.0 | 1.3 ± 1.3 | 146 ± 8 | 75 ± 36 |
| H | 4: 2 15N CSA + 2 15N-H DC | 15.8 ± 1.6 | 23.9 ± 2.0 | 127 ± 109 | 30 ± 81 |
Every fit was performed using the data obtained at 33% hydration (water/(water + lipid + peptide), w/w). The fits result from uniform distributions (see comment 4 in numbered list in text for Gaussian distributions). The uncertainty of each parameter (3σ value) was computed by the Monte Carlo approach (36) as described in Materials and Methods.
The simulated 15N-H DCs were computed using the dynamical parameters obtained from solution A. SSD is the standard deviation.
Results and Discussion
Secondary structure and aggregation state of WALP23
For interpretation of the solid-state NMR data in terms of a structural and dynamic model of WALP23, it first is important to know whether under the experimental conditions used, the peptide can be considered as a monomeric and stable helix, diffusing freely in the fluid bilayer.
Different studies have shown the backbone structure of the WALP peptides to be remarkably robust, even withstanding considerable mismatch. This is most clear from FTIR measurements, which showed, at the amide I position of a range of different-length WALP peptides, an intense narrow band that did not shift or broaden upon either increasing or decreasing bilayer thickness (9). Also, CD measurements (9,38) showed an α-helical structure under a range of conditions, and fits of 2H NMR data on Ala-d4-labeled peptides suggested significant distortion only at conditions of extreme positive mismatch (39). In addition, a variety of MD simulations suggested a rather perfect and robust α-helix (11,40). With respect to the aggregational behavior, it should be mentioned that different lines of experimental evidence suggest that WALP23 by itself has no tendency to self-associate in a lipid bilayer. For example, the lipid flip-flop-promoting effect of the peptides was found to be perfectly linear over a wide range of peptide concentrations (41). Also, self-quenching of pyrene-labeled peptides was found to occur only at peptide concentrations significantly higher than those in this study (42), and the same holds for sucrose density gradient centrifugation experiments, which showed homogeneous peptide/lipid mixtures up to very high peptide/lipid ratios (38). So far, only in the gel phase of DPPC and other saturated lipids aggregates have been observed by AFM. However, these were linear aggregates, supporting in fact the notion that the peptides have a strong preference for interaction with lipids (43). Nevertheless, to avoid any risk of peptide aggregation, we performed our analysis on samples with a relatively low peptide/lipid ratio of 1:100.
Static 13C and 15N CSA tensor parameters computed by quantum chemistry
Static 13C and 15N CSAs and asymmetry parameters were found to be close to the experimental values obtained on dry peptide. Consequently, we can be confident of their correctness and, in particular, of the quality of the eigenvectors (tensor orientation relative to the molecular frame (see Table 1)). To check the accuracy of our theoretical chemical-shielding eigenvectors, we can define the tensor orientation relative to the peptide plane (Fig. 2 and Table 1).
Table 1.
Chemical shift tensor orientation relative to the peptide plane computed for WALP23 peptide
| 13C | αC | βC | γC |
|---|---|---|---|
| Ala11 | 32.2 | 2.1 | 2.1 |
| Ala13 | 31.8 | 2.4 | 2.0 |
| 15N | αN | βN | γN |
| Leu12 | 20.9 | 17.9 | 19.2 |
| Leu14 | 20.7 | 17.7 | 19.5 |
See text for details regarding definition of angles.
Figure 2.
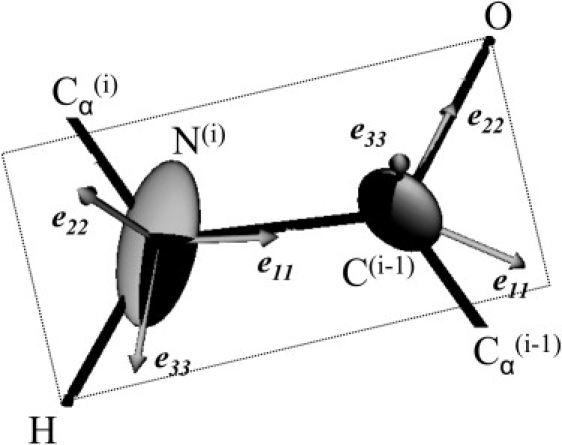
Schematic representation of eigenvector orientation with respect to the peptide plane for 13C1 and 15N CSA tensors.
For the 15N tensor, αN is the angle between e11 and the Ni-Ci−1 peptide bond, βN the angle between e22 and the normal to the peptide plane, and γN the angle between e33 and the N-H bond, where eii is the eigenvector associated with the σii eigenvalue (see Fig. 3 for eii representation). For the 13C1 tensor, αC is the angle between e11 and the Ni-Ci−1 peptide bond, βC the angle between e22 and the C-O bond, and γC the angle between e33 and the normal to the peptide plane. The angles obtained on both peptides (see Table 1) are in agreement with the standard values used to describe 15N and 13C chemical-shift tensor eigenvectors (44–46). In particular, 15N the angle between e33 and the N-H bond is equal to 19° and the e11 is tilted by ∼21° from the peptide plane. For 13C1, the αC angle is close to 32°, e22 is almost colinear to the carbonyl C-O bond, and e33 is perpendicular to the peptide plane.
Figure 3.
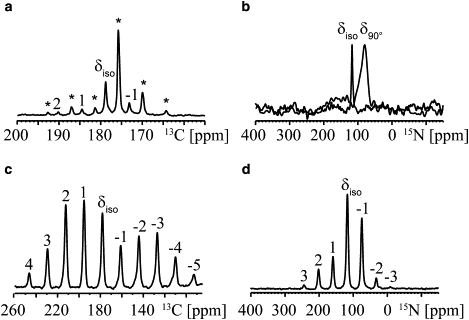
13C and 15N MAS NMR spectra for CSA determination for WALP23(2H3-Ala13,13C1-Ala11,15N-Leu12) peptide in DMPC liposomes (molar ratio 1:100, wt % water of 33%) and and as a dry peptide. (a) 13C1-Ala11 spinning side-band spectrum measured at 1 kHz spinning frequency. The peaks labeled with stars correspond to signals from lipid carbonyls. (b) Overlay of 15N-Leu12 10 kHz spinning MAS NMR spectrum (black) and 15N-Leu12 NMR powder spectrum (gray) of liposomes, displaying the highest intensity for bilayers oriented at 90°. Similar data were obtained for WALP23(2H3-Ala7,13C1-Ala13,15N-Leu14) peptide in DMPC liposomes (data not shown). (c) 13C1-Ala11 spinning side-band spectrum at 4 kHz spinning frequency, on dry peptide. (d) 15N-Leu12 spinning side-band spectrum measured at 3 kHz spinning frequency on dry peptide. Similar data were obtained for WALP23(2H3-Ala7,13C1-Ala13,15N-Leu14) peptide (data not shown).
MAS NMR experiments on peptides inserted into membranes
To ensure compatibility of the existing 2H NMR data with the new NMR data, a deuterium-labeled alanine was included in each peptide, which made it possible to check the sample conditions by deuterium NMR. For both triple-labeled peptides, the quadrupolar splittings found were identical to previously published values (6) (data not shown). In addition, each sample permitted the determination of the 15N amide chemical shift and averaged CSA for one leucine, 13C carbonyl chemical shift and averaged CSA for one alanine (Fig. 3), and one 15N-13C dipolar coupling linked to the dynamics and orientation of the N-C1 peptide bond (data not shown).
Isotropic chemical shifts were determined at a spinning frequency of 10 kHz on both peptides inserted into bilayers. The carbonyl 13C line widths were equal to 0.3 ppm, which is a good indication of structural homogeneity. The isotropic 13C chemical shift, 178.9 ppm in both cases, was typical for a peptide in a helical conformation, confirming that the peptide's dynamics preserves a helical conformation at positions 11 and 13.
To determine dynamically averaged CSAs, two strategies were applied, depending on spectral overlap and sensitivity of each nucleus. For 13C, dynamically averaged CSAs were obtained from slow-spinning side-band MAS spectral analysis, which makes it possible to resolve resonances arising from the peptides and those arising from natural-abundance lipid carbonyls (see Fig. 3 a). For 15N dynamically averaged CSAs, the most efficient method, due to the low signal intensity, appeared to be a comparison between the isotropic chemical shift measured under MAS conditions and the powder spectrum measured under static conditions. The most intense shoulder of the powder spectrum provides a value of δ90° (Fig. 3 b), as was already proposed by Cady et al. (47) for axially diffusing peptides. The magic-angle hole can be seen in 15N static spectra and results from the colinearity of the chemical-shift tensor and the dipolar-coupling tensor in uniaxially mobile molecules (48).
The CSA values for both 15N and 13C confirm the dynamical behavior of the peptide, since they are significantly lower in liposomes than in peptide powder (compare, for example, Fig. 3, a and c, and Fig. 3, b and d, and values in Table 2). Moreover, the asymmetry parameters extracted from spinning side-band analysis of the 13C spectra were found to be null for both peptides in liposomes, in agreement with dynamics involving fast axial diffusion along the bilayer normal, as was already demonstrated in previous 2H NMR experiments (5,6). The resulting part of the anisotropic interaction is directly related to the peptide's motion in terms of tilt (τ0), rotation (ρ0), wobbling (Δτ), and oscillation (Δρ).
All the computed static and experimentally determined static and dynamically averaged CSAs are summarized in Table 2, together with quadrupolar and dipolar splittings.
Data analysis and extraction of dynamical parameters
Six new anisotropic constraints have been determined, and these were combined with the previously published six quadrupolar splittings determined on peptides including deuterated alanines. As described in Materials and Methods, we then explored the entire space of possible solutions in terms of a four-parameter dynamical model involving tilt, τ0, rotation, ρ0, wobbling, Δτ, and oscillations, Δρ. In the fitting procedure, the principal axis orientations were taken from computed values, whereas the principal values were the experimental ones, measured on the dry peptide powder by standard spinning side-band analysis (see Table 2). The complementary orientations of the anisotropic tensors allowed for identification of a well-defined region of agreement between simulations and experiment, as illustrated in Fig. 4 and Table 3 (line A).
Figure 4.
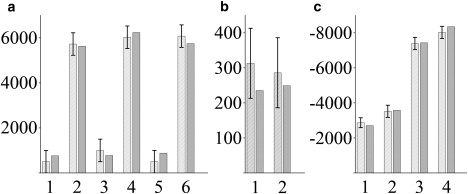
Best fit (dark gray bars) to the 12 experimental anisotropic constraints obtained from NMR (light gray bars) corresponding to results on line A of Table 1. (a) CD3 quadrupolar couplings (Hz) of Ala7,9,11,13,15,17 (1–6, respectively). (b) 15N-13C1 dipolar couplings (Hz) of Ala11-Leu12 (1) and Ala13-Leu14 (2). (c) CSAs of 13C1-Ala11 (1) and Ala13 (2) (Hz) and 15N-Leu12 (3) and Leu14 (4) (Hz). The experimental values are summarized in Table 2.
Several important conclusions can be drawn from this analysis:
-
1.
The complete set of anisotropic constraints is sufficient to extract the complete orientation and dynamics of the WALP peptide in DMPC (Table 3, line A). The tilt and rotation angles are determined with good accuracy (20.8 ± 1.4° and 146 ± 7°, respectively), which agrees well with the tilt angle of 23° obtained recently using a fluorescence approach (49) and confirms the trends indicated in molecular dynamics simulations (11). Although the qualitative agreement with fluorescence and MD simulation is interesting, it should be realized that these two approaches each have their own limitations, and we believe that the NMR approach presented here is more accurate. In particular, the MD work is on a completely different timescale, and the motions seen by MD may or may not accurately reflect the population examined on the timescale of the NMR experiment. Our NMR analysis furthermore shows that both the tilt and rotation angles are very stable among the various subsets of constraints (Table 3, lines A–E). The wobbling and oscillation amplitudes are also determined with reasonable accuracy, although slightly less precisely than tilt and rotation. The oscillation amplitude, Δρ, is very large (±84°), confirming that data analysis based on a semistatic, two-parameter model is inappropriate. The order of magnitude of Δρ is in complete agreement with the molecular dynamics simulation (11,50). It should be stressed that, compared with the GALA analysis, the rotation angle, ρ0, initially determined is not modified. The angle of 155°, as determined in Ozdirekcan et al. (11,50) corresponds to 145° in the molecular frame used in this article, and hence corresponds well with the value of 146° obtained here. These results show that dynamical averaging mostly affects the tilt angle determination.
-
2.
15N-13C dipolar couplings obtained for this highly dynamic peptide are weak (∼300 Hz). The accuracy is relatively low due to the low signal/noise ratio. Removal of these constraints does not significantly change the outcome of the fit (Table 3, line B), suggesting that one should proceed with the analysis without parameters from the fairly time-consuming REDOR experiments.
-
3.
Adding 15N-H dipolar couplings such as those that can be measured from the (also time-consuming) PISEMA experiment does not add much to the data analysis (Table 3, line C). This can be understood from the fact that the main tensor orientations of 15N-H dipolar coupling and 15N CSA interactions are similar, i.e., almost parallel to the helix axis. Both are very sensitive to the helix tilt (τ0 and Δτ) and less sensitive to helix rotation (ρ and Δρ). Therefore, using only these two interactions is not sufficient for accurate data analysis (see Table 3, line H, where τ0, Δτ, and ρ0 are different, and where Δρ is not determined), but using one of them is necessary (and sufficient) to separate the two pairs of variables. Accordingly, using only the six CD3 quadrupolar splittings and two 15N CSA values gives a good solution to the problem (Table 3, line E), whereas six quadrupolar splittings alone are not sufficient (Table 3, lines F1 and F2; note that the best fitting in this case is obtained for F1, i.e., for a very small tilt angle). It should be stressed that the PISEMA experiment offers the advantage of a 2D experiment in terms of increased resolution, and thus makes it possible to obtain exploitable anisotropic interactions on uniformly 15N-labeled peptides.
-
4.
About exploring various motional models: largely similar conclusions were derived when a Gaussian distribution was used instead of a uniform distribution. In the case of the Gaussian distributions, a global minimum was found for τ0 = 16.2° ± 1.6°, Δτ = 12.0° ± 1.6°, ρ0 = 146.6° ± 15.4°, Δρ = 98.2° ± 5.5°. Hence, a slightly smaller tilt angle was found, and the other parameters were well within the experimental error. Although MD simulations show that the helical structure is very stable (11,40,51), internal dynamics of the peptide is also expected to influence the data analysis to some extent. This point is currently under examination by assessing such “internal order parameters” from MD simulations and by incorporating them into the data fitting process.
-
5.
In the search for an optimal strategy to do a dynamical analysis of a transmembrane peptide (with fast axial diffusion around the bilayer normal), we propose to use six anisotropic interactions that allow optimal sampling of different tensor orientations by selecting specific labeling positions along the peptide helix (Table 3, line D). Thus, accurate dynamical analysis is already possible by synthesizing only two different triply labeled peptides, each of which includes a 2H (CD3 on Ala11 or Ala13), a 13C1 (Ala11 or Ala13) and a 15N (Leu12 or Leu14) label, which would require ∼3–4 days of measurement time on a 500- to 700-MHz solid-state NMR spectrometer.
-
6.
Regarding description of the motion in terms of order parameters: using the notation of Goormaghtigh et al. (52), one can convert the values of τ0 and Δτ into values of order parameters. The distribution of helix axis can be described by the product of two order parameters: Shelix = Shelix angle × Shelix order
Shelix angle = < 3cos2(τ0) − 1 > /2 is related to the average tilt angle, τ0
Shelix order = < 3cos2(τ1) − 1 >/2 is related to the wobbling ( brackets indicate the average over τ1 ∈ [0,Δτ]) (see Supporting Material for angle definitions).
For the uniform distribution, the values τ0 = 20.8° and Δτ = 12.2° translate into Shelix angle = 0.81, Shelix order = 0.97, and Shelix = 0.78. This value of Shelix order is significantly higher than the molecular order parameter Szz of a DPPC (53) or DMPC (54) lipid in the fluid phase, which is around 0.6. This is not surprising considering that the peptide is anchored on both sides of the bilayer, whereas the lipid molecules extend over only half of the bilayer.
Several methods have been used to study structural properties of membrane proteins and to determine tilt angle of transmembrane peptides, including solid-state NMR, EPR, CD, ATR-FTIR, and fluorescence spectroscopy (for a recent review, see Holt and Killian (55). All these techniques, like 2H GALA NMR, are powerful and simple to interpret in terms of an averaged orientation of the helices (τ0 and ρ0). However, the existence of distributions of orientation (characterized by a wobbling, Δτ, and oscillation, Δρ, parameters) and averaging effects make things more difficult. For instance, it is clearly stated in the very comprehensive review of Goormaghtigh et al. (52) that in the usual data treatment of ATR infrared spectroscopy, “Shelix order cannot be evaluated independently from Shelix angle”, so that usually Shelix order is set to 1 (i.e., Δτ = 0). Moreover, in the case of WALP23, one has to deal with the existence of a nonaxially symmetric distribution of the rotation angle, ρ, and the associated amplitude of oscillations (Δρ), and this had never been included in the data analysis for a tilted peptide, to the best of our knowledge.
Conclusions
In this work, we analyzed a data set of different NMR anisotropic interactions (quadrupolar couplings, dipolar couplings, and CSAs) with an extended, dynamical four-parameter (τ0, Δτ, ρ0, Δρ) peptide model. An averaged tilt angle of 21° for WALP23 peptides inserted into DMPC has been determined. This is much larger than the tilt angle of 5.2° obtained by 2H GALA NMR analysis based on a quasistatic model and follows the trends toward larger tilt angles indicated from MD simulations. It also provides an accurate determination of rotation angle, wobbling, and oscillation amplitudes. It does not, however, give any indication of the various timescales of these motions. These could be obtained in principle by analyzing solid-state deuterium NMR inversion-recovery and Jeener-Broekaert relaxation data in a similar way to the one proposed by Prosser et al. for lipids (53) or for the peptide gramicidin (14). 1H, 13C, and 15N relaxation studies in the rotation frame under magic-angle spinning and with static-oriented samples could also be used to characterize the motional model (56). It should be stressed, however, that for this particular case, the motional model should include at least three timescales, characterizing diffusion along the bilayer normal, wobbling, and oscillations (not counting slow ensemble motions of the bilayer).
In the strategy, the combination of at least three different anisotropic tensors possessing complementary orientations proved to be essential to solve the inherent under determination brought by the explicit introduction of dynamical averaging and to analyze the orientation and dynamics of transmembrane peptides in a precise and efficient way. This approach should be very generally applicable to any transmembrane peptide and will allow for assessment of the influence of hydrophobic mismatch not only on tilt angles but also on the amplitudes of wobbling and oscillations.
Supporting Material
Additional text, references, and figure are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00145-1.
Supporting Material
Acknowledgments
The authors thank the CALcul en MIdi-Pyrénées (CALMIP) computing center for generous allocations of computer time, and Rutger Staffhorst, Jacques Doux, and Lucie Khemtémourian for peptide synthesis. The NMR spectra were recorded on spectrometers financed with the help of European Structural funds, Région Midi-Pyrénées, and CNRS.
We acknowledge financial support from the European Marie Curie program (A.H.) (BIOMEM, Contract MEST-CT 2004-007931) and Conseil Régional Midi-Pyrénées and PRES Université de Toulouse (L.R.).
Footnotes
A. Holt's present address is The Netherlands Cancer Institute, Amsterdam, The Netherlands.
References
- 1.Hong M. Structure, topology, and dynamics of membrane peptides and proteins from solid-state NMR spectroscopy. J. Phys. Chem. B. 2007;111:10340–10351. doi: 10.1021/jp073652j. [DOI] [PubMed] [Google Scholar]
- 2.Nyholm T.K.M., Özdirekcan S., Killian J.A. How protein transmembrane segments sense the lipid environment. Biochemistry. 2007;46:1457–1465. doi: 10.1021/bi061941c. [DOI] [PubMed] [Google Scholar]
- 3.Cross T.A., Opella S.J. Solid-state NMR structural studies of peptides and proteins in membranes. Curr. Opin. Struct. Biol. 1994;4:574–581. [Google Scholar]
- 4.Jones D.H., Barber K.R., Grant C.W. Epidermal growth factor receptor transmembrane domain: 2H NMR implications for orientation and motion in a bilayer environment. Biochemistry. 1998;37:16780–16787. doi: 10.1021/bi981520y. [DOI] [PubMed] [Google Scholar]
- 5.van der Wel P.C.A., Strandberg E., Koeppe R.E. Geometry and intrinsic tilt of a tryptophan-anchored transmembrane α-helix determined by 2H NMR. Biophys. J. 2002;83:1479–1488. doi: 10.1016/S0006-3495(02)73918-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strandberg E., Özdirekcan S., Killian J.A. Tilt angles of transmembrane model peptides in oriented and non-oriented lipid bilayers as determined by 2H solid-state NMR. Biophys. J. 2004;86:3709–3721. doi: 10.1529/biophysj.103.035402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vostrikov V.V., Grant C.V., Koeppe R.E. Comparison of “Polarization inversion with spin exchange at magic angle” and “geometric analysis of labeled alanines” methods for transmembrane helix alignment. J. Am. Chem. Soc. 2008;130:12584–12585. doi: 10.1021/ja803734k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Planque M.R.R., Kruijtzer J.A., Killian J.A. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane α-helical peptides. J. Biol. Chem. 1999;274:20839–20846. doi: 10.1074/jbc.274.30.20839. [DOI] [PubMed] [Google Scholar]
- 9.de Planque M.R.R., Goormaghtigh E., Killian J.A. Sensitivity of single membrane-spanning α-helical peptides to hydrophobic mismatch with a lipid bilayer: effects on backbone structure, orientation, and extent of membrane incorporation. Biochemistry. 2001;40:5000–5010. doi: 10.1021/bi000804r. [DOI] [PubMed] [Google Scholar]
- 10.Esteban-Martín S., Salgado J. The dynamic orientation of membrane-bound peptides: bridging simulations and experiments. Biophys. J. 2007;93:4278–4288. doi: 10.1529/biophysj.107.113043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Özdirekcan S., Etchebest C., Fuchs P.F. On the orientation of a designed transmembrane peptide: toward the right tilt angle? J. Am. Chem. Soc. 2007;129:15174–15181. doi: 10.1021/ja073784q. [DOI] [PubMed] [Google Scholar]
- 12.Straus S.K., Scott W.R., Watts A. Assessing the effects of time and spatial averaging in 15N chemical shift/15N-1H dipolar correlation solid state NMR experiments. J. Biomol. NMR. 2003;26:283–295. doi: 10.1023/a:1024098123386. [DOI] [PubMed] [Google Scholar]
- 13.Strandberg E., Esteban-Martín S., Ulrich A.S. Orientation and dynamics of peptides in membranes calculated from 2H-NMR data. Biophys. J. 2009;96:3223–3232. doi: 10.1016/j.bpj.2009.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prosser R.S., Davis J.H. Dynamics of an integral membrane peptide: a deuterium NMR relaxation study of gramicidin. Biophys. J. 1994;66:1429–1440. doi: 10.1016/S0006-3495(94)80933-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ten Kortenaar P.B.W., Vandijk B.G., Tesser G.I. Rapid and efficient method for the preparation of Fmoc-amino acids starting from 9-fluorenylmethanol. Int. J. Pept. Protein Res. 1986;27:398–400. [Google Scholar]
- 16.Gullion T., Schaefer J. Rotational-echo double-resonance NMR. J. Magn. Reson. 1989;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Gullion T., Schaefer J. Detection of weak heteronuclear dipolar coupling by rotational-echo double-resonance nuclear magnetic resonance. Adv. Magn. Reson. 1989;13:57–83. [Google Scholar]
- 18.Razdow A. MathSoft; Needham, MA: 1986. Mathcad. [Google Scholar]
- 19.Gullion T., Baker D.B., Conradi M.S. New, compensated Carr-Purcell sequences. J. Magn. Reson. 1990;89:479–484. [Google Scholar]
- 20.Frisch M.J., Pople J.A. Gaussian, Inc.; Wallingford, CT: 2004. Gaussian 03 Revision E.01. [Google Scholar]
- 21.Maseras F., Morokuma K. IMOMM a new integrated ab-initio plus molecular mechanics geometry optimization scheme of equilibrium structures and transition-states. J. Comput. Chem. 1995;16:1170–1179. [Google Scholar]
- 22.Vreven T., Morokuma K. On the application of the IMOMO (integrated molecular orbital plus molecular orbital) method. J. Comput. Chem. 2000;21:1419–1432. [Google Scholar]
- 23.Becke A.D. Density-functional thermochemistry. 3. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 24.Lee C., Yang W., Parr R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 25.Foresman A.E., Frisch A. Second edition. Gaussian; Pittsburgh: 1996. Exploring Chemistry with Electronic Structure Methods. [Google Scholar]
- 26.Dewar M.J.S., McKee M.L., Rzepa S. MNDO parameters for third-period elements. J. Am. Chem. Soc. 1978;100:3607. [Google Scholar]
- 27.Dewar M.J.S., Thiel W. MINDO/3 study of addition of singlet oxygen δ-G-1o-2 to 1,3-butadiene. J. Am. Chem. Soc. 1977;99:2338–2339. [Google Scholar]
- 28.Anders E., Koch R., Freunscht P. Optimization and application of lithium parameters for PM3. J. Comput. Chem. 1993;14:1301–1312. [Google Scholar]
- 29.Wolinski K., Sadlej A.J. Self-consistent perturbation-theory: open-shell states in perturbation-dependent non-orthogonal basis-sets. Mol. Phys. 1980;41:1419–1430. [Google Scholar]
- 30.Wolinski K., Hinton J.F., Pulay P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical-shift calculations. J. Am. Chem. Soc. 1990;112:8251–8260. [Google Scholar]
- 31.Ditchfield R. Self-consistent perturbation theory of diamagnetism I. A gauge-invariant LCAO method for NMR chemical shifts. Mol. Phys. 1974;27:789–807. [Google Scholar]
- 32.McWeeny R. Perturbation theory for the Fock-Dirac density matrix. Phys. Rev. 1962;126:1028–1034. [Google Scholar]
- 33.London F.J. Théorie quantique des courants interatomiques dans les combinaisons aromatiques. J. Phys. Radium. 1937;8:397–409. [Google Scholar]
- 34.Schmidt-Rohr K., Spiess H.W. Harcourt Brace; London: 1994. Multidimensional solid-state NMR and polymers. 444–452. [Google Scholar]
- 35.Czaplicki J., Cornélissen G., Halberg F.F. GOSA, a simulated annealing-based program for global optimization of nonlinear problems, also reveals transyears. J. Appl. Biomed. 2006;4:87–94. [PMC free article] [PubMed] [Google Scholar]
- 36.Bevington P., Robinson D.K. McGraw-Hill; New York: 2003. Data Reduction and Error Analysis for the Physical Sciences. 212–216. [Google Scholar]
- 37.Hahn T. CUBA: a library for multidimensional numerical integration. Comput. Phys. Commun. 2005;168:78–95. [Google Scholar]
- 38.Killian J.A., Salemink I., Greathouse D.V. Induction of nonbilayer structures in diacylphosphatidylcholine model membranes by transmembrane α-helical peptides: importance of hydrophobic mismatch and proposed role of tryptophans. Biochemistry. 1996;35:1037–1045. doi: 10.1021/bi9519258. [DOI] [PubMed] [Google Scholar]
- 39.Daily A.E., Greathouse D.V., Koeppe R.E. Helical distortion in tryptophan- and lysine-anchored membrane-spanning α-helices as a function of hydrophobic mismatch: a solid-state deuterium NMR investigation using the geometric analysis of labeled alanines method. Biophys. J. 2008;94:480–491. doi: 10.1529/biophysj.106.097543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ulmschneider J.P., Doux J.P.F., Ulmschneider M.B. Peptide partitioning and folding into lipid bilayers. J. Chem. Theory Comput. 2009;5:2202–2205. doi: 10.1021/ct900256k. [DOI] [PubMed] [Google Scholar]
- 41.Kol M.A., van Laak A.N.C., de Kruijff B. Phospholipid flop induced by transmembrane peptides in model membranes is modulated by lipid composition. Biochemistry. 2003;42:231–237. doi: 10.1021/bi0268403. [DOI] [PubMed] [Google Scholar]
- 42.Sparr E., Ash W.L., Killian J.A. Self-association of transmembrane α-helices in model membranes: importance of helix orientation and role of hydrophobic mismatch. J. Biol. Chem. 2005;280:39324–39331. doi: 10.1074/jbc.M502810200. [DOI] [PubMed] [Google Scholar]
- 43.Sparr E., Ganchev D.N., de Kruijff B. Molecular organization in striated domains induced by transmembrane α-helical peptides in dipalmitoyl phosphatidylcholine bilayers. Biochemistry. 2005;44:2–10. doi: 10.1021/bi048047a. [DOI] [PubMed] [Google Scholar]
- 44.Hartzell C.J., Whitfield M., Drobny G.P. Determination of the N-15 and C-13 chemical-shift tensors of L–C-13 alanyl-L- N-15 alanine from the dipole-coupled powder patterns. J. Am. Chem. Soc. 1987;109:5966–5969. [Google Scholar]
- 45.Marassi F.M. NMR of peptides and proteins in oriented membranes. Concepts Magn. Reson. 2002;14:212–224. [Google Scholar]
- 46.Wu C.H., Ramamoorthy A., Opella S.J. Simultaneous characterization of the amide H-1 chemical shift,H-1-N-15 dipolar, and N-15 chemical-shift interaction tensors in a peptide bond by 3-dimensional solid-state NMR spectroscopy. J. Am. Chem. Soc. 1995;117:6148–6149. [Google Scholar]
- 47.Cady S.D., Goodman C., Hong M. Determining the orientation of uniaxially rotating membrane proteins using unoriented samples: a 2H, 13C, and 15N solid-state NMR investigation of the dynamics and orientation of a transmembrane helical bundle. J. Am. Chem. Soc. 2007;129:5719–5729. doi: 10.1021/ja070305e. [DOI] [PubMed] [Google Scholar]
- 48.Yamaguchi S., Huster D., Hong M. Orientation and dynamics of an antimicrobial peptide in the lipid bilayer by solid-state NMR spectroscopy. Biophys. J. 2001;81:2203–2214. doi: 10.1016/S0006-3495(01)75868-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holt A., Koehorst R.B.M., Killian J.A. Tilt and rotation angles of a transmembrane model peptide as studied by fluorescence spectroscopy. Biophys. J. 2009;97:2258–2266. doi: 10.1016/j.bpj.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Özdirekcan S., Rijkers D.T.S., Killian J.A. Influence of flanking residues on tilt and rotation angles of transmembrane peptides in lipid bilayers. A solid-state 2H NMR study. Biochemistry. 2005;44:1004–1012. doi: 10.1021/bi0481242. [DOI] [PubMed] [Google Scholar]
- 51.Shi L., Cembran A., Veglia G. Tilt and azimuthal angles of a transmembrane peptide: a comparison between molecular dynamics calculations and solid-state NMR data of sarcolipin in lipid membranes. Biophys. J. 2009;96:3648–3662. doi: 10.1016/j.bpj.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goormaghtigh E., Raussens V., Ruysschaert J.M. Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim. Biophys. Acta. 1999;1422:105–185. doi: 10.1016/s0304-4157(99)00004-0. [DOI] [PubMed] [Google Scholar]
- 53.Prosser R.S., Davis J.H., Kothe G. Deuterium NMR relaxation studies of peptide-lipid interactions. Biochemistry. 1992;31:9355–9363. doi: 10.1021/bi00154a005. [DOI] [PubMed] [Google Scholar]
- 54.Mayer C., Grobner G., Kothe G. Orientation-dependent deuteron spin-lattice relaxation times in bilayer membranes: characterization of the overall lipid motion. Chem. Phys. Lett. 1990;165:155–161. [Google Scholar]
- 55.Holt A., Killian J.A. Orientation and dynamics of transmembrane peptides: the power of simple models. Eur. Biophys. J. 2010;39:209–621. doi: 10.1007/s00249-009-0567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fares C., Qian J., Davis J.H. Magic angle spinning and static oriented sample NMR studies of the relaxation in the rotating frame of membrane peptides. J. Chem. Phys. 2005;122:194908. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.