

Abstract
Limb-girdle muscular dystrophy type 2I (LGMD-2I) is caused by mutations in fukutin-related protein gene (FKRP) that lead to abnormal glycosylation of α-dystroglycan in skeletal muscle. Heart involvement in LGMD-2I is common, but little is known about underlying cardiac pathology. Here, we describe two patients with LGMD-2I (homozygous FKRP mutation c.826C>A, p.Leu276Ile) who developed severe congestive heart failure requiring cardiac transplantation. The dystrophic pathology and impairment of α-dystroglycan glycosylation were severe in the heart but mild in the skeletal muscle, underscoring the lack of correlation between cardiac and skeletal muscle involvement in some LGMD-2I patients.
Keywords: LGMD-2I, heart, pathology, α-dystroglycan, glycosylation, FKRP
Neuromuscular disorders caused by abnormal glycosylation of α-dystroglycan are both genotypically and phenotypically diverse. Mutations in fukutin-related protein gene (FKRP), which encodes a putative Golgi-based glycosyltransferase, are one of the genetic causes of abnormal α-dystroglycan glycosylation. The clinical spectrum associated with FKRP mutations is wide; most patients fall into one of two categories – congenital muscular dystrophy type 1C (MDC1C), a severe muscle disease with onset in the first few weeks of life,1 or limb-girdle muscular dystrophy type 2I (LGMD-2I), a much milder phenotype with onset in the first to third decade of life2. Rare FKRP patients may have structural eye and/or brain abnormalities reminiscent of muscle-eye-brain disease or Walker-Warburg syndrome3, while others have only hyperCKemia4. In the majority of patients of European descent, the LGMD-2I phenotype is associated with a relatively conservative c.826C>A (p.Leu276Ile) mutation, either in the homozygous or compound heterozygous state. In addition to muscular dystrophy, many LGMD-2I patients show cardiac involvement that varies from subclinical to severe: 16–89% of LGMD-2I patients have cardiac disease. The reported frequency depends on the series, diagnostic criteria and the diagnostic modality used5–10. Some patients even present with cardiomyopathy in the absence of overt skeletal muscle symptoms11,12. However, little is known about the pathology underlying the functional heart abnormalities. Here, we present cardiac pathology in two patients with LGMD-2I who underwent heart transplantation following development of severe congestive heart failure.
CASE REPORTS
Clinical history
Patient #1 was born at term via normal spontaneous vaginal delivery following an uncomplicated pregnancy. She met all early motor milestones including rolling over by 4 months and sitting alone by 6 months. At seven months of age, she began to develop cough and increased rate of breathing. After a chest X-ray demonstrated prominent cardiomegaly (Fig. 1A), she was admitted to the hospital, where myocardial biopsy led to a diagnosis of idiopathic cardiomyopathy. During the hospitalization, the patient had a cardiac arrest that required resuscitation; subsequently, she developed a seizure and was treated with phenobarbital. She underwent cardiac transplantation at age eight months. There were no significant postoperative complications, and she was discharged home at nine months; phenobarbital was gradually discontinued. At age ten months, the patient was hospitalized for possible rejection and was treated with high dose steroid therapy. She continued to be followed in the cardiology clinic and underwent routine endomyocardial biopsies, which were all normal.
Figure 1.

Chest X-rays showing severe cardiomegaly in patient #1 (panel A) and patient #2 (panel B).
On routine serum testing at age nine years, the patient was noted to have persistently elevated serum transaminase values and elevated creatine kinase level (5942 IU/L). At the time, she was in the second grade and was performing at grade level academically. She has had no cramping or muscle pain, and her parents report no concerns about weakness or clumsiness. While she was active in several recreational sports including soccer, volleyball, basketball, and gymnastics, the parents note that she is slower than her peers. The patient has had no respiratory symptoms, but complains of intermittent fatigue with exercise. Neurologic examination shows normal cranial nerve function, strong neck flexion and extension, but mild proximal weakness in deltoids, biceps, triceps, infraspinatus and iliopsoas muscles. She runs with a narrow based gait, albeit somewhat slowly for her age.
Patient #2 was also born at term via spontaneous vaginal delivery following a normal pregnancy. His parents and two siblings have no history of neurologic or cardiac disease. The patient’s early development was normal; he learned to walk at 10 months of age. At 19 months of age, he developed difficulties with walking and climbing that were accompanied by highly elevated creatine kinase levels (11,000 U/L). After a viral illness at the age of 4 years, the patient had difficulty raising his arms, but the symptoms gradually improved. According to his parents, a muscle biopsy performed at the time was abnormal, but it was negative for Duchenne’s muscular dystrophy; the slides and tissue were not available for review. The weakness progressed slowly. It affected predominantly the proximal lower extremities and showed exacerbations following illness or exertion. The patient was able to play sports until age 15 as a baseball pitcher. However, he could not run well, and between ages 15 and 17 he developed difficulty climbing stairs.
At the age of 17, the patient developed shortness of breath, paroxysmal nocturnal dyspnea, and right upper quadrant abdominal pain following a flu-like illness. Cardiac examination showed regular rate and rhythm, and he had a 2/6 systolic murmur at the apex that radiated to the axilla. Chest X-ray showed cardiomegaly with mild interstitial edema, but no pericardial effusion (Fig. 1B). Echocardiogram showed severe left ventricular enlargement and moderate left ventricular hypertrophy, reduction in left systolic function with an ejection fraction below 20%, global hypokinesis with some regional variation, and a moderate degree of mitral and tricuspid regurgitation. Neurologic examination showed good strength of neck flexion and extension. In the upper extremities, muscle strength was normal except for slight weakness of deltoid and infraspinatus muscles. In the lower extremities, there was mild weakness of hip flexion, abduction and adduction, but good strength of hip extension. The patient needed help to rise from a chair; however, he had no quadriceps weakness on manual examination. Below the knees, the strength was normal. The patient’s heart failure worsened, and he underwent cardiac transplantation one year after development of the first cardiac symptoms (at age 18).
Skeletal muscle pathology
Patient #1 underwent biopsy of the left deltoid at age 9; it showed mild myopathic features (scattered necrotic and regenerating muscle fibers, mild endomysial fibrosis, and moderate variation in muscle fiber diameters; Fig. 2A). Immunofluorescence staining of cryosections demonstrated normal staining for merosin (not shown), dystrophin (carboxy terminus antibody; Fig. 2D), β-dystroglycan (Fig. 2P), and α-dystroglycan core domain (GT20ADG antibody13; Fig. 2L). Immunostains for sarcoglycans, desmin, emerin, caveolin, and dysferlin were also normal (not shown). In contrast, staining with two different α-dystroglycan glycoepitope antibodies (IIH6 and VIA4-1) showed a mosaic pattern of absent to nearly normal staining that is characteristic of the milder dystroglycanopathies (Fig. 2H and not shown). Direct sequencing of the FKRP gene detected the common point mutation c.826C>A (resulting in p.Leu276Ile amino acid change) in both alleles, thus establishing the diagnosis of LGMD-2I.
Figure 2.

Skeletal muscle and cardiac cryosections from patient #1 and control patients were stained with H&E and a panel of antibodies using immunofluorescence methodology. While the muscle pathology in patient #1 is mild for LGMD 2I (A), the heart shows features of cardiomyopathy (variation in myocyte diameter, variation in nuclear size and shape, and fibrosis) that are relatively severe (B). Staining for dystrophin, β-dystroglycan, and the core domain of α-dystroglycan is normal in both the skeletal muscle and the heart of patient #1. In contrast, staining for glycosylated α-dystroglycan ranges from negative to normal in a mosaic pattern in patient #1’s skeletal muscle and is absent in her heart. Shown here are representative micrographs of staining with H&E (A–B) and dystrophin (C–F), glycosylated α-dystroglycan (VIA4-1, G–H; IIH6, I–J), core α-dystroglycan (GT20ADG, K–N) and β-dystroglycan antibodies (O–R). Control skeletal muscle, panels C, G, K and O. Patient #1’s skeletal muscle, panels A, D, H, L and P. Control heart, panels E, I, M and Q. Patient #1’s heart, panels B, F, J, N and R. Scale bars, 100 μm.
Patient #2 underwent a repeat biopsy of the left quadriceps femoris at age 17. It showed myopathic features consistent with a mild muscular dystrophy: there was moderate variation in muscle fiber diameters, scattered necrotic and regenerating muscle fibers, frequent split fibers, mild-moderate endomysial fibrosis, and a significant increase in central nuclei (Fig. 3A). While myopathic features were more severe than in patient #1, immunofluorescence staining of cryosections demonstrated the same staining pattern: normal staining for dystrophin (rod domain and carboxy terminus), sarcoglycans, utrophin, merosin, β-dystroglycan, and core domain of α-dystroglycan (Figs. 3D and 3E, or not shown), and a mosaic pattern of absent to nearly normal staining with IIH6 and VIA4-1 antibodies (Figs. 3B and 3C). As in the case of patient #1, direct sequencing of the FKRP gene detected the common point mutation c.826C>A (p.Leu276Ile) in both alleles.
Figure 3.

Skeletal muscle cryosections from patient #2 were stained with H&E and a panel of antibodies using immunofluorescence methodology. The muscle pathology is relatively mild for LGMD-2I (A), but more severe than in patient #1 (compare with Fig. 2A). Staining for the core domain of α-dystroglycan (D) and β-dystroglycan (E) is normal. In contrast, staining for glycosylated α-dystroglycan ranges from negative to normal in a mosaic pattern that is characteristic for LGMD-2I (VIA4-1 antibody, panel B; IIH6 antibody, panel C). Scale bars, 200 μm.
Cardiac pathology
The explanted heart of patient #1 showed cardiomegaly with marked left ventricular dilatation (heart weight, 78 g [expected weight, 41±7 g]; left ventricular wall thickness, 0.7 cm). There was significant muscle fiber hypertrophy and moderate interstitial fibrosis (cryosection, Fig. 2B; paraffin-embedded section, Fig. 4C). Immunofluorescence staining of cryosections demonstrated normal labeling for dystrophin (Fig. 2F), β-dystroglycan (Fig. 2R), and α-dystroglycan core domain (Fig. 2N). In contrast, staining with α-dystroglycan glycoepitope antibodies (IIH6 and VIA4-1) showed a complete absence of labeling (Fig. 2J and not shown). Similarly, immunoperoxidase staining of paraffin sections following proteinase K-mediated antigen retrieval showed positive dystrophin (not shown) and absent IIH6 staining (Fig. 4D).
Figure 4.
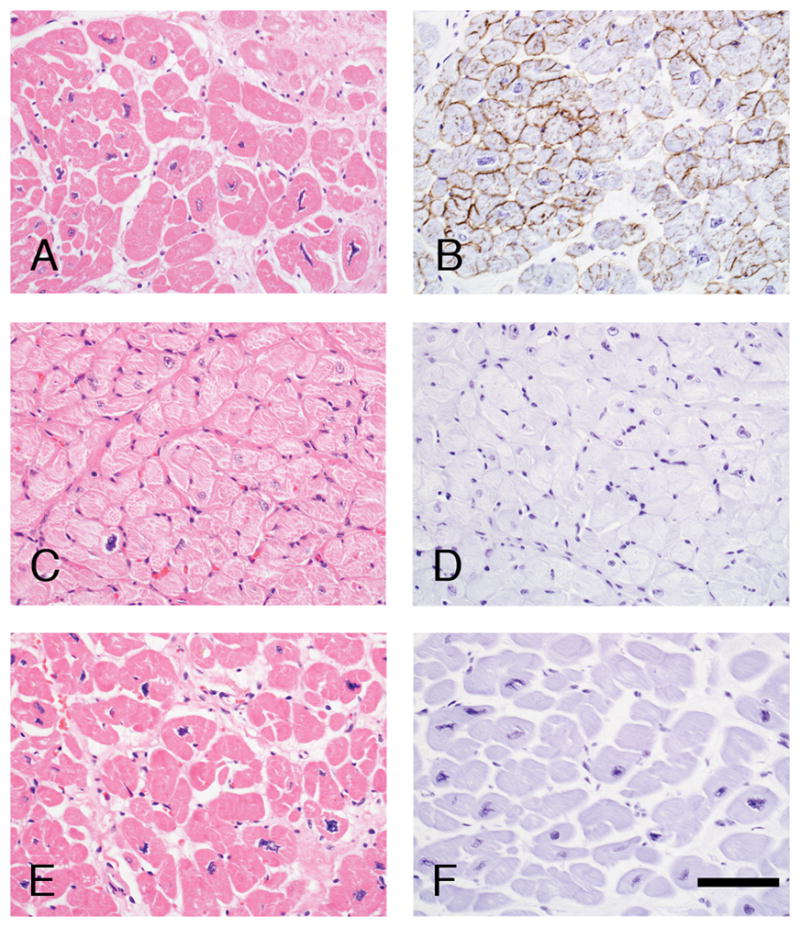
Paraffin sections from a control patient who was transplanted for treatment of severe ischemic cardiomyopathy (panels A and B), patient #1 (panels C and D), and patient #2 (panels E and F) were stained with H&E (panels A, C and F) or the α-dystroglycan glycoepitope specific antibody, IIH6, using an immunoperoxidase methodology (panels B, D and F). While nearly all cardiac myocytes stain strongly in the ischemic cardiomyopathy heart, α-dystroglycan is negative in both LGMD-2I patients. Scale bar, 100 μm.
The explanted heart of patient #2 showed biventricular hypertrophy and dilatation (heart weight, 570 g [expected weight, 320±40 g]; ventricular wall thickness, 1.3 cm on the left and 0.7 cm on the right); valves and coronary arteries were normal. There was marked endocardial sclerosis, myocyte hypertrophy, and interstitial fibrosis with extensive fatty replacement (Fig. 4E). Frozen heart tissue was not available for analysis. However, immunoperoxidase staining of paraffin sections following proteinase K-mediated antigen retrieval showed positive dystrophin (not shown) and absent IIH6 staining (Fig. 4F) – a pattern similar to, but more severe, than the one observed in the patient’s skeletal muscle and essentially the same as the one seen in the heart of patient #1.
DISCUSSION
The two LGMD-2I patients described in this report developed mildly progressive muscular dystrophy in childhood and severe congestive heart failure in infancy (patient #1) or the mid-late teens (patient #2). In concordance with this clinical presentation, the cardiac pathology was much more severe than the skeletal muscle pathology in both cases. There was prominent cardiac myocyte hypertrophy, interstitial fibrosis, and fatty replacement leading to ventricular hypertrophy and dilatation. Cardiac muscle fibers also showed a more severe reduction in immunoreactive glycosylated α-dystroglycan than skeletal muscle fibers. Interestingly, however, frank myocyte necrosis was observed only in the skeletal muscle specimens from both patients. Although patient #1 developed heart failure much earlier in life than patient #2, the pathologic findings and α-dystroglycan staining pattern (normal with the core antibody but absent with the glycoepitope-specific antibodies) were the same in both patients.
The lack of correlation between the severity of cardiac and skeletal muscle involvement in LGMD-2I was noted in two recent clinical series9,10. In addition, clinical disparity between skeletal muscle and cardiac function was recently reported in another LGMD-2I patient who was homozygous for p.Leu276Ile mutation in FKRP gene; he underwent cardiac transplantation for treatment of severe dilated cardiomyopathy at the age of eight12. At the time of report, that patient was 20 years old and had normal skeletal muscle bulk and strength, but showed subclinical dystrophic features on lower extremity imaging. The minimal skeletal muscle involvement in this patient and our patient #1 (the two patients who underwent cardiac transplantation very early in life) raises the possibility that skeletal muscle pathology might have been mitigated by immunosuppressive therapy, particularly given the responsiveness to steroids noted in some dystroglycanopathies14.
While the risk for development of cardiac complications has been associated with some FKRP gene mutations (for example, c.1364C>A15), it is currently not clear whether the homozygous or heterozygous state for the common c.826A>C (p.Leu276Ile) mutation results in a higher risk of cardiac involvement. In one recent series7, it was reported that heterozygous patients were at higher risk, with 66% of heterozygotes but only 4% of homozygotes developing cardiac involvement by 20 years of age. In contrast, another recent study5 reported a higher risk of dilated cardiomyopathy in homozygous rather than heterozygous patients (19 vs. 9%, respectively), while two other studies9,10 found no difference between the two genotypes. Interestingly, the three LGMD-2I patients who required cardiac transplantation for treatment of severe heart failure (one described by D’Amico et al.12 and two in the current report) were homozygous for the common European mutation (p.Leu276Ile). Together, these data raise the possibility that cardiac involvement in homozygous patients is less frequent but more severe than in their heterozygous counterparts. However, a larger series will be required to clarify whether this represents a statistically significant trend.
In summary, there are now three cases that demonstrate that LGMD-2I patients who develop dilated cardiomyopathy have impairment of α-dystroglycan glycosylation and pathology in the cardiac muscle that is more severe than in the skeletal muscle.
Supplementary Material
Acknowledgments
This work was supported by NIH grant NS053672. GT20ADG and IIH6 antibodies were a gift from Kevin P. Campbell, The University of Iowa.
ABBREVIATIONS
- CK
creatine kinase
- FKRP
fukutin-related protein
- LGMD-2I
limb-girdle muscular dystrophy type 2I
Footnotes
A part of this work was presented at the Annual Meeting of the American Association of Neuropathologists in San Diego (April 2008).
References
- 1.Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. Am J Hum Genet. 2001;69:1198–1209. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brockington M, Yuva Y, Prandini P, Brown SC, Torelli S, Benson MA, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet. 2001;10:2851–2859. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- 3.Beltran-Valero de Bernabe D, Voit T, Longman C, Steinbrecher A, Straub V, Yuva Y, et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. 2004;41:e61. doi: 10.1136/jmg.2003.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Paula F, Vieira N, Starling A, Yamamoto LU, Lima B, de Cassia Pavanello R, et al. Asymptomatic carriers for homozygous novel mutations in the FKRP gene: the other end of the spectrum. Eur J Hum Genet. 2003;11:923–930. doi: 10.1038/sj.ejhg.5201066. [DOI] [PubMed] [Google Scholar]
- 5.Sveen ML, Schwartz M, Vissing J. High prevalence and phenotype-genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann Neurol. 2006;59:808–815. doi: 10.1002/ana.20824. [DOI] [PubMed] [Google Scholar]
- 6.Mercuri E, Brockington M, Straub V, Quijano-Roy S, Yuva Y, Herrmann R, et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol. 2003;53:537–542. doi: 10.1002/ana.10559. [DOI] [PubMed] [Google Scholar]
- 7.Poppe M, Bourke J, Eagle M, Frosk P, Wrogemann K, Greenberg C, et al. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann Neurol. 2004;56:738–741. doi: 10.1002/ana.20283. [DOI] [PubMed] [Google Scholar]
- 8.Gaul C, Deschauer M, Tempelmann C, Vielhaber S, Klein HU, Heinze HJ, et al. Cardiac involvement in limb-girdle muscular dystrophy 2I: Conventional cardiac diagnostic and cardiovascular magnetic resonance. J Neurol. 2006;253:1317–1322. doi: 10.1007/s00415-006-0213-0. [DOI] [PubMed] [Google Scholar]
- 9.Sveen ML, Thune JJ, Kober L, Vissing J. Cardiac involvement in patients with limb-girdle muscular dystrophy type 2 and Becker muscular dystrophy. Arch Neurol. 2008;65:1196–1201. doi: 10.1001/archneur.65.9.1196. [DOI] [PubMed] [Google Scholar]
- 10.Wahbi K, Meune C, Hamouda EH, Stojkovic T, Laforet P, Becane HM, et al. Cardiac assessment of limb-girdle muscular dystrophy 2I patients: an echography, Holter ECG and magnetic resonance imaging study. Neuromuscul Disord. 2008;18:650–655. doi: 10.1016/j.nmd.2008.06.365. [DOI] [PubMed] [Google Scholar]
- 11.Muller T, Krasnianski M, Witthaut R, Deschauer M, Zierz S. Dilated cardiomyopathy may be an early sign of the C826A Fukutin-related protein mutation. Neuromuscul Disord. 2005;15:372–376. doi: 10.1016/j.nmd.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 12.D’Amico A, Petrini S, Parisi F, Tessa A, Francalanci P, Grutter G, et al. Heart transplantation in a child with LGMD2I presenting as isolated dilated cardiomyopathy. Neuromuscul Disord. 2008;18:153–155. doi: 10.1016/j.nmd.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 13.Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 14.Godfrey C, Escolar D, Brockington M, Clement EM, Mein R, Jimenez-Mallebrera C, et al. Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophy. Ann Neurol. 2006;60:603–610. doi: 10.1002/ana.21006. [DOI] [PubMed] [Google Scholar]
- 15.Kefi M, Amouri R, Chabrak S, Mechmeche R, Hentati F. Variable cardiac involvement in Tunisian siblings harboring FKRP gene mutations. Neuropediatrics. 2008;39:113–115. doi: 10.1055/s-2008-1081465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.