

Abstract
Connexin26 is a ubiquitous gap junction protein that serves critical homeostatic functions. Four single-site mutations found in the transmembrane helices (M1–M4) cause different types of dysfunctional channels: 1), Cx26T135A in M3 produces a closed channel; 2), Cx26M34A in M1 severely decreases channel activity; 3), Cx26P87L in M2 has been implicated in defective channel gating; and 4), Cx26V84L in M2, a nonsyndromic deafness mutant, retains normal dye coupling and electrophysiological properties but is deficient in IP3 transfer. These mutations do not affect Cx26 trafficking in mammalian cells, and make normal-appearing channels in baculovirus-infected Sf9 membranes when imaged by negative stain electron microscopy. Upon dodecylmaltoside solubilization of the membrane fraction, Cx26M34A and Cx26V84L are stable as hexamers or dodecamers, but Cx26T135A and Cx26P87L oligomers are not. This instability is also found in Cx26T135A and Cx26P87L hemichannels isolated from mammalian cells. In this work, coexpression of both wild-type Cx26 and Cx26P87L in Sf9 cells rescued P87L hexamer stability. Similarly, in paired Xenopus oocytes, coexpression with wild-type restored function. In contrast, the stability of Cx26T135A hemichannels could not be rescued by coexpression with WT. Thus, T135 and P87 residues are in positions that are important for oligomer stability and can affect gap junction gating.
Introduction
Gap junctions (GJs) serve important functions for direct intercellular communication between most cell types in all metazoan species. They play dynamic roles in developmental regulation and signal transduction pathways, providing a low-resistance pathway for metabolites and ions. Small molecules and ions diffuse passively through GJ channels that span the bilayers of both cells and the extracellular space that separate them. The proteins that make up the GJ channels are called connexins (Cxs), and the sequence of each isoform influences which specific signals can pass. The expression and function of Cxs within their native cellular environment involve a highly regulated process. Mutations in Cx genes are the cause of several hereditary diseases (connexinopathies). These mutations can disrupt intercellular communication by affecting Cx synthesis, trafficking, docking, and functionality.
Connexin26 (Cx26) is the second smallest of the GJ protein family and one of the most prevalent members. It is highly expressed in many major organ systems, such as the liver, brain, kidney, intestines, and skin, where it is thought to function as part of many homeostatic mechanisms. Point mutations derived from the genomic DNA of many patients occur throughout the entire Cx26 sequence and are the major cause of nonsyndromic sensorineural deafness and ectodermal dysplasia (also known as keratitis-ichthyosis-deafness syndrome) (1), a rare disorder affecting both the inner ear and skin. Cx26 mutations account for over half of all congenital cases of hearing impairment (2). It has been proposed that defective GJ communication results in a malfunctioning of K+ recycling that leads to apoptosis of the endothelial cells underlying the hair cells in the inner ear, impairment of ionic transmission in the sensory neurons, and disruption of the endocochlear potential (3). In a previous study (3), targeted ablation of Cx26 in the inner ear of mice resulted in normal formation of the inner ear; however, inner hair cells began to die 14 days postnatally, presumably due to sound stimulation in combination with insufficient molecular transfer of K+, Ca2+, or glutamate via GJs. The lethality of the Cx26 knockout in mice illustrates the critical role that Cx26 plays during pregnancy and embryogenesis (particularly placenta formation) in this species (4).
Cxs assemble as hexamers, either homo- or heteromerically, to form connexons or hemichannels within each cell, which upon docking in turn form intercellular GJ channels. Comparisons of the amino acid sequences of various Cxs have shown that the four membrane-spanning domains and the two extracellular loops are the most conserved domains, and the most variable sequences are found in the cytoplasmic central loop and C-terminal (CT) domains (5). The cytoplasmic domains are highly flexible and play important roles in gating and binding of cytoplasmic proteins (5). The four membrane-spanning segments (M1–M4) form a four-α-helical bundle, so upon oligomerization, a closed cylindrical structure is obtained with one major helix (and possibly part of a second) from each subunit contributing to the pore lining of the channel. Two published structures resolved at 7–10 Å (6,7) and a recent x-ray crystal structure resolved at 3.5 Å (8) contain four transmembrane rods corresponding to these α-helices (6,7). The N-terminus (NT), the connecting loop between the M2 and M3 helices (referred to as the cytoplasmic loop (CL)) and the CT comprise the cytoplasmic domains. Each Cx contributes two extracellular loops (referred to as E1 and E2) to the extracellular domain, which mediates docking between cells.
In this study, we examined four mutations found in the transmembrane α-helices: one in M1 (M34A), two in M2 (V84L and P87L), and one in M3 (T135A). These mutations were chosen because they form GJ structures in mammalian cells but function differently from the wild-type (WT) and each other. These functions include 1), forming a completely closed channel (T135A (9)); 2), forming a channel with aberrant gating properties distinct from WT (M34A (10); M. M. Toloue, I. M. Skerrett, and B. J. Nicholson, unpublished results); 3), forming a nonfunctional homotypic channel, but functional rectifying channel, when paired with WT (P87L (11)); and 4), eliminating IP3 signaling while maintaining dye transfer and normal electrophysiological gating properties (12). In addition, Cx26V84L causes some cases of nonsyndromic hereditary deafness (13), and two of these mutations are correlates of disease mutations found in other isoforms. Cx43T154A causes cases of oculo-dento-digital dysplasia (ODDD) (14), and Cx50P88S contributes to congenital cataracts in some patients (15). The M34A mutation in Cx26 is a correlate of a deafness mutant, M34T, which has been characterized in depth (16) and is the one from which an electron microscopy (EM) crystal structure to 10 Å has been determined. This same mutation is also found in Cx32 in some cases of Charcot-Marie-Tooth X-linked disease (CMTX) (17). In this work, we sought to determine how these mutations affect hemichannel stability, and, in the case of any detected instabilities, whether they can be rescued by heteromeric interactions with WT subunits (of relevance to the dominant or recessive nature of the diseases caused by these mutants). The results contribute to our knowledge base of Cx26 intra- and intersubunit amino acid interactions, which are important for channel assembly and function.
Materials and Methods
For details of the materials and methods used in this work, see the Supporting Material.
Results
Mutants within the transmembrane regions of Cxs have been associated with various diseases and diverse phenotypes. We investigated a select group of mutants in Cx26 that are functionally aberrant in different ways from WT and each other to ascertain whether there are common themes in terms of channel assembly and function. These four mutants express well at the plasma membrane with little or no intracellular accumulations (7,9,12,18). We focused on obtaining specimens for WT Cx26 channels and hemichannels, and four single-site mutations (Fig. S1):
-
1.
Cx26T135A: A closed pore mutation in the third transmembrane helix (9).
-
2.
Cx26M34A: A mutation in the first transmembrane helix that causes dramatically reduced conductance but no change in voltage gating. Analogous mutations have been identified in nonsyndromic hereditary deafness and CMTX.
-
3.
Cx26P87L: A mutant that disrupts channel gating by transjunctional voltage such that it is nonfunctional homotypically but forms a rectifying channel when paired heterotypically with Cx26WT. This site-specific mutation is in a central position in the second transmembrane helix (11). An analogous mutant in Cx50 (P88S) causes cataracts as a result of mistrafficking, forming large GJ-like accumulations in the cytoplasm (15).
-
4.
Cx26V84L: A nonsyndromic deafness pore mutant in the second transmembrane helix that eliminates the passage of a signaling molecule, IP3, but still retains normal dye coupling, conductance, and voltage gating (12).
All four mutants form GJs or single connexon layers in Sf9 membranes
As part of this study, we developed a hybrid protocol from two other protocols we commonly use to purify GJs from cells using a His6 tag genetically appended to the CT. A previous protocol developed in our laboratory for isolating GJs from tissue culture cells relied solely on buoyant density gradient centrifugation after the use of three different detergents (Sarcosyl, Brij 58, and Saponin (19)) to separate GJs from other cellular material. However, the quantity and quality of the preparations are strongly dependent on high expression from cells, and the preparations contain other membranes that have the same density as GJ plaques or single membranes containing connexons. The new protocol we developed is a hybrid of the procedure developed by Hand et al. (19), followed by the Ni-NTA bead purification scheme typically used for our protein purification procedure (9,10). In this procedure, the detergent-insoluble fraction from the Saponin treatment is incubated with Ni-NTA beads overnight and allowed to bind. This batch approach significantly increases the yield of Cx26-V5-His6 membranes compared to that obtained using a preformed Ni-NTA column. Beads are separated from non- or weakly attached material by several low-speed spins in a noneluting buffer, and collected after incubation in an eluting buffer containing a high concentration of imidazole.
The micrographs shown in Fig. 1 A are typical images of Sf9 membranes containing Cx26WT and the four Cx26 mutants. These membranes were stained with 2% uranyl acetate and imaged with transmission EM. Cx26WT (Fig. 1 A, top left) and mutant channels (V84L, M34A, T135A, and P87L; Fig. 1 A) appear as doughnut-like structures in a close-packed arrangement. As we noted previously (9), Sf9-derived membranes containing overexpressed Cx26 channels tend to be more disordered than those obtained in mammalian cells using similar procedures, although the channels are still easily identified (Fig. 1 A, insets). At this level of resolution and under high electron beam irradiation, the mutant channels are indistinguishable from WT hexamers or dodecamers. The purity of the preparation is illustrated in a silver-stained gel and equivalent Western blot of a Cx26V84L preparation that shows that the primary bands are specific for Cx26 (Fig. 1 A, lower left). A comparison of WT Cx26 material prepared by our previously published method (19) is shown for comparison in Fig. 1 B. Note the increased number of nonspecific bands that were not identified in the Cx26 Western blot.
Figure 1.
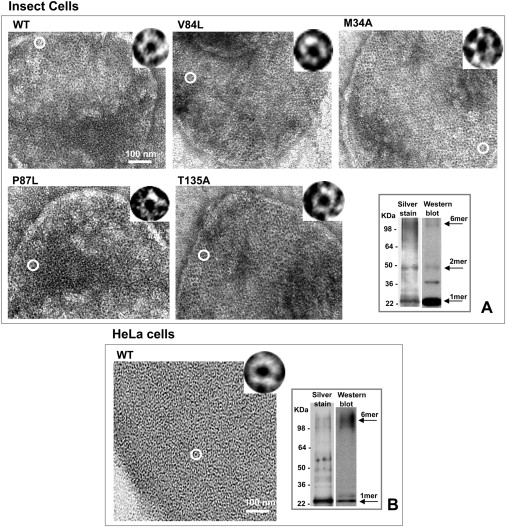
Isolated membranes from Sf9 insect cells infected with Cx26 WT and mutant baculoviruses. (A) Electron micrographs of negatively stained membrane fraction show that the WT and pore mutants are able to form GJ channels. At this resolution and high-dose conditions, the mutant channels are indistinguishable from WT. The circle indicates a single channel in each membrane that is 2× magnified in the inset at the top of each micrograph. A protein gel and Western blot from a Cx26V84L preparation show good purity (A, bottom right) as compared to a more nonspecific protocol using untagged Cx26WT in HeLa cells, seen in negative stain and on a gel and Western blot (B, left and right, respectively).
Cx26M34A and Cx26V84L connexons are stable, but Cx26P87L and Cx26T135A are not, after detergent solubilization and purification from Sf9 cells
Since these Cx26 proteins assemble into channels and hemichannels, we investigated whether hexamer stability is important for maintaining channel function. It was previously shown that Cx26M34A connexons are stable upon detergent solubilization (10), but those of Cx26T135A are not (9). Here, we also tested the stability of two other Cx26 hemichannels with mutations in the M2 helix. We used two methods to assess the stability of hemichannels: visual inspection of electron micrographs of negatively stained preparations (Fig. 2 A), and Blue native (BN) gel analysis of oligomeric associations (Fig. 2 B). Detergent-stable WT, Cx26V84L, and Cx26M34A all appear as homogeneous, doughnut-like structures, with the stain in the pore being particularly clear (Fig. 2 A, top row). These particles were previously reported to be mainly hexameric connexons (20). The BN gel Western blot analysis of these three preparations contains only two bands—a stronger one at the hexamer position and a slightly weaker one at the dodecamer position (Fig. 2 B, top row)—indicating that, although the hexamer may be the predominant species, there remain some dodecameric associations. In contrast, electron micrographs of Cx26P87L and Cx26T135A contain a variety of structures, ranging from units that look similar to those of stable mutants, to much smaller doughnut-like structures and unidentifiable aggregates (Fig. 2 A, bottom row). The BN gel banding patterns, which contain several bands ranging from monomer to dodecamer, confirm this heterogeneity.
Figure 2.
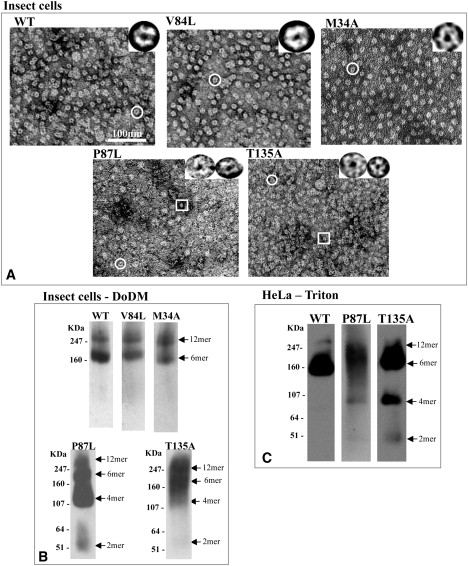
Connexons purified from Sf9 membrane lysate by His6-tag and nickel resin purification after solubilization with 2% DoDM. (A) Cx26 T135A and P87L mutants form partial connexons and aggregates as visualized by EM, whereas WT, V84L, and M34A form stable, classic doughnut-shaped structures. A circle highlights a representative connexon structure, and a rectangle indicates a smaller oligomer that is only visible in the mutants that are unstable in DoDM. These are shown at higher magnification in the insets in the panel A micrographs. (B) Western blots of BN gels confirm the stability or instability of the hexamer seen in the electron micrographs. The two bands for the WT, M34A, and V84L mutants correspond to the hexameric and dodecameric forms, whereas the ladder of bands seen in the T135A and P87L mutant connexons indicate that they are unstable. (C) Western blots of BN gels of Cx26 channels from HeLa cells after solubilization with 1% Triton.
Cx26P87L and Cx26T135A instability is confirmed in mammalian membranes using 1% Triton X-100 as the solubilizing detergent
We also isolated membranes from HeLa cells stably expressing rCx26WT, rCx26P87L, or rCx26T135A to determine whether the instability of Cx26P87L and Cx26T135A is also evident in mammalian cells. We prepared the membranes using the same procedure employed for the insect cells. The mammalian membrane preparations were not completely solubilized with 2% dodecyl-maltoside (DoDM), but were soluble in 1% Triton X100. This solubility difference is mostly likely a reflection of the unique lipid composition of GJs in mammalian cells (21). BN gel and Western blots of Cx26P87L and Cx26T135A from Triton X-100 solubilized mammalian cells showed increased evidence, compared to WT, of oligomeric forms smaller than hexamers (Fig. 2 C), as was the case in DoDM extracts from Sf9 cells. Cx26P87L preparations showed less-intense, lower-molecular-weight bands on these gels than Cx26T135A, indicating that it may be more stable than Cx26T135A, but still not as stable as Cx26WT.
Assembly and degradation of rCx26T135A-4C and rCx26P87L-4C mutants are similar to those of WT proteins when stably expressed in HeLa cells
To determine whether hexamer instabilities have an effect on GJ trafficking, we studied the assembly and degradation of rCx26T135A and rCx26P87L stably expressed in communication-deficient HeLa cells and compared them with those of a cell line exogenously expressing rCx26WT. An optical pulse-chase technique was used to image different spatiotemporal protein populations (22). Both WT and mutant proteins tagged with the tetracysteine tag 4C (23) accumulated on the cell surface and concentrated at cell-cell interfaces in the punctate distribution characteristic of GJ plaques. The labeling obtained with profluorescent FlAsH-EDT2 (green) or profluorescent ReAsH-EDT2 (red) was specific, as indicated by colocalization with the antibody staining (data not shown). HeLa cells stably expressing rCx26WT-4C, rCx26T135A-4C, or rCx26P87L-4C were stained with FlAsH-EDT2 (green), incubated for 7 h in regular medium (chase time), and then labeled with ReAsH-EDT2 (red), fixed, and imaged (Fig. 3, A–C). Both WT and mutant proteins exhibited the typical GJ formation/removal when new GJ channels were added at the edges of the plaque adjacent to older ones. Reversing the temporal order of FlAsH-EDT2 (green) and ReAsH-EDT2 (red) applications produced a similar distribution, but with a reversed color pattern (data not shown). Regardless of the temporal order of labeling, we observed that the segregation of green- and red-labeled Cxs occurred predominantly asymmetrically, such that one side of the plaque elongated more than the opposite portions.
Figure 3.
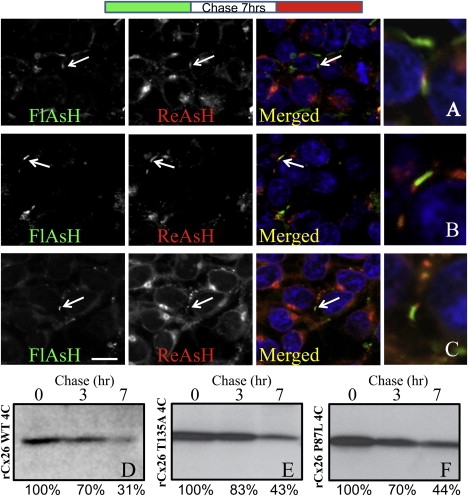
Trafficking analysis of tetracysteine-tagged rCx26T135A and rCx26P87L as compared to rCx26WT. Optical pulse-chase analysis of (A) rCx26WT-4C, and (B) rCx26T135A-4C and (C) rCx26P87L-4C mutants stably expressed in HeLa cells. (Diagram at top) Cells were labeled with FlAsH-EDT2, followed by a chase time of 7 h (white), and then labeled with ReAsH-EDT2 (red). Individual fluorophores are shown as grayscale images, but merged images are colorized with FlAsH-EDT2 as green and ReAsH-EDT2 as red. White arrows indicate the newer proteins (red) added at the edges of the plaques adjacent to the older ones (green), and a larger-magnification image of these plaques is shown. For an analysis of GJ degradation, these same HeLa cell lines were labeled with Sulfo-NHS-SS-Biotin for 30 min at 37°C and then chased for 0, 3, or 7 h. (D–F) rCx26 was immunoprecipitated from equal volumes of total cell lysate (and therefore from equal numbers of cell equivalents) and analyzed by SDS-PAGE. There were no significant differences in trafficking kinetics for synthetic and degradative pathways between Cx26WT and these two mutants.
To assess the level of degradation of Cx26WT and mutant Cxs from the plasma membrane, we used cell-surface biotinylation with a pulse-chase approach (24). In brief, HeLa cell monolayers were incubated at 4°C with a membrane-impermeant protein biotinylation reagent. After 30 min, the biotinylation reaction was quenched and the cells were chased at 37°C with tissue culture medium for various periods of time. As shown in Fig. 3, D–F, the two rCx26 mutant proteins at the cell surface degraded at a rate similar to that observed for the WT protein (with half-lives of ∼5–6 h). Considering that the mutants are properly assembled in plaques but functionally defective, we hypothesize that the two mutations cause only a structural instability that likely affects movement of subunits in opening or closing the channels. Thus, although the oligomer stability affects the function of the channel, the trafficking patterns are unaffected.
Hexamers can be rescued by expression of Cx26P87L with WT to form heteromeric oligomers
We further explored the question of how one of these unstable mutants, Cx26P87L, can affect hexamer stability by coinfecting Sf9 cells with Cx26P87L and increasing amounts of untagged WT (Cx26WT). In this rescue assay, only Cxs containing the V5-His6 tag were purified, which in this case were either monomers, oligomers of Cx26P87L, or heteromeric Cx26P87L/Cx26WT associations. As a control experiment, only untagged Cx26WT was expressed and purified from infected Sf9 cells. Electron micrographs of these preparations did not show any connexon-like structures or protein aggregates, and a Western blot BN gel or denaturing polyacrylamide gel electrophoresis (PAGE) analysis contained no specific Cx26 bands at all (data not shown). Therefore, we are confident that this rescue assay is a reliable indicator that we isolated only proteins containing the His6 tag.
We used the ratio of the mutant and WT multiplicity of infection (MOI) values as the variable for testing whether heteromeric hemichannel structure can be rescued with WT. The MOI is used as a diagnostic for the level of protein expression in the baculovirus/Sf9 system. The results of a typical rescue experiment are shown in Fig. 4. For MOI ratios < 1 (Fig. 4, A–C), full rescue was not possible. EM images show a variety in the size of the structures, confirming the presence of lower oligomeric states (middle left column) seen also in the nondenaturing gels (middle right column). Only at MOI ratios of ∼≥1 did we see a recovery of connexon structures in both electron micrographs and BN gels (Fig. 4, D and E). However, these preparations do not look as homogeneous as the WT or stable mutant preparations shown in Fig. 4 A (top row) and may be more susceptible to negative stain drying or the low pH of the uranyl acetate, which could cause some aggregation.
Figure 4.
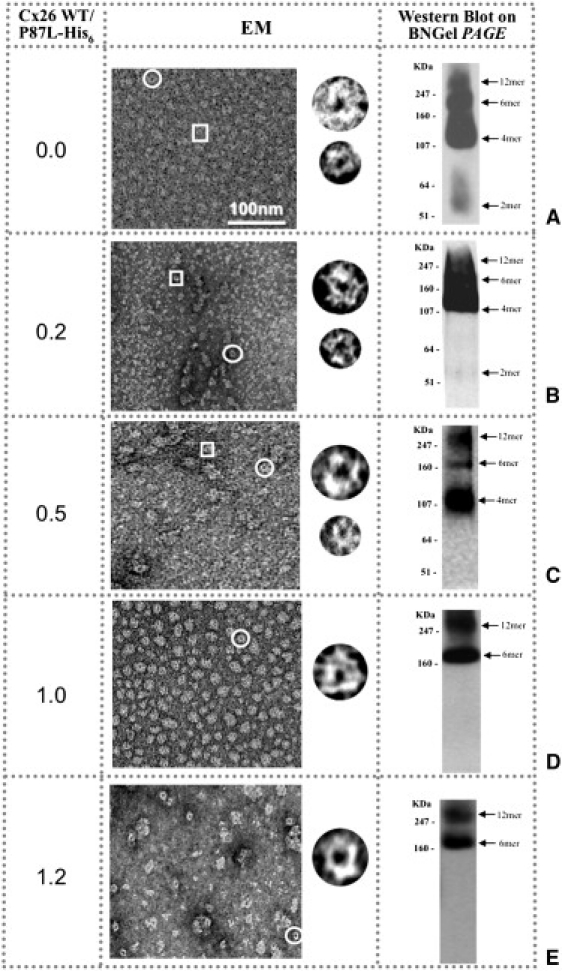
Cx26P87L mutant channels can be rescued by coexpression with Cx26WT to form recognizable doughnut-like structures. Visualizations with EM (middle column) and Western blot BN gel PAGE (BNgel, right column) are used as diagnostics of hemichannel stability. (A–C) The His6-tagged-based affinity protein purification reveals that hemichannels formed by Cx26WT and Cx26P87L-V5-His6 are unstable when the ratio of the WT MOI to Cx26P87L is formed primarily from this mutant. (D and E) The hemichannel structure is maintained when the ratio of the MOI for Cx26WT and Cx26P87L is ∼ or > 1:1. Circles indicate larger doughnut-shaped structures, and rectangles highlight smaller ones. Examples of both are shown in the column to the right of the micrograph fields.
Functionality is restored by coexpression of Cx26P87L with Cx26WT in Xenopus oocytes
Since Cx26WT was able to rescue the Cx26P87L hexamer structure, we utilized Xenopus oocytes to test whether Cx26WT-Cx27P87L heteromers were functional. Cx26WT GJ channels close symmetrically in response to high transjunctional voltages (Fig. 5 A) and also show a slight sensitivity to transmembrane voltage (11). Cx26WT-Cx26P87L pairs form heterotypic junctions with conductances 50-fold lower than that of WT-WT pairs (Fig. 5 F). This conductance is lowest at resting and negative potentials, and is activated when positive transjunctional voltages are applied relative to the Cx26P87L expressing cell (Fig. 5 D). This is the reverse response to WT channels. Homotypic pairings of Cx26P87L do not yield detectable currents, because with either voltage polarity, the hemichannel at the negative voltage pole will always be in the closed configuration.
Figure 5.
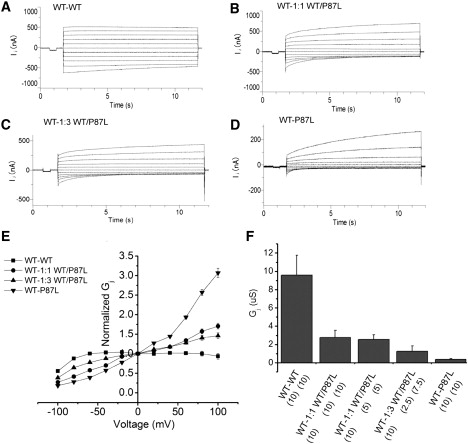
Cx26WT rescues Cx26P87L channel function. (A–D) Superimposed traces of transjunctional currents between pairs of oocytes in response to 10 s voltage pulses ranging from −100 mV to +100 mV in 20 mV increments. One oocyte was injected with 10 ng Cx26WT cRNA and paired with oocytes injected with (A) 10 ng WT cRNA, (B) 10 ng 1:1 WT/P87L cRNAs, (C) 10 ng 1:3 WT/P87L cRNAs, or (D) 10 ng P87L cRNA alone. (E) The normalized steady-state transjunctional conductances of WT-WT (squares), WT-1:1 WT/P87L (circles), WT-1:3 WT/P87L (triangles), and WT-P87L (inverted triangles) are plotted against the transjunctional voltage (E). The steady-state conductance is normalized by dividing the current amplitude at the end of the 10 s voltage step by the initial current amplitude. Voltage polarity is defined as positive for depolarizing voltage steps in WT-WT pairs, or with respect to depolarization in the non-WT side for other pairs. The conductance of each pair was measured by a 10 mV pulse before the incrementing voltage steps (F) transjunctional conductance (Gj). Mean ± SE of five to six pairs in each group.
To study the function of heteromeric channels of Cx26WT and Cx26P87L, we paired oocytes injected with 10 ng Cx26WT with oocytes coinjected with mixtures of Cx26WT and Cx26P87L RNAs at ratios of 1:1 and 1:3 (10 ng total injected RNA). The heteromeric mixtures formed by Cx26WT and P87L produced gating properties consistent with a weighted average of mutant and WT channels (Fig. 5, B and C). This is most evident in the plots of transjunctional voltage against junctional conductance in Fig. 5 E. At first glance, it appears that a mixture of homomeric WT and mutant hemichannels could explain this result. Since WT channels have much higher open probability, and hence higher conductance than mutant channels, the WT properties would be expected to dominate. However, the gating properties of the channels, even in 1:1 mixes of WT and mutant, show a significant mutant phenotype (Fig. 5, B and C), which suggests that they must reflect heteromeric channels with both subunits, consistent with the previous biochemical analysis.
The fact that the mutant gating phenotype is evident even when WT subunits are expressed in excess demonstrates that mutant subunits exert a partially dominant effect, at least in terms of voltage gating properties (Fig. 5, B and C). A dominant negative effect on functional expression levels is also evident from the 70% drop in conductance that is obtained when 10 ng of RNA encoding mutant subunits is added to an oocyte containing 10 ng RNA encoding WT subunits (Fig. 5 F, first two bars). Although the mutant clearly has dominant negative effects on the WT subunits, it is also evident that the WT subunits serve to partially rescue mutant function, based on the steady increase in conductance of cell pairs as more WT subunits are expressed with mutant subunits (Fig. 5 F). The gating profiles of this increased conductance, as discussed above, clearly show that it cannot simply be attributed to assembly of homomeric WT channels. Thus, the electrophysiology data are consistent with a rescue of mutant channel assembly by WT subunits.
Heteromeric hexamers are not rescued by expression of Cx26WT with Cx26T135A
In a previous work (9), we showed that preparations of His6-tagged, purified Cx26T135A homomeric hemichannels were unstable. We also noted that the banding patterns of this mutant were highly variable depending on the preparation, with no one oligomeric state predominating. These preparations also became increasingly unstable over time. To determine whether this variability reflected a mix of oligomeric forms in situ, or differential stability to detergent extraction, we subjected the Cx26T135A membrane preparation to cross-linking with DSP before detergent solubilization. This stabilized the hexameric and dodecameric forms, as indicated by the BN gel (cf. gels in Fig. S2, A and B). These preparations also had a distinct appearance by EM from Cx26T135A preparations (Fig. S2 B), forming structures similar to Cx26WT (Fig. 2 A), although, as is common with cross-linked samples, there was some hemichannel/channel aggregation. We also found that Cx26P87L hemichannels could be stabilized by chemical cross-linking, as evidenced by doughnut-like structures in the electron micrographs, and only hexameric and dodecameric bands on the Western blots (data not shown).
Although the dominant-negative effects of Cx26T135A (11) did not allow functional tests of heteromeric mutant/WT channels, structural comparisons indicated that there was no rescue of Cx26T135A detergent instability by coexpression of WT (Fig. 6, A–D). In fact, this mutation seemed to be more destabilizing of Cx26WT oligomerization, as evidenced by a highly heterogeneous population of aggregates with increasing MOI ratios. Even at the very high ratio of 2.6, where one would expect heteromers containing possibly four to five Cx26WT subunits (Fig. 6 D), we saw no bands on BN gels. EM images of this sample contained mainly blank areas or, rarely, areas containing detergent micelles (Fig. 6 D, EM image) and perhaps misfolded or aggregated protein seen at the top of the gel.
Figure 6.

T135A hemichannels cannot be rescued by WT. Even with increasing amounts of Cx26WT, no stable hemichannel structures were obtained. (A–D) As the ratio of MOIs increases from 0 to ∼2.6, we see few definitive hemichannel structures (middle column) or bands on the BN gels (BNgel, right column). When the MOI ratio of Cx26WT to Cx26T135A is 0.5 (B), we observed a very heterogeneous population of hemichannel sizes in the electron micrograph and the Blue Native gel Western blot. In panel C, aggregates and a smaller fraction of stable connexons are visible. The Western blot confirms this instability. (D) At the highest ratio of ∼2.6 WT to 1 Cx26T135A, where higher expression of Cx26WT as compared to Cx26T135A would promote hexamers containing predominantly WT, we see aggregated protein and micelles in the EM images and no detectable oligomer-specific bands on the BN gels.
The stability of two other T135 mutants indicates that the hydroxyl group in threonine plays a role in Cx stability
The Thr in the M3 helix is conserved in all α and β Cxs (9). We originally speculated that this Thr might play an important role in maintaining the M3 helix by size and/or shape, or via hydrogen (H)-bonding from the OH group of the Thr residue to an acceptor in an adjacent helix. We hypothesized that the substitution of a Val or Ser for this Thr would distinguish whether the β-branching and size, or H-bonding capability were more critical for preserving channel function.
We constructed T135S and T135V baculoviruses, expressed them in Sf9 cells, and then purified them for EM and BN gel analysis. As shown previously, T135A is very unstable (Fig. 7 A, left) in both EM and BN gels (Fig. 7 B, left). Electron micrographs of negatively stained preparations of purified T135V mutant protein show a similar heterogeneous population of Cx-bearing structures (Fig. 7 A, middle), which is confirmed by the BN gel shown in Fig. 7 B (middle). In contrast, T135S protein preparations contain many more hemichannel structures (circles) but also some small oligomeric structures (rectangle) (Fig. 7 A, right). The BN gel confirms that the majority of this mutant protein forms hexamers and dodecamers to a greater extent than T135A or T135V (Fig. 7 B, right). Therefore, it seems likely that it is the H-bonding that is the critical variable in determining hexamer stability at this site. However, the minor population of smaller oligomeric structures indicates that the fit between Thr and its H-bonding partner is not perfect, perhaps because the β-branched side chain of T135 restricts its conformation for optimal orientation of the H-bond donor-acceptor pair.
Figure 7.
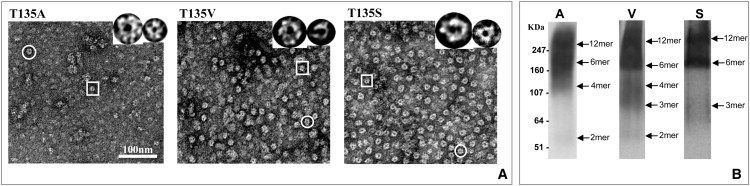
Comparison of T135 mutants for hexamer stability. T135A, T135V, and T135S connexons were purified and examined by (A) EM and (B) BN gel PAGE for connexon structure and oligomeric composition. Of the three mutants, T135S is the most stable, suggesting that the Ser hydroxyl group promotes better hexamer stability than Val, which is similar in shape to Thr. Circles indicate larger doughnut-shaped structures, and rectangles highlight smaller ones. Examples of both are shown in panel A.
Discussion
In this study, we investigated four unique mutations of Cx26 that differ in their functional phenotype and amino acid position within the transmembrane domains. A summary of the experiments in this study, and others, is shown in Table S1. We also note that these mutations have disease-causing correlates in other Cxs. These connexinopathies include various forms of hereditary deafness (Cx26), CMTX (Cx32), and ODDD (Cx43). As a way of understanding the molecular basis of disease based on single site-specific amino acid mutations, we explored whether oligomer stability plays a role in their dysfunction. M34T and V84L in Cx26 are both recessive, nonsyndromic deafness mutations, whereas Cx43T154A is a dominant mutation in ODDD (14), causing small eyes, underdeveloped teeth, syndactyly, and finger malformations. Cx32P87L causes CMTX, producing axonal loss, clusters of regenerating fibers, and paranodal demyelination (25). In terms of channel function, V84L has the least effect of these mutations, affecting only IP3 transfer, whereas Cx43T154A, which is at the opposite end of the functional spectrum, eliminates channel function entirely. Previous EM studies of the Cx26M34A structure suggested that mutations at M34 eliminate a close interaction with the NT, creating a plug of density found in the pore (7,8). The expectation is that M34T might cause a similar displacement of the NT into the pore, consistent with its reduced conductance.
In the most lethal of these mutations, Cx26T135A, all function was eliminated in homotypic and heterotypic combinations in paired Xenopus oocytes, and was not recoverable on mixing with Cx26WT (indeed, the mutant showed a marked dominant-negative effect) (9). Consistent with this, stable oligomers were never recovered, even with high coexpression of Cx26WT. For the other three mutations for which aberrant functions could be measured, we found that either the homomeric hemichannel was stable, in the cases of the more mild mutations, or stability was recovered by coexpression with WT, in the case of the more severe phenotype.
Conservation of these four residues among Cx species
The strict conservation of amino acids and positions among Cx isoforms has led to the theory that they play pivotal roles in channel functionality. In examining the sequences of 77 Cxs from various species, we find that these four residues tend to be highly conserved among the Cx family. As we showed in our earlier publication (9), the Thr in equivalent positions in the M3 helix is identical among α and β subgroups. The equivalent position to M34 in M1 of Cx26 is a Met in only some of the β members of the Cx family (human and mammalian Cx26 and Cx32, chick Cx31, human and rodent Cx30, Xenopus Cx30, and Croaker Cx32.2), with Leu being found in ∼66% of Cxs at this position. In contrast, the proline in the M2 helix, corresponding to P87 in Cx26, is 100% identical in these sequences, and V84 is ∼90% identical. In the latter case, the majority of the substitutions at this position are ones with strong similarity to Val (i.e., Ile and Leu). In fact, among 20 human Cxs, the amino acid composition at this residue is considered as conserved by the sequence alignment and analysis program, ClustalW2 (26). Therefore, based on sequence conservation alone, these positions would seem to be important.
Possible roles played by these four residues in intra- or intermolecular interactions
We sought to determine whether the spatial arrangement of these amino acids in current molecular structure models would provide insights into to why these amino acid positions are important and how these site mutations render Cx26 channels nonfunctional. Fig. S3 highlights the position of these four mutations and H94 on the coordinates of the recently published x-ray crystal structure (8). Studies in other membrane channels have suggested that sites facing the aqueous pore or lipid bilayer would be more tolerant of mutagenesis than sites at interhelical packing interfaces. Thus, Cx26M34A (in M1) and Cx26V84L (in M2), which form stable hexamers and dodecamers, might be expected to face the aqueous pore or lipid bilayer, but not other helices. In fact, both have been postulated, based on cysteine scanning accessibility studies, to be pore-lining residues, either in Cx46 hemichannels in the case of M34 (27,28), or in Cx32 GJs in the case of V84 (29). The latter study concluded that in GJ channels of Cx32, the M34 site is actually located in an aqueous extracellular crevice, but this would also be consistent with the current findings of its tolerance for mutagenesis. The effects of a deafness-associated mutation at the V84 site are also consistent with it lining the pore, given the highly selective effects of this mutant on pore selectivity (12). These results contrast with the recently published x-ray crystal structure of the open channel (8), where M34 is in the pore but forms an interaction with the indole ring of Trp3 in the NT, helping to anchor it against the pore wall. V84 is proposed to be at the M2/M3 helix interface. Although it does not seem to interact with any specific amino acid chain on M3, it is difficult to explain the access of thiol reagents to this site. In the x-ray-crystal-based structural model, V84 is ∼20 Å from the nearest ordered amino acid in the narrowest part of the GJ pore (amino acid 2 Asn), so it is difficult to understand how a mutation at this site could directly impede the passage of IP3, unless it were through an indirect effect. However, the specificity of the effect on IP3, and not on dye passage or conductance, makes this less likely. These comparisons are currently limited by the resolution of the x-ray crystal structure, which does not allow individual side chains to be directly visualized. Instead, it relies on model fitting onto the traced α-carbon backbone, and entails the usual caveats for dealing with structural interpretations based on mutagenesis. Nonetheless, based on the effects on oligomer stability, our current results are consistent with previous cysteine accessibility studies.
The P87 amino acid is in a central position within the M2 helix in all Cxs. The four transmembrane segments visualized in the Cx26WT x-ray crystal crystallographic structure (8) and in the two electron crystallographic structures of truncated Cx43 (6) and Cx26M34A (7) form a four α-helix bundle. The main effect of having a proline in the middle of an α-helix would be to cause a kink, and this kink can be seen at the P87 position in the 3.5 Å x-ray crystal structure (8). This proline was shown to be important for the voltage gating of the channel (11), and Monte Carlo simulations further suggested that the helix may undergo a change in bend angle from 37° to 20° during gating as the result of a broken H-bond (30). Within the x-ray crystal structure, P87 is on the same helix face as V84 (Fig. S3), and its sensitivity to mutagenesis may be mostly a product of its role in channel gating. This is emphasized by the observation that the gating phenotype of the Cx26P87L mutation appears to be dominant in heteromeric channels when mixed 1:1 with WT. Not surprisingly, the changes in the relative stability of the open and closed states induced by this mutation are also reflected in the stability of the hexamers in detergent. The mutant hexamer is stable with a 1:1 heteromeric combination of Cx26P87L and WT monomers, consistent with a possible dimeric association of connexin subunits, such that the change in bend angle in Cx26P87L may be compensated for by a neighboring WT monomer in terms of channel stability, whereas the mutant still exerts a dominant effect on gating. Therefore, our experiments, together with published structural and functional studies, indicate that the P87L mutation causes a straightening of the M2 helix, which then causes a disruption or distortion of subunit-subunit interactions that are critical for both oligomer stability and gating.
In contrast, the observations that Cx26T135A hemichannels are unstable suggest that this residue lies at either a subunit-subunit or a helix-helix boundary. It is interesting to note that an absolutely conserved Trp residue is located next to the Thr, which could play a role in restricting its motility. Although the packing of T135 with adjacent subunits is critical for channel function, the hexamer is stable within the membrane and can be isolated by cross-linking early during the isolation. Its instability is evident in the presence of detergent over time. The similar lack of stability of the T135V mutant (even though the size and β-branch of the side chain are conserved) and the greater stability of the T135S mutant (which preserves the OH group) indicate that H-bonding at this position is likely to play a role in stabilizing the connexon. A possible candidate for H-bonding is H94, which, like T135, is conserved in most Cxs. In previous studies, Ek et al. (31) examined the nonfunctionality of substitutions at the equivalent position in Cx43 (H95) and the restoration of function when this histidine was moved to positions 1 or 2 amino acids from Cx43H95. As shown in Fig. S3, in the x-ray crystal structure, T135 is located on the side of the helix facing M2 and most likely forms an H-bonding pair with H94. The distance between the T135 and H94 donor and acceptor atoms is approximately that needed for an H-bond. Weak forces, such as H-bonding, would explain why the hemichannels are stable when first isolated, yet dissociate with time.
Conclusions
Previous studies of Cx mutations have focused on mistrafficking, disruption of docking, or aberrations in channel properties, such as gating. By monitoring connexon stability after detergent extraction in this work, we were able to show that certain mutations can cause instability in the hexameric connexons by disrupting subunit interactions. We hypothesize that these mutations can cause instability directly, as in the M3 T135 mutations, or indirectly by mutation of the critical P87 in M2. Thus, for Cx26T135A and Cx26P87L, we find that rescue of function and rescue of stability by coexpression with Cx26WT are related.
Supporting Material
Methods, one table, and three figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00151-7.
Supporting Material
Acknowledgments
We thank Dr. Angela Cone for assistance with some experiments.
This work was supported by grants from the National Institutes of Health (GM065937 and GM072881 to G.E.S., and GM055437 and CA048049 to B.J.N.) and the National Science Foundation (MCB-0131425 to G.E.S.). Most of the work presented here was conducted at the National Center for Microscopy and Imaging Research at San Diego, which is supported by National Institutes of Health grant RR04050 (awarded to Dr. Mark Ellisman). B.J.N. acknowledges support of the National Cancer Institute-supported Cancer Therapy and Research Center at the University of Texas Health Science Center at San Antonio.
References
- 1.Richard G., Rouan F., Russell L. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am. J. Hum. Genet. 2002;70:1341–1348. doi: 10.1086/339986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rabionet R., Zelante L., Estivill X. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum. Genet. 2000;106:40–44. doi: 10.1007/s004390051007. [DOI] [PubMed] [Google Scholar]
- 3.Cohen-Salmon M., Ott T., Petit C. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr. Biol. 2002;12:1106–1111. doi: 10.1016/s0960-9822(02)00904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gabriel H.D., Jung D., Willecke K. Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. J. Cell Biol. 1998;140:1453–1461. doi: 10.1083/jcb.140.6.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sosinsky G.E., Nicholson B.J. Structural organization of gap junction channels. Biochim. Biophys. Acta. 2005;1711:99–125. doi: 10.1016/j.bbamem.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Unger V.M., Kumar N.M., Yeager M. Three-dimensional structure of a recombinant gap junction membrane channel. Science. 1999;283:1176–1180. doi: 10.1126/science.283.5405.1176. [DOI] [PubMed] [Google Scholar]
- 7.Oshima A., Tani K., Sosinsky G.E. Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc. Natl. Acad. Sci. USA. 2007;104:10034–10039. doi: 10.1073/pnas.0703704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maeda S., Nakagawa S., Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- 9.Beahm D.L., Oshima A., Sosinsky G.E. Mutation of a conserved threonine in the third transmembrane helix of α- and β-connexins creates a dominant-negative closed gap junction channel. J. Biol. Chem. 2006;281:7994–8009. doi: 10.1074/jbc.M506533200. [DOI] [PubMed] [Google Scholar]
- 10.Oshima A., Doi T., Fujiyoshi Y. Roles of Met-34, Cys-64, and Arg-75 in the assembly of human connexin 26. Implication for key amino acid residues for channel formation and function. J. Biol. Chem. 2003;278:1807–1816. doi: 10.1074/jbc.M207713200. [DOI] [PubMed] [Google Scholar]
- 11.Suchyna T.M., Xu L.X., Nicholson B.J. Identification of a proline residue as a transduction element involved in voltage gating of gap junctions. Nature. 1993;365:847–849. doi: 10.1038/365847a0. [DOI] [PubMed] [Google Scholar]
- 12.Beltramello M., Piazza V., Mammano F. Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat. Cell Biol. 2005;7:63–69. doi: 10.1038/ncb1205. [DOI] [PubMed] [Google Scholar]
- 13.Bruzzone R., Veronesi V., White T.W. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003;533:79–88. doi: 10.1016/s0014-5793(02)03755-9. [DOI] [PubMed] [Google Scholar]
- 14.van Es R.J., Wittebol-Post D., Beemer F.A. Oculodentodigital dysplasia with mandibular retrognathism and absence of syndactyly: a case report with a novel mutation in the connexin 43 gene. Int. J. Oral Maxillofac. Surg. 2007;36:858–860. doi: 10.1016/j.ijom.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Berthoud V.M., Minogue P.J., Beyer E.C. Loss of function and impaired degradation of a cataract-associated mutant connexin50. Eur. J. Cell Biol. 2003;82:209–221. doi: 10.1078/0171-9335-00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skerrett I.M., Di W.L., Nicholson B.J. Aberrant gating, but a normal expression pattern, underlies the recessive phenotype of the deafness mutant connexin26M34T. FASEB J. 2004;18:860–862. doi: 10.1096/fj.03-0763fje. [DOI] [PubMed] [Google Scholar]
- 17.Tan C.C., Ainsworth P.J., MacLeod P.M. Novel mutations in the connexin 32 gene associated with X-linked Charcot-Marie-Tooth disease. Hum. Mutat. 1996;7:167–171. doi: 10.1002/(SICI)1098-1004(1996)7:2<167::AID-HUMU14>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 18.Thönnissen E., Rabionet R., Ott T. Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression. Hum. Genet. 2002;111:190–197. doi: 10.1007/s00439-002-0750-2. [DOI] [PubMed] [Google Scholar]
- 19.Hand G.M., Müller D.J., Sosinsky G.E. Isolation and characterization of gap junctions from tissue culture cells. J. Mol. Biol. 2002;315:587–600. doi: 10.1006/jmbi.2001.5262. [DOI] [PubMed] [Google Scholar]
- 20.Stauffer K.A., Kumar N.M., Unwin N. Isolation and purification of gap junction channels. J. Cell Biol. 1991;115:141–150. doi: 10.1083/jcb.115.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Locke D., Harris A.L. Connexin channels and phospholipids: association and modulation. BMC Biol. 2009;7:52. doi: 10.1186/1741-7007-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaietta G., Deerinck T.J., Ellisman M.H. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- 23.Martin B.R., Giepmans B.N., Tsien R.Y. Mammalian cell-based optimization of the biarsenical-binding tetracysteine motif for improved fluorescence and affinity. Nat. Biotechnol. 2005;23:1308–1314. doi: 10.1038/nbt1136. [DOI] [PubMed] [Google Scholar]
- 24.Musil L.S., Goodenough D.A. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 1993;74:1065–1077. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- 25.Kuntzer T., Dunand M., Bogousslavsky J. Phenotypic expression of a Pro 87 to Leu mutation in the connexin 32 gene in a large Swiss family with Charcot-Marie-Tooth neuropathy. J. Neurol. Sci. 2003;207:77–86. doi: 10.1016/s0022-510x(02)00394-5. [DOI] [PubMed] [Google Scholar]
- 26.Larkin M.A., Blackshields G., Higgins D.G. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 27.Kronengold J., Trexler E.B., Verselis V.K. Single-channel SCAM identifies pore-lining residues in the first extracellular loop and first transmembrane domains of Cx46 hemichannels. J. Gen. Physiol. 2003;122:389–405. doi: 10.1085/jgp.200308861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou X.W., Pfahnl A., Dahl G. Identification of a pore lining segment in gap junction hemichannels. Biophys. J. 1997;72:1946–1953. doi: 10.1016/S0006-3495(97)78840-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skerrett I.M., Aronowitz J., Nicholson B.J. Identification of amino acid residues lining the pore of a gap junction channel. J. Cell Biol. 2002;159:349–360. doi: 10.1083/jcb.200207060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ri Y., Ballesteros J.A., Bargiello T.A. The role of a conserved proline residue in mediating conformational changes associated with voltage gating of Cx32 gap junctions. Biophys. J. 1999;76:2887–2898. doi: 10.1016/S0006-3495(99)77444-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ek J.F., Delmar M., Taffet S.M. Role of histidine 95 on pH gating of the cardiac gap junction protein connexin43. Circ. Res. 1994;74:1058–1064. doi: 10.1161/01.res.74.6.1058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.