

Abstract
Linking experiments with the atomistic resolution provided by molecular dynamics simulations can shed light on the structure and dynamics of protein-disordered states. The sampling limitations of classical molecular dynamics can be overcome using metadynamics, which is based on the introduction of a history-dependent bias on a small number of suitably chosen collective variables. Even if such bias distorts the probability distribution of the other degrees of freedom, the equilibrium Boltzmann distribution can be reconstructed using a recently developed reweighting algorithm. Quantitative comparison with experimental data is thus possible. Here we show the potential of this combined approach by characterizing the conformational ensemble explored by a 13-residue helix-forming peptide by means of a well-tempered metadynamics/parallel tempering approach and comparing the reconstructed nuclear magnetic resonance scalar couplings with experimental data.
Main Text
Molecular dynamics (MD) simulations are a valuable tool for studying complex biomolecular systems because they can provide an atomistic description of their structure and dynamics. This is particularly important for protein conformationally disordered states (1,2). Characterizing the structure and the dynamics of these states with standard experimental tools is challenging, as the heterogeneous nature of these states can only be represented by ensembles of conformations. Much is to be gained by combining MD simulations with experiments. This can be achieved either by restraining the MD simulations to reproduce the experimental data (3) or by calculating the experimental observables from extensive MD sampling. Unfortunately, MD predictive power is severely limited due to the complexity of the biomolecule free energy landscape, which prevents exhaustive sampling by means of standard MD. Many methods have been proposed to overcome the sampling limitations; here we focus on metadynamics (4,5), which has been successfully applied to a variety of complex biomolecular processes (6–8). Metadynamics relies on the introduction of a history-dependent potential acting on a selected number of slow degrees of freedom, the collective variables (CVs). If properly applied, metadynamics can greatly speed-up sampling and reconstruct the free energy surface (FES) associated to the CVs through the relation
| (1) |
where s are the CVs, V(s, t) is the bias potential, T is the system temperature, and ΔT is an input parameter representing an effective sampling temperature of the CV space (5). The power of metadynamics can be further enhanced by combining it with parallel tempering (PT) (9): the combined method (PTMetaD) has been demonstrated to greatly improve the performance of both PT and single-replica metadynamics (10,11). The introduction of metadynamics bias potential leads to a correct (see Eq. 1) probability distribution along the CVs but distorts that of the other degrees of freedom. Because CVs often do not correspond to experimental observables, this feature has severely limited the possibility for quantitatively comparing metadynamics simulations to experiments. Very recently, we have been able to solve this problem devising a method to recover, on-the-fly, the correct Boltzmann distribution from metadynamics trajectories (12).
Here, we show that the conformational ensemble spanned by a C-peptide, corresponding to the first 13 N-terminal residues of RNase A, can be easily characterized by means of PTMetaD. We validate our results via comparison of the reconstructed nuclear magnetic resonance (NMR) scalar couplings with experiments. This peptide has attracted considerable attention due to its propensity to form α-helical structures, which is unusual for such a short chain. Circular dichroism (CD) shows that the average helicity of C-peptide depends on both temperature and pH and exhibits a maximum at pH 5.25 and T = 276 K (13). Even under these optimal conditions, NMR data suggested the presence of multiple conformations of different helical content (14).
We choose a mutant sequence of C-peptide whose 3JHNHα scalar coupling constants and CD spectra were determined at T = 277 K and pH = 4.5 (15). The peptide was solvated in 1192 TIP3P (16) water molecules and the AMBER99SB (17) force field was adopted. The system was simulated with a PTMetaD approach for 20 ns using GROMACS4 (18) and PLUMED (19). We used 64 replicas spanning the temperature interval between 274 and 650 K. The stochastic thermostat of Bussi et al. (20) was used for this. Additional technical details can be found in the Supporting Material.
The metadynamics bias was applied to a single CV that measures the population of α-helical backbone H-bonds, defined as
| (2) |
where d(Oi, HNi+4) is the distance between the backbone oxygen atom of residue i and the backbone amide hydrogen of residue i + 4. It must be stressed that Sα does not represent the only slow mode of our system. However, combining metadynamics with PT greatly accelerates the sampling of degrees of freedom that are transverse to the CVs (10). To assess the reliability of the simulation protocol, we performed two PTMetaD simulations, starting from different initial configurations. A first run (i.e., Open) started from a random coil structure, whereas the second one (i.e., Closed) started from an α-helical configuration corresponding to ribonuclease A NMR structure (see Supporting Material).
In both of the simulations, the entire CV range (from Sα ≃ 0 to Sα ≃ 9) was rapidly explored and the estimate of F(Sα) converged in ∼10 ns. We can arbitrarily subdivide the Sα space into three regions: an almost random coil state (AC, Sα < 4); a state of an α-helicity roughly similar to that of the ribonuclease NMR structure (PH, 4 < Sα < 7); and an almost fully α-helical state (FH, Sα > 7). In Fig. 1 the estimate of the free-energy differences among those states as a function of the simulation time is reported for replica with T = 278 K. The free-energy differences indicate that AC is the most stable state. However, configurations with partially formed α-helices are significantly populated (ΔGPH-AC ≃ 0.5 kcal/mol), whereas fully folded α-helical structures are less likely (ΔGFH-AC ≃ 2 kcal/mol). Such findings are in qualitative agreement with the average helicity measured with CD spectroscopy (13). NMR experiments performed on wild-type C-peptide suggested that in similar conditions three sets of conformations were populated: a set of largely extended conformations; a set of largely helical forms; and a set of partially α-helical structures with a salt-bridge between the Glu2 and Arg10 side chains (14). Such interaction is believed to induce a kink in the backbone preventing the N-terminal residues from adopting an α-helical structure. To get a deeper insight into the interplay between α-helix formation and Glu2-Arg10 side-chain interaction, we used the reweighting algorithm for reconstructing the FES as a function of both Sα and the salt-bridge coordinate SSB.
Figure 1.
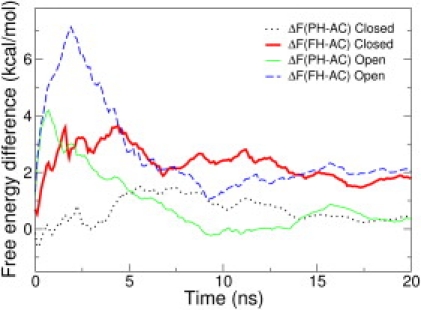
Estimates of free-energy difference among the three Sα regions (see text) as a function of the simulation time for two independent PTMetaD simulations (open and closed).
The resulting FES is shown in Fig. 2. The landscape reconstructed from the simulations provides a detailed picture of the conformational ensemble which remarkably matches the hypothesis of Osterhout et al. (14). Indeed, the suggested sets of configurations correspond to local free-energy minima. The free-energy minimum at (Sα ≃ 6, SSB ≃ 0.8) corresponds to conformations with a formed salt-bridge and whose helicity is slightly less than NMR structure. Fully α-helical configurations require breaking the salt-bridge and are less stable.
Figure 2.
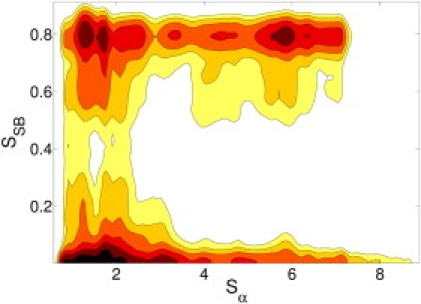
FES as a function of Sα and SSB from the open simulation. SSB = [1 – (d/5.0)6]/[1 – (d/5.0)12], where d is the distance in Ångstroms between the Cδ of Glu2 and the central carbon atom in the guanidine group of Arg10. Contour lines are plotted every kBT.
To make quantitative comparison with experimental data, we focus here on NMR scalar couplings. These observables are related to dihedral angles by the classical Karplus relations (21). In particular, 3JHNHα values can be expressed as a function of the Φ backbone angle J(Φ) = Acos2(Φ + Δ) + Bcos(Φ + Δ) + C, where A, B, C, and Δ are empirical parameters. Different parameters have been proposed, but here we focus on two empirical sets: the parameters calibrated on ubiquitin (22) (UBIQ) and another set that was calibrated on flavodoxin (23) (FLAV). As done in other studies (24–26), we measured the agreement between experiments and simulations using
| (3) |
where σ = 0.91 is the estimated systematic error (25,26). The results for the two PTMetaD simulations are reported in Table 1. Although the agreement is in general good, its quality depends on the parameter choice. However, the scalar couplings obtained in the two simulations are in good agreement within the same parameter set (χ2(Jclosed, Jopen) < 0.09). This makes us confident that sampling has been adequate and the remaining error is due to force-field inaccuracies and/or J(Φ) parameterization.
Table 1.
χ2 values
| UBIQ | FLAV | |
|---|---|---|
| Open | 1.71 | 0.88 |
| Closed | 1.88 | 0.92 |
| MD | 4.56 | 3.29 |
χ2 values using UBIQ (22) and FLAV (23) parameter set for the PTMetaD simulations and for a standard 20-ns MD simulation averaged over eight independent runs (see Supporting Material).
In this letter, we presented the characterization of the disordered structural ensemble explored by a mutant of C-peptide by means of PTMetaD simulation. Using a reweighting algorithm, we have been able to compare our results to NMR data. Such comparison allowed us both to validate our simulations and to gain a deeper insight into the conformationally averaged information provided by the experiments. We believe that such a combined approach may provide a much better understanding of the protein conformationally heterogeneous states. Future developments will point toward the reconstruction of the kinetics relative to the conformational transitions by means of stochastic dynamics simulations on the reconstructed FESs.
Supporting Material
One figure and one table are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00198-0.
Supporting Material
Acknowledgments
The authors thank “Centro Enrico Fermi” for providing the computational resources.
References and Footnotes
- 1.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Vendruscolo M. Determination of conformationally heterogeneous states of proteins. Curr. Opin. Struct. Biol. 2007;17:15–20. doi: 10.1016/j.sbi.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Laio A., Parrinello M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA. 2002;99:12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barducci A., Bussi G., Parrinello M. Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008;100:020603. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- 6.Parrinello M. Physical Biology: From Atoms to Medicine. In: Zewail A.H., editor. Imperial College Press; London, UK: 2008. [Google Scholar]
- 7.Laio A., Gervasio F.L. Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008;71:126601. [Google Scholar]
- 8.Pfaendtner J., Branduardi D., Voth G.A. Nucleotide-dependent conformational states of actin. Proc. Natl. Acad. Sci. USA. 2009;106:12723–12728. doi: 10.1073/pnas.0902092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansmann U.H.E. Parallel tempering algorithm for conformational studies of biological molecules. Chem. Phys. Lett. 1997;281:140–150. [Google Scholar]
- 10.Bussi G., Gervasio F.L., Parrinello M. Free-energy landscape for β-hairpin folding from combined parallel tempering and metadynamics. J. Am. Chem. Soc. 2006;128:13435–13441. doi: 10.1021/ja062463w. [DOI] [PubMed] [Google Scholar]
- 11.Bonomi M., Gervasio F.L., Parrinello M. Insight into the folding inhibition of the HIV-1 protease by a small peptide. Biophys. J. 2007;93:2813–2821. doi: 10.1529/biophysj.107.106369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonomi M., Barducci A., Parrinello M. Reconstructing the equilibrium Boltzmann distribution from well-tempered metadynamics. J. Comput. Chem. 2009;30:1615–1621. doi: 10.1002/jcc.21305. [DOI] [PubMed] [Google Scholar]
- 13.Shoemaker K.R., Kim P.S., Baldwin R.L. Tests of the helix dipole model for stabilization of α-helices. Nature. 1987;326:563–567. doi: 10.1038/326563a0. [DOI] [PubMed] [Google Scholar]
- 14.Osterhout J.J., Baldwin R.L., Wright P.E. 1H NMR studies of the solution conformations of an analogue of the C-peptide of ribonuclease A. Biochemistry. 1989;28:7059–7064. doi: 10.1021/bi00443a042. [DOI] [PubMed] [Google Scholar]
- 15.Caballero-Herrera A., Nordstrand K., Berndt K. Effect of urea on peptide conformation in water: molecular dynamics and experimental characterization. Biophys. J. 2005;89:842–857. doi: 10.1529/biophysj.105.061978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 17.Hornak V., Abel R., Simmerling C. Comparison of multiple AMBER force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 19.Bonomi M., Branduardi D., Parrinello M. PLUMED: a portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009;180:1961–1972. [Google Scholar]
- 20.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 21.Karplus M. Contact electron-spin coupling of nuclear magnetic moments. J. Chem. Phys. 1959;30:11–15. [Google Scholar]
- 22.Hu J.S., Bax A. Determination of φ and χ1 angles in proteins from C13-C13 three-bond J couplings measured by three-dimensional heteronuclear NMR. how planar is the peptide bond? J. Am. Chem. Soc. 1997;119:6360–6368. [Google Scholar]
- 23.Schmidt J.M., Blumel M., Ruterjans H. Self-consistent 3J coupling analysis for the joint calibration of Karplus coefficients and evaluation of torsion angles. J. Biomol. NMR. 1999;14:1–12. doi: 10.1023/A:1008345303942. [DOI] [PubMed] [Google Scholar]
- 24.Best R.B., Buchete N.V., Hummer G. Are current molecular dynamics force fields too helical? Biophys. J. 2008;95:L07–L09. doi: 10.1529/biophysj.108.132696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Best R.B., Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J. Phys. Chem. B. 2009;113:9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wickstrom L., Okur A., Simmerling C. Evaluating the performance of the ff99SB force field based on NMR scalar coupling data. Biophys. J. 2009;97:853–856. doi: 10.1016/j.bpj.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.