

Abstract
The serotonin 5-HT3 receptor (5-HT3R) is a member of the Cys-loop ligand-gated ion channel family. We used a combination of site-directed mutagenesis, homology modeling, and ligand-docking simulations to analyze antagonist-receptor interactions. Mutation of E236, which is near loop C of the binding site, to aspartate prevents expression of the receptor on the cell surface, and no specific ligand binding can be detected. On the other hand, mutation to glutamine, asparagine, or alanine produces receptors that are expressed on the cell surface, but decreases receptor affinity for the competitive antagonist d-tubocurarine (dTC) 5-35-fold. The results of a double-mutant cycle analysis employing a panel of dTC analogs to identify specific points of interactions between the dTC analogs and E236 are consistent with E236 making a direct physical interaction with the 12 –OH of dTC. dTC is a rigid molecule of known three-dimensional structure. Together with previous studies linking other regions of dTC to specific residues in the binding site, these data allow us to define the relative spatial arrangement of three different residues in the ligand-binding site: R92 (loop D), N128 (loop A), and E236 (near loop C). Molecular modeling employing these distance constraints followed by molecular-dynamics simulations produced a dTC/receptor complex consistent with the experimental data. The use of the rigid ligands as molecular rulers in conjunction with double-mutant cycle analysis provides a means of mapping the relative positions of various residues in the ligand-binding site of any ligand-receptor complex, and thus is a useful tool for delineating the architecture of the binding site.
Introduction
The serotonin type 3 receptor (5-HT3R) is a member of the Cys-loop ligand-gated ion channel family, which includes the muscle and neuronal nicotinic acetylcholine receptors (AChRs), the glycine receptor (GlyR), and the γ-aminobutyric acid type A (GABAAR) and ρ (GABAAρR) receptors (1,2). Two different subunits, 5-HT3A and 5-HT3B, have been shown to be present in functional 5-HT3Rs (3). Expression of the 5-HT3A subunit (4) results in 5-HT-gated channels with a pharmacology appropriate for 5-HT3Rs. However, there are some differences between the properties of the expressed homomeric receptors and 5-HT3Rs in some, but not all, neurons. The most significant difference is that the single-channel conductance of the homomeric receptors is in the subpicosecond range, whereas that of the receptors in some peripheral (but not central nervous system) neurons is in the range of 9–19 pS (5).
The 5-HT3B subunit does not form functional receptors by itself; however, coexpression with the 5-HT3A subunit produces heteromeric receptors with a single-channel conductance of 16 pS (6). The expression patterns of the 5-HT3A and 5-HT3B subunits suggest that both 5-HT3A homomers and 5-HT3A/5-HT3B heteromers exist in both the central and peripheral nervous systems (7). Despite the differences in their single-channel properties, these two types of receptors have very similar ligand-binding properties (8). For example, inclusion of the rat 5-HT3B subunit produces receptors with only an ∼2-fold increase in IC50 for d-tubocurarine (dTC) inhibition of currents (9). Thus, 5-HT3A homopentamers are an appropriate model for the structure of the ligand-binding domain of native 5-HT3Rs, regardless of whether they are 5-HT3A homomers or 5-HT3A/5-HT3B heteromers.
Previous studies have used a combination of site-directed mutagenesis and molecular modeling to probe the architecture of the ligand-binding domain of the 5-HT3R (10–16). In this approach, the effects of introduced mutations on ligand binding and/or agonist-elicited currents are used to identify residues that may play a role in ligand-receptor interactions. The data are then analyzed in terms of a structural model for the extracellular domain of the receptor constructed using the structure of the molluscan acetylcholine binding protein (AChBP) (17) as the template for modeling, and the models that are most consistent with the data are then used to guide further experiments.
In most of these studies, only the effects of receptor mutations were monitored. In this work, we combined the introduction of mutations in the receptor with alterations in ligand structure to more fully probe ligand-receptor interactions using double-mutant cycle analysis (18). To take full advantage of the double-mutant cycle analysis, we employed a panel of ligands with a number of defined small changes in structure. One ligand that can be altered in a number of ways is dTC, a competitive antagonist of both AChRs (19) and 5-HT3Rs (20). dTC has a 1- to 2000-fold higher affinity for the murine 5-HT3R than the human receptor, and it was shown that the regions responsible for this species difference are located in the amino terminal extracellular domain (21). We subsequently showed that a major determinant for this difference is located in loop F of the binding site (22). Using a series of dTC analogs, we demonstrated that the same regions of dTC that are important for high-affinity binding to the AChR (23,24) are also important for binding to the 5-HT3R (25). In a subsequent study, we showed that N128 in the 5-HT3R interacted with the 2′N of dTC, and that R92 most likely interacted with the 2N of dTC (15).
In this study, we map an additional residue in the putative ligand-binding site (E236) onto the dTC structure, and then use the rigid three-dimensional structure of dTC to provide relative spatial distances between three separate residues in the ligand-binding domain. These distances then become spatial restraints in the subsequent modeling process. The use of a rigid molecular ruler to obtain experimentally derived spatial positions of residues in the binding site thus provides a general approach for delineating the architecture of a portion of the ligand-binding site.
Materials and Methods
Molecular biology and transfection
A cDNA clone corresponding to the short form of the murine 5-HT3A subunit (26) isolated from a neuroblastoma N1E-115 cell line cDNA library (27) was used in these studies. Site-directed mutagenesis was carried out using the QuickChange system (Stratagene, La Jolla, CA), and the entire coding region of the mutant subunit was sequenced to ensure that only the desired mutation was present. Since the amino terminus of the mature 5-HT3A subunit is unknown, the amino acid numbering system used here includes the signal sequence and starts from the initial methionine. tsA201 cells were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 U/mL streptomycin. Cultures at 50–60% confluence were transfected with 10 μg receptor cDNA per 100 mm dish using Fugene transfection reagent (Roche Diagnostics, Indianapolis, IN). Maximal expression was obtained 36–72 h after transfection.
Ligand-binding assays
Transfected cells were processed as previously described (15) and membranes were incubated for 2 h at 37°C in 154 mM NaCl, 20 mM Tris-HCl, pH 7.4, containing the appropriate concentrations of the competing unlabeled ligand (e.g., dTC) and radioligand ([3H]granisetron, 85 Ci/mmol; PerkinElmer, Waltham, MA). Binding was terminated by rapid vacuum filtration onto GF/B filters. Nonspecific binding was defined as that which was not displaced by 10 μM m-chlorophenyl biguanide. IC50 values for the various dTC analogs were determined by fitting the data to the following equation:
| (1) |
where θ is the fractional amount of [3H]granisetron bound in the presence of the antagonist at concentration [I] compared with that in the absence of antagonist, IC50 is the concentration of antagonist at which θ = 0.5, and n is the apparent Hill coefficient. Ki values were calculated from the IC50 values and the Kd for [3H]granisetron using the Cheng-Prusoff relation (28):
| (2) |
where [L] is the concentration of [3H]granisetron used to determine the IC50 value in the experiment, and Kd is the dissociation constant for [3H]granisetron. Error estimates of ΔΔGint values calculated from Ki values were obtained through analysis of propagation of errors (29).
dTC analogs
The structures of the dTC analogs used in this study are shown in Fig. 1. Two of the compounds—dTC (Sigma, St. Louis, MO) and metocurine (Diosynth, Chicago, IL)—were obtained commercially, and the others were obtained from Dr. Steen Pedersen of Baylor University College of Medicine (23,24). The purity of all compounds was checked by high-performance liquid chromatography both before use and after prolonged incubation with the assay buffers.
Figure 1.
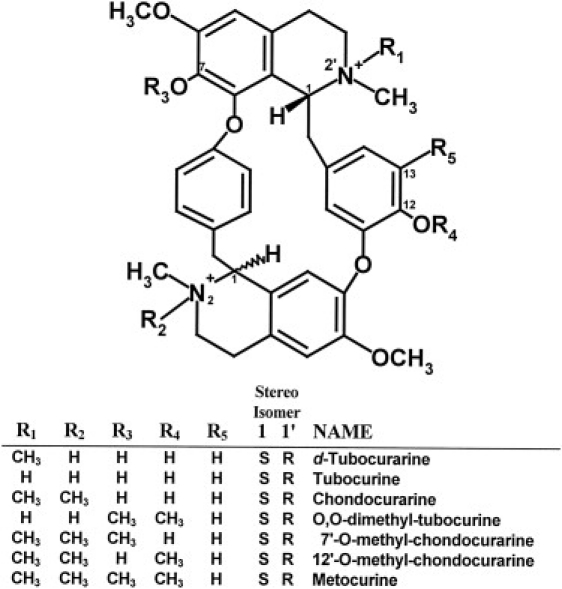
dTC analogs used in this study.
Immunofluorescence analysis of receptor expression
tsA201 cells on poly-L-lysine-coated coverslips were fixed with 4% paraformaldehyde in Tris-buffered saline (0.1 M Tris, 154 mM NaCl, pH 7.4) for 45 min, followed by three washes with phosphate-buffered saline (PBS). Intracellular receptor expression was determined by incubation with PBS containing 0.1% Triton X-100 for 30 min, and Triton X-100 was omitted to visualize receptor surface expression. Nonspecific antibody binding was blocked with 1% bovine serum albumin in PBS for 5 min, followed by incubation with the primary rabbit 5-HT3A polyclonal antibody pAb120 (30) for 90 min. Cells were washed extensively with bovine serum albumin/PBS before incubation with a secondary antibody blocking buffer containing 10% goat serum (Invitrogen; Carlsbad, CA) in PBS for 10 min. Cells were incubated with FITC-conjugated goat anti-rabbit IgG (Jackson Immunoresearch Laboratories, West Grove, PA) for 90 min, followed by extensive washing with goat serum/PBS. Stained preparations were mounted and analyzed using a Nikon PCM 2000 laser-scanning confocal imaging system. All images were recorded using threshold black levels set from imaging nontransfected tsA201 control cell preparations.
Molecular modeling and ligand docking
A model of the extracellular domain of 5-HT3A pentamers was generated using MODELLER 9v5 (31,32). The structures of the Aplysia AChBP in the apo (PDB ID: 2BYN) and methyllycaconitine-bound (PDB ID: BYR) forms were used as templates because they are representative of the structure in the resting state and with a small-molecule antagonist bound (both forms have similar conformations (33)). The sequence alignment between the templates and 5-HT3A monomers was performed with the SALIGN function of MODELLER, which uses a variable gap-opening penalty that depends on the three-dimensional structure of the template. All five subunits were modeled simultaneously to ensure structural integrity between subunit interfaces, and polar hydrogens were included to allow for main-chain hydrogen bonding. When additional experimentally derived distance restraints were included in the modeling process, the distance was harmonically restrained to be around the specified value ± a standard deviation of 0.5 Å. A set of 30–50 models was generated, and ProSA (34) was used to evaluate the generated models and to identify regions that might need further refinement by manually adjusting the alignment. The model that was ranked highest by ProSA was chosen for ligand docking.
Ligand docking was performed using AutoDock4 (35). The scoring functions used in AutoDock can discriminate between near-native and misdocked conformations of the ligand, and the conformations of ligands docked in a binding site agree with bound conformations in crystal structures of ligand-protein complexes (36). Docking was performed on a 30 × 30 × 40 Å grid with a spacing of 0.375 Å. The size of the grid ensures that the ligand has sufficient freedom to be docked in all possible orientations but is not allowed to move far outside of the binding site. We performed 256 separate simulations and chose the one that was most consistent with our experimentally derived criteria (i.e., 2′N of dTC near N128, 2N near R92, and 12 –OH near E236).
Molecular-dynamics simulations
For molecular-dynamics (MD) simulations, the pentameric 5-HT3A extracellular domain with bound dTC obtained from AutoDock was placed in a periodic box of approximate dimensions 10 nm × 10 nm × 10 nm. Counterions were added to provide overall charge neutrality, and ∼28,000 water molecules were added. The simulation system contained ∼100,000 atoms. The protein was modeled with the AMBER parm99SB force field (37) and dTC was modeled with the AMBER gaff force field (38) with charge assignment from the AM1-BCC model (39,40). Water molecules were modeled with the TIP3P force field (41). All simulations were conducted with NAMD v2.6 (42) with a time step of 2 fs. The simulations were run at a constant temperature of 310 °K and pressure of 1.013 bar using a Langevin thermostat with a damping coefficient of 5 ps−1 and a Langevin piston with a period of 100 fs and a decay of 50 fs. All simulations were carried out to ∼5 ns; for each simulation, the root mean-square deviation saturated at ∼2.5 ns.
Results
E236 is located in the N-terminal portion of the β10 strand of loop C. Previous work on this residue has produced conflicting results. Schreiter et al. (43) reported that replacement of E236 with aspartate (E236D) abolished radioligand binding, and electrophysiological responses were reduced to <1% of that seen for wild-type (WT) receptors, whereas the E236Q substitution produced functional receptors but reduced the apparent affinity for agonists and antagonists >20-fold (please note that in their publication, Schreiter et al. refer to this residue as E235). Furthermore, the E236D receptors that were synthesized were mostly trapped in intracellular compartments. On the other hand, Thompson et al. (13) did not detect any significant difference in [3H]granisetron affinity relative to WT receptors for E236D receptors. Based on these results, Schreiter et al. proposed that E236 plays a significant role in both receptor trafficking and affinity, whereas Thompson et al. did not propose a role for this residue. The proposed location of E236 and the conflicting results concerning the E236D mutation led us to reexamine this residue.
We examined the interaction of E236D, E236Q, E236N, and E236A receptors with competitive antagonists. In agreement with Schreiter et al. (43) and in contrast to Thompson et al. (13), we find that the E236D mutation abolishes [3H]granisetron binding. Furthermore, using confocal microscopy to examine the subcellular distribution of receptors, E236D receptors were not expressed on the cell surface and were trapped in intracellular compartments (Fig. 2), also in agreement with Schreiter et al. (Thompson et al. did not examine the subcellular distribution of E236D receptors.). E236Q, E236N, and E236A receptors were transported to the cell surface and show small (3- to 8-fold) decreases in granisetron affinity (WT Kd = 2.2 nM, mutant receptor Kd = 6–18 nM).
Figure 2.
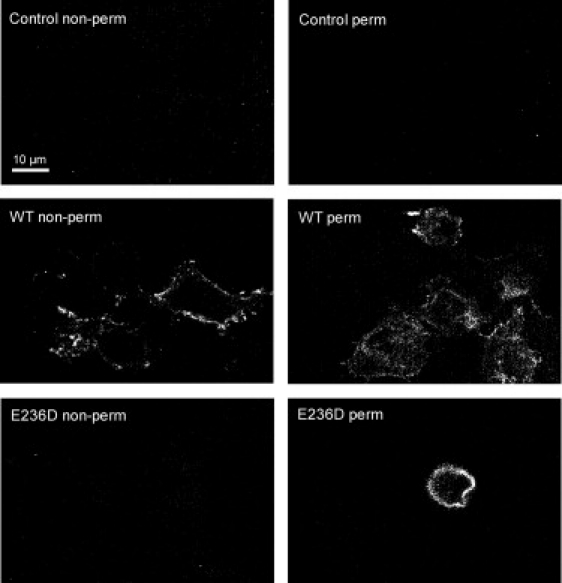
Subcellular localization of WT and mutant 5-HT3Rs. Confocal images of immunofluorescent-labeled permeabilized and nonpermeabilized cells expressing WT or E236D 5-HT3Rs are shown. Surface expression is only visible in nonpermeabilized cells expressing WT 5-HT3Rs. Intracellular receptor expression of WT and E236D 5-HT3Rs is observed in permeabilized cell preparations. No fluorescence is visible in untransfected control cells. Scale bar: 10 μm.
We examined the interaction of a panel of dTC analogs (Fig. 1) with WT and mutant receptors. Table 1 shows estimates of the affinity of WT, E236Q, E236N, and E236A receptors for the dTC analogs. All three mutations reduced the affinity for dTC 5- to 35-fold, with the greatest decrease in affinity observed for the E236N receptor. Fig. 3 shows inhibition curves for dTC, chondocurarine, and 12′-O-methyl-chondocurarine (12′-OMCC) for WT, E236Q, and E236N receptors. Chondocurarine differs from dTC by the presence of an additional methyl group at the 2N position, whereas 12′-OMCC differs from dTC by a second methyl group at the 2N and replacement of the 12 –OH to a –OCH3 group (Fig. 1); the latter change is also the only difference between chondocurarine and 12′-OMCC. For WT and E236Q receptors, replacement of the 12 –OH by a –OCH3 group resulted in a large decrease in apparent affinity relative to dTC and chondocurarine, whereas the same replacement had only a small effect on affinity for the E236N receptor. These data suggest that E236 interacts with the 12 –OH in dTC.
Table 1.
Affinity of dTC analogs for mutant and WT 5-HT3Rs
| Ligand | WT pKi ± SD | E236Q pKi ± SD | E236N pKi ± SD | E236A pKi ± SD |
|---|---|---|---|---|
| dTC | 7.20 ± 0.02 | 6.11 ± 0.05∗ | 5.65 ± 0.06∗ | 6.53 ± 0.07∗ |
| tubocurine | 7.03 ± 0.03 | 6.18 ± 0.08∗ | 5.67 ± 0.06∗ | 6.79 ± .06 |
| chondocurarine | 6.63 ± 0.06 | 5.45 ± 0.07∗ | 5.24 ± 0.04∗ | 6.33 ± 0.04∗ |
| metocurine | 5.34 ± 0.03 | 4.39 ± 0.05∗ | 5.43 ± 0.09 | 6.13 ± 0.07∗ |
| O,O-dimethyltubocurine | 5.67 ± 0.05 | 4.46 ± 0.09∗ | 5.08 ± 0.08∗ | 6.47 ± 0.08∗ |
| 7′-O-methylcondocurarine | 7.19 ± 0.02 | 5.67 ± 0.04∗ | 5.20 ± 0.09∗ | 6.66 ± 0.07∗ |
| 12′-O-methylchondocurarine | 5.51 ± 0.02 | 4.45 ± 0.05∗ | 5.06 ± 0.06 | 5.93 ± 0.05∗ |
Estimates of pKi values were calculated from experimentally determined pIC50 values for the inhibition of [3H]granisetron binding to WT or mutant receptors as described in the Materials and Methods section. Errors represent the error determined by the Levenberg-Marquardt regression routine used in the fitting. Values for the mutant receptors marked with ∗ are statistically different from WT at a 95% confidence level using Student's t-test.
Figure 3.
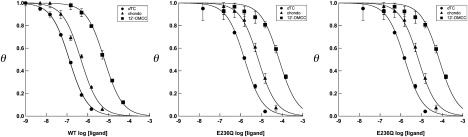
Effects of mutations at E236 on dTC, chondocurarine, and 12′-O-methylchondoocurine affinity. The concentration dependence of inhibition of [3H]granisetron binding to WT, E236Q, and E236N 5-HT3Rs by dTC (•), chondocurarine (▴), and 12′-OMCC (■) are shown. Each data point represents the mean ± SE of three determinations. The solid curves are drawn according to Eq. 1 using log(IC50) values of −6.90 (WT, dTC), −6.33 (WT, chondocurarine), −5.25 (WT,12′-OMCC), −5.81 (E236Q, dTC), −5.15 (E236Q, chondocurarine), −4.14 (E236Q,12′-OMCC), −5.34 (E236N, dTC), −4.91 (E236N, chondocurarine), and −4.76 (E236N,12′-OMCC).
To further examine the interaction of dTC with E236, we analyzed the effects of substitutions at various positions in dTC using double-mutant cycle analysis (18). The underlying logic of this approach is that if residue x in the binding site interacts with substituent y on the ligand, then the effect of mutating x should depend on whether substituent y in the ligand is changed or not. The free energy of interaction, ΔΔGint, is calculated from the Ki values as
| (3) |
where , W is the WT receptor, M is the mutant receptor, and L1 and L2 are the two ligands being compared. The absolute value of ΔΔGint is used in comparisons, as the sign of ΔΔGint depends on which ligand is chosen as L1 and which is chosen as L2. This approach has been applied to identify points of contact between peptide toxins and K+ channels (44,45), AChRs and α-neurotoxins (46) and dTC analogs (47), and 5-HT3R and granisetron (16) and dTC (15). |ΔΔGint| values ≥ 1 kcal/mol are consistent with a small spatial separation between the residue under investigation and the portion of the ligand that is altered (48).
Fig. 4 shows mutant cycles examining the effects of the E236N and E236Q mutations on chondocurarine, 7′-OMCC, and 12′-OMCC affinity. These cycles examine the effects of replacement of the –OH group by a –OCH3 group at either the 7 position (the chondocurarine/7′-OMCC cycles) or the 12 position (the chondocurarine/12′-OMCC cycles). The only cycle that shows a |ΔΔGint| ≥ 1 kcal/mol is the WT/E236N/chondocurarine/12′-OMCC cycle, with |ΔΔGint| = 1.6 ± 0.3 kcal/mol. Table 2 gives the results of 15 different mutant cycles constructed for various WT/mutant receptor pairs. In all cases where |ΔΔGint| > 1 kcal/mol, the substituent at the 12 position differs between L1 and L2, whereas for all cases where |ΔΔGint| < 1, the substituent at this position is the same for the two ligands in the cycle (either –OH or –OCH3). These data are consistent with E236 making a physical interaction with the 12 position of dTC.
Figure 4.
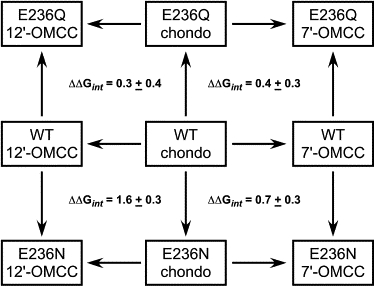
Double-mutant cycles for WT, E236Q, and E236N receptors and chondocurarine, 12′-OMCC, and 7′-OMCC. The interaction coefficient, ΔΔGint, for each combination of the receptors and ligands was determined from the Ki values of each ligand for each receptor. The ΔΔGint value of 1.6 ± 0.3 kcal/mol for the WT/E236N/chondocurarine/12′-OMCC cycle indicates that an interaction between E236 and the 12 OH of the ligand is altered by the E236N mutation.
Table 2.
ΔΔGint values in kcal/mol for various ligand pairings
| Ligand pair | Differences | E236Q ΔΔGint | E236N ΔΔGint | E236A ΔΔGint |
|---|---|---|---|---|
| dTC/metocurine | 2N, 7, 12 | 0.1 ± 0.2 | 2.2 ± 0.3 | 1.9 ± 0.3 |
| dTC/tubocurine | 2′N | 0.3 ± 0.4 | 0.3 ± 0.4 | 0.6 ± 0.3 |
| dTC/O,O-DMTC | 2′N, 7, 12 | -0.2 ± 0.3 | 1.6 ± 0.3 | 2.3 ± 0.3 |
| metocurine/tubocurine | 2N, 2′N, 7, 12 | 0.2 ± 0.4 | -1.9 ± 0.4 | -1.3 ± 0.3 |
| metocurine/O,O-DMTC | 2N, 2′N | -0.3 ± 0.3 | -0.6 ± 0.4 | 0.4 ± 0.3 |
| O,O-DMTC/tubocurine | 7, 12 | 0.5 ± 0.5 | -1.4 ± 0.4 | -1.7 ± 0.3 |
| dTC/chondocurarine | 2N | -0.2 ± 0.3 | 0.1 ± 0.3 | 0.3 ± 0.3 |
| dTC/7′-OMCC | 2N, 7 | -0.6 ± 0.2 | 0.6 ± 0.3 | 0.2 ± 0.3 |
| chondocurarine/7′-OMCC | 7 | -0.4 ± 0.3 | -0.6 ± 0.3 | 0.1 ± 0.3 |
| O,O-DMC/7′-OMCC | 7, 12 | -0.4 ± 0.3 | -2.2 ± 0.3 | -2.1 ± 0.3 |
| dTC/12′-OMCC | 2N, 12 | 0.2 ± 0.3 | 1.7 ± 0.3 | 1.5 ± 0.2 |
| 7′-OMCC/12′-OMCC | 7, 12 | 0.8 ± 0.3 | 2.3 ± 0.3 | 1.3 ± 0.2 |
| metocurine/12′-OMCC | 7 | 0.1 ± 0.3 | -0.5 ± 0.4 | -0.4 ± 0.3 |
| chondocurarine/12′OMCC | 12 | 0.5 ± 0.4 | 1.7 ± 0.4 | 1.2 ± 0.3 |
| metocurine/7′-OMCC | 12 | -0.7 ± 0.2 | -2.8 ± 0.3 | -1.7 ± 0.3 |
ΔΔGint values were determined for double-mutant cycles using WT and mutant receptors, and the indicated ligand pairs from the Ki values according to Eq. 3.
Discussion
It is extremely challenging to interpret data from site-directed mutagenesis studies in terms of protein structure. A number of residues in loops A–F of 5-HT3R have been implicated as playing potential roles in ligand-receptor interactions (11–13,15,16,21,22,27,43,49–52). However, cataloging residues that may interact with ligands is the first step in elucidating the architecture of the ligand-binding site. In this report, we focus on E236, which is within loop C in the ligand-binding domain. Previous work on this residue has produced conflicting results regarding the effect of the E236D mutation. One group reported that E236D receptors were severely compromised, with cell-surface expression levels and electrophysiological responses reduced >100-fold relative to WT (43), whereas the other reported that E236D receptors had radioligand affinity similar to WT receptors (13). Our data are consistent with the former and suggest that E236 plays a role in proper assembly and/or folding of the receptor. It is unclear why a conservative replacement (E → D) has such a profound effect on receptor assembly when other replacements (Q, N, and A) do not. We have made the E236D mutant several different times, sequencing the entire coding region each time, and obtained the same result each time. Although the effect is real, and confirms the independent finding by another group (43), the underlying mechanism is unclear.
Residues homologous to E236 in other Cys-loop ligand-gated ion channels and the AChBP have also been implicated in ligand-receptor interactions. In the α-subunit of the AChR, dTC protects cysteine-substituted αD200 (αD200C) from alkylation (53). In the β-subunit of the GABAAR, βR207 faces into the binding site and stabilizes GABA binding to the receptor (54). In the Aplysia AChBP, the homologous residue (D197) forms a salt bridge with the antagonist α-conotoxin ImI (33). It is clear that this residue is located in the ligand-binding domain and plays a role in receptor function.
Double-mutant cycle analysis allows the effects of mutations to be interpreted in terms of specific ligand-receptor interactions. This type of analysis allows one to determine which parts of the ligand interact with which residues in the binding site. Mapping these points of interaction onto the three-dimensional structure of ligands provides information about the spatial orientation of residues in the ligand-binding site. One can use these distance measurements as additional constraints in the modeling process and further refine a model for the binding site.
Our data suggest that E236 makes a physical interaction with the 12 –OH of dTC. The data are consistent with a direct physical interaction; however, the effect could be due to either an interaction between the ligand and the receptor or a conformational rearrangement in the receptor induced by the mutation. Although it is not possible a priori to determine which mechanism applies, there are guidelines one can apply to discriminate between these two possibilities.
If there is a direct interaction between part of the ligand and a specific residue in the receptor, they must be physically close. In a study of barnase/barstar interactions, Schreiber and Fersht (48) showed that |ΔΔGint| values ≥ 1 kcal/mol were correlated with the residues in question being within ≤ 4 Å of each other, whereas lower free energies of interaction were correlated with greater spatial separation. The larger the |ΔΔGint| value, the more likely it is that the effect is due to a direct physical interaction with the region of the ligand that has been modified. We employ |ΔΔGint| ≥ 1 as our cutoff for identifying an interaction.
On the other hand, if the observed coupling is due to a conformational rearrangement in the receptor induced by the mutation, it is unlikely that the coupling would be limited to a single portion of the ligand. Analysis of 15 different mutant cycles for each receptor employing seven different dTC analogs shows that the coupling localizes to a single part of dTC (the 12 –OH), which is what one would expect for disruption of a specific interaction. Furthermore, cycles employing two very different substitutions at the same position (E236N and E236A) show the same coupling. It is unlikely that these two mutations would produce exactly the same conformational change in the receptor. Thus, the data are most consistent with the notion that E236 is in close physical contact with the 12 –OH of dTC.
The mutant cycles indicate coupling between E236 and the 12 –OH of dTC for E236N and E236A, but not E236Q, receptors. WT and E236Q receptors show a large (30- to 60-fold) decrease in affinity relative to dTC for those compounds with a 12 –OCH3 (metocurine, OODMC, and 12′-OMCC), whereas E236N and E236A receptors show smaller decreases (<4-fold) for the same changes. The N and A side chains have smaller volumes than either E or Q, with N being 20.7 Å3 smaller than E, A being 49.8 Å3 smaller than E, and Q being 5.5 Å3 larger than E (55). Introduction of the bulky –OCH3 substitution may create a steric clash with the E and Q side chains, but not the smaller N and A side chains, explaining the differential effects of the mutations on analogs with a 12 –OH versus a 12 –OCH3.
The sign of the ΔΔGint values for the mutant cycles (positive for cycles in which the –OH is replaced by –OCH3; negative for the converse) indicates that replacement of the –OH with –OCH3 either removes a favorable interaction or creates an unfavorable one. The |ΔΔGint| values obtained from the E236A and E236N cycles are 1.3–2.8 kcal/mol, within the range for a hydrogen bond. Both E and Q can make a hydrogen bond with 12 –OH; however, the E236A and E236N mutants cannot, due to either their chemical nature (E236A) or smaller size (E236N). Conversion of the –OH to –OCH3 could disrupt this interaction and affect WT and E236Q receptors equally, consistent with the fact that the ΔΔGint values for E236Q cycles do not meet our criteria for significance.
However, all mutants show a reduction in affinity for dTC relative to WT receptors. The smallest reduction in affinity is seen for the E236A receptor, which is not consistent with a hydrogen bond between E236 and the 12 –OH. Rather than the removal of a favorable interaction (e.g., a hydrogen bond), it may be the introduction of an unfavorable steric interaction by conversion of the –OH to the bulkier –OCH3 substituent that the mutant cycle analysis detects. E236 and E236Q are approximately the same size and exhibit similar differential sensitivity to –OCH3 versus –OH substituents, whereas the smaller E236N and E236A do not. This appears to be a more reasonable interpretation of the results. However, regardless of the underlying mechanism for the coupling, the double-mutant cycle analysis clearly supports the notion that E236 and the 12 –OH are in close physical contact.
Previous work in our laboratory suggests that N128 (loop A, (+)-face of the binding site) interacts with the 2′N and R92 (loop D, (−)-face) interacts with the 2N of dTC (15). Another group has proposed that N128 faces away from the ligand-binding site (56). This conclusion was based on the observation that the N128A mutation has no effect on [3H]granisetron affinity, which was interpreted to mean that this residue played no role in antagonist-receptor interaction. We also reported that although the N128A mutation has no effect on [3H]granisetron affinity, it does have a significant effect on dTC affinity (15). This formed the basis for the double-mutant cycle analysis, which led to the conclusion that N128 interacts with the 2′N of dTC. Since the conclusion of the other group was based on a negative result obtained with the use of a single ligand, and ours was based on the use of multiple ligands and a demonstrated effect of the mutation on antagonist-receptor interactions, we believe our placement of N128 as forming part of and facing into the binding site is correct.
The identification of a third residue (E236, within loop C, (+)-face) that interacts with a specific portion of dTC gives us an opportunity to delineate the spatial relationships among these three residues in the binding site. dTC is a constrained molecule of known three-dimensional structure (57,58). MD simulations of dTC in solution show that the 2N-2′N distance during a 5 ns simulation is relatively constant. The two nitrogens are separated by 10.4 ± 0.2 Å during the entire simulation period (data not shown), supporting the notion that dTC is a rigid molecule. By mapping residues in the binding site to the points of interaction with dTC, we can determine spatial relationships between specific residues. Assuming that atoms are hard spheres (with dimensions determined by their van der Waals radii (59)) that make close contact with each other, we obtain estimates of the distances between the guanidinium of R92, the amide of N128, and the carboxylate of E236. The N128:E236 distance is ∼9 Å, the N128:R92 distance is ∼16 Å, and the R92:E238 distance is ∼12 Å. These distances become additional restraints in the homology modeling process, allowing refinement of the model with experimental data. The refined homology model derived with the use of these experimentally obtained distance restraints are then used for ligand docking with AutoDock.
One weakness of homology modeling is that although it does a good job in predicting the main-chain conformation, it is weaker for side-chain conformation (60). Although MD simulations are far more computationally intensive than homology modeling and ligand docking, they can be used to refine the static structures obtained by homology modeling and ligand docking through relaxation to a more thermodynamically stable conformation governed by interactions within the protein, between the protein and the ligand, and between the complex and its environment. This approach has been used with the AChBP (61,62), AChR (63–65), and GlyR (66). Eriksson and Roux (67) performed homology modeling of the Shaker K+ channel followed by MD-driven docking of agitoxin2 with imposition of spatial relationships/restraints obtained from double-mutant cycle analysis (45). The resulting simulations produced channel-toxin complexes that were in strong agreement with the experimental data.
We used the dTC-receptor complex obtained from docking simulations, using the model refined by employing the distance restraints described above as the initial starting state, and performed MD simulations for 5 ns. The structures relaxed to a stable state within 2.5 ns, and the stable conformation is shown in Fig. 5. Also shown in the figure are the locations of five other residues that have been implicated in 5-HT3R/dTC interactions on the basis of effects of mutations on dTC affinity: one on the (+)-face (D229, loop C) (21)) and four on the (−)-face (W90, loop D (27); Y141, loop E (52); Y153, loop E (52); and I207, loop F (22)). Examination of this complex shows that most of the residues cluster around docked dTC. Furthermore, R92, N128, and E236 are positioned to make the interactions with dTC predicted from experimental data, indicating that this approach can provide a more realistic picture of the ligand-receptor complex than the more static docking procedures used previously.
Figure 5.
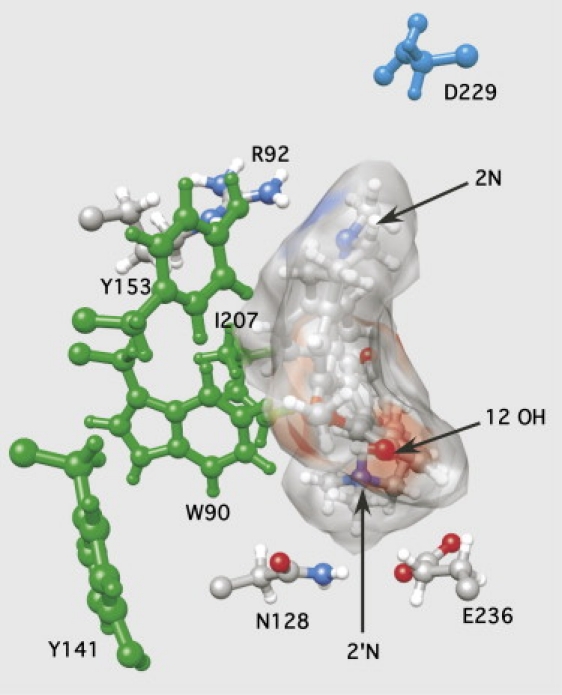
Representation of dTC in the 5-HT3R binding site looking down from the top (i.e., facing the synapse) of the receptor. A model of the dTC/5-HT3R complex was obtained using MD calculations as described in the text. Residues used in the mapping (R92, N128, and E236) are shown in standard colors, and additional residues implicated in receptor/dTC interactions are shown in cyan ((+)-face) or green ((−)-face)). Note that R92 is close to the 2N, N128 is close to the 2′N, and E236 is close to the 12 –OH.
This study shows the power of combining double-mutant cycle analysis with the use of a rigid ligand to probe ligand-receptor interactions in such a way as to allow different portions of the ligand to be mapped onto specific residues in the receptor. This approach is applicable to any ligand-receptor system in which a conformationally constrained ligand that can be modified is employed. The ligand can be either a small-molecule compound like dTC, with various substituents, or recombinant peptide toxins such as scorpion toxins and conotoxins, which assume fairly compact structures constrained by disulfide bonds. These peptide toxins target a number of ion channel families, including voltage-gated K+ channels (e.g., agitoxin2 (45)), voltage-gated Na+ channels (e.g., scorpion β-toxins (68)), voltage-gated Ca2+ channels (e.g., ω-conotoxins (69)), and various nicotinic AChR subtypes (e.g., α-conotoxins (70)). In conjunction with molecular modeling studies, this molecular ruler approach provides an iterative process for modeling and experimentally testing models, which in turn can accelerate the process of mapping the three-dimensional architecture of a ligand-binding domain. It provides an alternative to resonance energy-transfer techniques to obtain spatial information in macromolecular complexes, and is advantageous because it does not require the use of reporter groups, which may alter receptor function when inserted into the ligand-binding site. Extensive mapping of the relative positions of residues in the binding site should allow the elucidation of the architecture of the ligand-binding domain, and thus provide useful information for the design of novel pharmacological agents with both high affinity and high specificity for use as therapeutic agents.
Acknowledgments
We thank Dr. Robert Nichols for the 5-HT3A-specific antibody pAb120 and guidance on the confocal microscopy experiments, Dr. Steen Pedersen for the dTC analogs, and Dr. Pat Loll for helpful discussions.
This work was supported in part by a grant from the American Heart Association Pennsylvania-Delaware Affiliate to M.M.W., and National Science Foundation grant DMR-0427643 to C.F.A.
References
- 1.Connolly C.N., Wafford K.A. The Cys-loop superfamily of ligand-gated ion channels: the impact of receptor structure on function. Biochem. Soc. Trans. 2004;32:529–534. doi: 10.1042/BST0320529. [DOI] [PubMed] [Google Scholar]
- 2.Lester H.A., Dibas M.I., Dougherty D.A. Cys-loop receptors: new twists and turns. Trends Neurosci. 2004;27:329–336. doi: 10.1016/j.tins.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Reeves D.C., Lummis S.C. The molecular basis of the structure and function of the 5-HT3 receptor: a model ligand-gated ion channel (review) Mol. Membr. Biol. 2002;19:11–26. doi: 10.1080/09687680110110048. [DOI] [PubMed] [Google Scholar]
- 4.Maricq A.V., Peterson A.S., Julius D. Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science. 1991;254:432–437. doi: 10.1126/science.1718042. [DOI] [PubMed] [Google Scholar]
- 5.Hussy N., Lukas W., Jones K.A. Functional properties of a cloned 5-hydroxytryptamine ionotropic receptor subunit: comparison with native mouse receptors. J. Physiol. 1994;481:311–323. doi: 10.1113/jphysiol.1994.sp020441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies P.A., Pistis M., Kirkness E.F. The 5-HT3B subunit is a major determinant of serotonin-receptor function. Nature. 1999;397:359–363. doi: 10.1038/16941. [DOI] [PubMed] [Google Scholar]
- 7.Jensen A.A., Davies P.A., Krzywkowski K. 3B but which 3B? And that's just one of the questions: the heterogeneity of human 5-HT3 receptors. Trends Pharmacol. Sci. 2008;29:437–444. doi: 10.1016/j.tips.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brady C.A., Stanford I.M., Barnes N.M. Pharmacological comparison of human homomeric 5-HT3A receptors versus heteromeric 5-HT3A/3B receptors. Neuropharmacology. 2001;41:282–284. doi: 10.1016/s0028-3908(01)00074-0. [DOI] [PubMed] [Google Scholar]
- 9.Hanna M.C., Davies P.A., Kirkness E.F. Evidence for expression of heteromeric serotonin 5-HT(3) receptors in rodents. J. Neurochem. 2000;75:240–247. doi: 10.1046/j.1471-4159.2000.0750240.x. [DOI] [PubMed] [Google Scholar]
- 10.Hazai E., Joshi P., Bikadi Z. A comprehensive study on the 5-hydroxytryptamine(3A) receptor binding of agonists serotonin and m-chlorophenylbiguanidine. Bioorg. Med. Chem. 2009;17:5796–5805. doi: 10.1016/j.bmc.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 11.Joshi P.R., Suryanarayanan A., Bikádi Z. Interactions of granisetron with an agonist-free 5-HT3A receptor model. Biochemistry. 2006;45:1099–1105. doi: 10.1021/bi051676f. [DOI] [PubMed] [Google Scholar]
- 12.Thompson A.J., Padgett C.L., Lummis S.C. Mutagenesis and molecular modeling reveal the importance of the 5-HT3 receptor F-loop. J. Biol. Chem. 2006;281:16576–16582. doi: 10.1074/jbc.M601265200. [DOI] [PubMed] [Google Scholar]
- 13.Thompson A.J., Price K.L., Lummis S.C. Locating an antagonist in the 5-HT3 receptor binding site using modeling and radioligand binding. J. Biol. Chem. 2005;280:20476–20482. doi: 10.1074/jbc.M413610200. [DOI] [PubMed] [Google Scholar]
- 14.Thompson A.J., Lochner M., Lummis S.C.R. Loop B is a major structural component of the 5-HT3 receptor. Biophys. J. 2008;95:5728–5736. doi: 10.1529/biophysj.108.135624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan D., Meyer J.K., White M.M. Mapping residues in the ligand-binding domain of the 5-HT(3) receptor onto d-tubocurarine structure. Mol. Pharmacol. 2006;70:571–578. doi: 10.1124/mol.106.024075. [DOI] [PubMed] [Google Scholar]
- 16.Yan D., White M.M. Spatial orientation of the antagonist granisetron in the ligand-binding site of the 5-HT3 receptor. Mol. Pharmacol. 2005;68:365–371. doi: 10.1124/mol.105.011957. [DOI] [PubMed] [Google Scholar]
- 17.Brejc K., van Dijk W.J., Sixma T.K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 18.Horovitz A. Double-mutant cycles: a powerful tool for analyzing protein structure and function. Fold. Des. 1996;1:R121–R126. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- 19.Jenkinson D.H. The antagonism between tubocurarine and substances which depolarize the motor end-plate. J. Physiol. 1960;152:309–324. doi: 10.1113/jphysiol.1960.sp006489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peters J.A., Malone H.M., Lambert J.J. Antagonism of 5-HT3 receptor mediated currents in murine N1E-115 neuroblastoma cells by (+)-tubocurarine. Neurosci. Lett. 1990;110:107–112. doi: 10.1016/0304-3940(90)90796-c. [DOI] [PubMed] [Google Scholar]
- 21.Hope A.G., Belelli D., Peters J.A. Molecular determinants of (+)-tubocurarine binding at recombinant 5-hydroxytryptamine3A receptor subunits. Mol. Pharmacol. 1999;55:1037–1043. doi: 10.1124/mol.55.6.1037. [DOI] [PubMed] [Google Scholar]
- 22.Zhang R., Wen X., Machu T. The role of loop F residues in determining differential d-tubocurarine potencies in mouse and human 5-hydroxytryptamine 3A receptors. Biochemistry. 2006;46:1194–1204. doi: 10.1021/bi0616100. [DOI] [PubMed] [Google Scholar]
- 23.Papineni R.V., Pedersen S.E. Interaction of d-tubocurarine analogs with the mouse nicotinic acetylcholine receptor. Ligand orientation at the binding site. J. Biol. Chem. 1997;272:24891–24898. doi: 10.1074/jbc.272.40.24891. [DOI] [PubMed] [Google Scholar]
- 24.Pedersen S.E., Papineni R.V. Interaction of d-tubocurarine analogs with the Torpedo nicotinic acetylcholine receptor. Methylation and stereoisomerization affect site-selective competitive binding and binding to the noncompetitive site. J. Biol. Chem. 1995;270:31141–31150. doi: 10.1074/jbc.270.52.31141. [DOI] [PubMed] [Google Scholar]
- 25.Yan D., Pedersen S.E., White M.M. Interaction of d-tubocurarine analogs with the 5HT3 receptor. Neuropharmacology. 1998;37:251–257. doi: 10.1016/s0028-3908(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 26.Hope A.G., Downie D.L., Burchell B. Cloning and functional expression of an apparent splice variant of the murine 5-HT3 receptor A subunit. Eur. J. Pharmacol. 1993;245:187–192. doi: 10.1016/0922-4106(93)90128-v. [DOI] [PubMed] [Google Scholar]
- 27.Yan D., Schulte M.K., White M.M. Structural features of the ligand-binding domain of the serotonin 5HT3 receptor. J. Biol. Chem. 1999;274:5537–5541. doi: 10.1074/jbc.274.9.5537. [DOI] [PubMed] [Google Scholar]
- 28.Cheng Y., Prusoff W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 29.Ku H. Notes on the use of propagation of error formulas. J. Res. Natl. Bur. Stand., C Eng. Instrum. 1966;70C:263–273. [Google Scholar]
- 30.Spier A.D., Wotherspoon G., Lummis S.C. Antibodies against the extracellular domain of the 5-HT3 receptor label both native and recombinant receptors. Brain Res. Mol. Brain Res. 1999;67:221–230. doi: 10.1016/s0169-328x(99)00055-8. [DOI] [PubMed] [Google Scholar]
- 31.Fiser A., Do R.K., Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 33.Hansen S.B., Sulzenbacher G., Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sippl M.J. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 35.Morris G.M., Huey R., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osterberg F., Morris G., Goodsell D. Automated docking to multiple target structures: Incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins. 2002;46:34–40. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- 37.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J., Wolf R.M., Case D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 39.Jakalian A., Bush B., Bayly C. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 40.Jakalian A., Jack D.B., Bayly C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 41.Jorgensen W., Chandrasekhar J., Klein M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 42.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schreiter C., Hovius R., Vogel H. Characterization of the ligand-binding site of the serotonin 5-HT3 receptor: the role of glutamate residues 97, 224, and 235. J. Biol. Chem. 2003;278:22709–22716. doi: 10.1074/jbc.M301801200. [DOI] [PubMed] [Google Scholar]
- 44.Hidalgo P., MacKinnon R. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 1995;268:307–310. doi: 10.1126/science.7716527. [DOI] [PubMed] [Google Scholar]
- 45.Ranganathan R., Lewis J.H., MacKinnon R. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 1996;16:131–139. doi: 10.1016/s0896-6273(00)80030-6. [DOI] [PubMed] [Google Scholar]
- 46.Malany S., Osaka H., Taylor P. Orientation of α-neurotoxin at the subunit interfaces of the nicotinic acetylcholine receptor. Biochemistry. 2000;39:15388–15398. doi: 10.1021/bi001825o. [DOI] [PubMed] [Google Scholar]
- 47.Willcockson I.U., Hong A., Pedersen S.E. Orientation of d-tubocurarine in the muscle nicotinic acetylcholine receptor-binding site. J. Biol. Chem. 2002;277:42249–42258. doi: 10.1074/jbc.M205383200. [DOI] [PubMed] [Google Scholar]
- 48.Schreiber G., Fersht A.R. Energetics of protein-protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- 49.Boess F.G., Steward L.J., Martin I.L. Analysis of the ligand binding site of the 5-HT3 receptor using site directed mutagenesis: importance of glutamate 106. Neuropharmacology. 1997;36:637–647. doi: 10.1016/s0028-3908(97)00044-0. [DOI] [PubMed] [Google Scholar]
- 50.Sullivan N.L., Thompson A.J., Lummis S.C. Defining the roles of Asn-128, Glu-129 and Phe-130 in loop A of the 5-HT3 receptor. Mol. Membr. Biol. 2006;23:442–451. doi: 10.1080/09687860600831539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suryanarayanan A., Joshi P.R., Schulte M.K. The loop C region of the murine 5-HT3A receptor contributes to the differential actions of 5-hydroxytryptamine and m-chlorophenylbiguanide. Biochemistry. 2005;44:9140–9149. doi: 10.1021/bi050661e. [DOI] [PubMed] [Google Scholar]
- 52.Venkataraman P., Venkatachalan S.P., Schulte M.K. Identification of critical residues in loop E in the 5-HT3ASR binding site. BMC Biochem. 2002;3:15. doi: 10.1186/1471-2091-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sullivan D., Chiara D.C., Cohen J.B. Mapping the agonist binding site of the nicotinic acetylcholine receptor by cysteine scanning mutagenesis: antagonist footprint and secondary structure prediction. Mol. Pharmacol. 2002;61:463–472. doi: 10.1124/mol.61.2.463. [DOI] [PubMed] [Google Scholar]
- 54.Wagner D.A., Czajkowski C., Jones M.V. An arginine involved in GABA binding and unbinding but not gating of the GABA(A) receptor. J. Neurosci. 2004;24:2733–2741. doi: 10.1523/JNEUROSCI.4316-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zamyatnin A.A. Amino acid, peptide, and protein volume in solution. Annu. Rev. Biophys. Bioeng. 1984;13:145–165. doi: 10.1146/annurev.bb.13.060184.001045. [DOI] [PubMed] [Google Scholar]
- 56.Price K.L., Bower K.S., Lummis S.C. A hydrogen bond in loop A is critical for the binding and function of the 5-HT3 receptor. Biochemistry. 2008;47:6370–6377. doi: 10.1021/bi800222n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Codding P., James M. The crystal and molecular structure of a potent neuromuscular blocking agent: d-tubocurarine dichloride pentahydrate. Acta Crystallogr. B. 1973;29:935–942. [Google Scholar]
- 58.Reynolds C., Palmer R. The crystal structure, absolute configuration and stereochemistry of (+)-tubocurarine dibromide methanol solvate: a potent neuromuscular blocking agent. Acta Crystallogr. B. 1976;32:1431–1439. [Google Scholar]
- 59.Bondi A. van der Waals volumes and radii. J. Phys. Chem. 1964;68:441–451. [Google Scholar]
- 60.Wallner B., Elofsson A. All are not equal: a benchmark of different homology modeling programs. Protein Sci. 2005;14:1315–1327. doi: 10.1110/ps.041253405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hibbs R.E., Radic Z., Johnson D.A. Influence of agonists and antagonists on the segmental motion of residues near the agonist binding pocket of the acetylcholine-binding protein. J. Biol. Chem. 2006;281:39708–39718. doi: 10.1074/jbc.M604752200. [DOI] [PubMed] [Google Scholar]
- 62.Hibbs R.E., Talley T.T., Taylor P. Acrylodan-conjugated cysteine side chains reveal conformational state and ligand site locations of the acetylcholine-binding protein. J. Biol. Chem. 2004;279:28483–28491. doi: 10.1074/jbc.M403713200. [DOI] [PubMed] [Google Scholar]
- 63.Henchman R.H., Wang H.-L., McCammon J.A. Ligand-induced conformational change in the α7 nicotinic receptor ligand binding domain. Biophys. J. 2005;88:2564–2576. doi: 10.1529/biophysj.104.053934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Henchman R.H., Wang H.-L., McCammon J.A. Asymmetric structural motions of the homomeric α7 nicotinic receptor ligand binding domain revealed by molecular dynamics simulation. Biophys. J. 2003;85:3007–3018. doi: 10.1016/S0006-3495(03)74720-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Law R.J., Henchman R.H., McCammon J.A. A gating mechanism proposed from a simulation of a human α7 nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 2005;102:6813–6818. doi: 10.1073/pnas.0407739102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Speranskiy K., Cascio M., Kurnikova M. Homology modeling and molecular dynamics simulations of the glycine receptor ligand binding domain. Proteins. 2007;67:950–960. doi: 10.1002/prot.21251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eriksson M.A., Roux B. Modeling the structure of agitoxin in complex with the Shaker K+ channel: a computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys. J. 2002;83:2595–2609. doi: 10.1016/S0006-3495(02)75270-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cohen L., Karbat I., Gurevitz M. Common features in the functional surface of scorpion β-toxins and elements that confer specificity for insect and mammalian voltage-gated sodium channels. J. Biol. Chem. 2005;280:5045–5053. doi: 10.1074/jbc.M408427200. [DOI] [PubMed] [Google Scholar]
- 69.Nielsen K.J., Schroeder T., Lewis R. Structure-activity relationships of ω-conotoxins at N-type voltage-sensitive calcium channels. J. Mol. Recognit. 2000;13:55–70. doi: 10.1002/(SICI)1099-1352(200003/04)13:2<55::AID-JMR488>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 70.Nicke A., Wonnacott S., Lewis R.J. α-Conotoxins as tools for the elucidation of structure and function of neuronal nicotinic acetylcholine receptor subtypes. Eur. J. Biochem. 2004;271:2305–2319. doi: 10.1111/j.1432-1033.2004.04145.x. [DOI] [PubMed] [Google Scholar]