

Abstract
We monitored the action of phospholipase A2 (PLA2) on L- and D-dipalmitoyl-phosphatidylcholine (DPPC) Langmuir monolayers by mounting a Langmuir-trough on a wide-field fluorescence microscope with single molecule sensitivity. This made it possible to directly visualize the activity and diffusion behavior of single PLA2 molecules in a heterogeneous lipid environment during active hydrolysis. The experiments showed that enzyme molecules adsorbed and interacted almost exclusively with the fluid region of the DPPC monolayers. Domains of gel state L-DPPC were degraded exclusively from the gel-fluid interface where the buildup of negatively charged hydrolysis products, fatty acid salts, led to changes in the mobility of PLA2. The mobility of individual enzymes on the monolayers was characterized by single particle tracking. Diffusion coefficients of enzymes adsorbed to the fluid interface were between 3.2 μm2/s on the L-DPPC and 4.9 μm2/s on the D-DPPC monolayers. In regions enriched with hydrolysis products, the diffusion dropped to ≈0.2 μm2/s. In addition, slower normal and anomalous diffusion modes were seen at the L-DPPC gel domain boundaries where hydrolysis took place. The average residence times of the enzyme in the fluid regions of the monolayer and on the product domain were between ≈30 and 220 ms. At the gel domains it was below the experimental time resolution, i.e., enzymes were simply reflected from the gel domains back into solution.
Introduction
The interplay between enzymes and the cell membrane is essential for the regulation of a wide range of biological processes. Phospholipase A2 (PLA2) enzymes play an important role in these regulatory processes inasmuch as they interact directly with the membrane by altering both its chemical composition and physical state, thereby controlling its function. By catalyzing the hydrolysis of sn-glycero-3-phospholipids at the sn-2 ester bond, they release 1-lysophosphatidylcholine and a free fatty acid, e.g., arachidonic acid, which takes part in cell signaling (1).
Secreted PLA2 constitutes small (14 kDa) calcium-dependent proteins found ubiquitously in the extracellular space of mammalians; e.g., blood and tear fluid (2), as well as in insect venoms (1). Previous studies have shown that the activity of both type IB (from, e.g., pancreas) and type IIA (from, e.g., snake venom) PLA2s are highly dependent on the state and composition of the lipid system (3–6). For instance, the activity toward phosphocholine (PC) vesicles is at a maximum near the gel-fluid phase transition temperature (4,7). At this temperature, gel and fluid state lipids coexist, and there is a maximum in both lipid state fluctuations and lipid lateral compressibility (4,8). The change in composition caused by lipid hydrolysis affects the phase behavior of the membranes. As biological membranes exist in a state slightly above their melting transition, phospholipases seem to play an important role in maintaining this state (8). A further intriguing feature of PLA2 kinetics is the frequently observed lag-burst phenomenon. This is especially pronounced on zwitterionic substrates such as PCs. It is characterized by slow initial hydrolysis (the lag period) followed by a sudden increase in activity by several orders of magnitude (the burst) (4,9,10). Various studies have shown that the lag phase can be practically annihilated by addition of hydrolysis products (4,9,10). This led to the notion that the burst is induced by phase separation (domain formation) of products accumulated in the membrane during the lag period. Furthermore, it has been suggested that the presence of negatively charged hydrolysis products (e.g., ionized free fatty acids) increases the electrostatic binding between the positively charged surface patch on PLA2 (referred to as the “i-face”) in which the entrance to the active site is located (11,12). Based on such observations, a substrate theory has evolved in which the thermodynamic state of the lipid structure, rather than the molecular structure of the individual lipids, determines the overall enzymatic activity (11).
Numerous studies have shown that PLA2 is interfacially activated, meaning that it only shows activity toward aggregated lipid structures, whereas it is virtually inactive on lipid monomers in solution (13). Molecular dynamics modeling suggests that even when tight binding is assumed between the enzyme and the phospholipid substrate, i.e., with PLA2 partially penetrating the lipid structure, a distance of ≈1.5 nm from the outer plane of the lipid structure to the active site of the enzyme still remains (14). This indicates a need for considerable protrusion of the individual lipid molecules from the aggregated structure in order to fit into the active site of the enzyme. Both lipid protrusion and enzyme penetration must be expected to be favored when the lipid state is highly fluctuating and lateral compressibility is at a maximum.
Despite its recognized importance, very few studies have directly investigated the dependence of heterogeneities and domain formation in the lipid structure on the activation of PLA2 (e.g., (3,12,15)). In a recent study (16), single enzyme diffusion of gel bilayer segments supported by glass was investigated. It is known, however, that glass perturbs the bilayer behavior. Further, the fluid-solid interface was not present in that study. To the best of our knowledge, direct visualization of single PLA2 molecules and quantification of the effect of the heterogeneous lipid environment has not been reported in the literature. It was the aim of this study to investigate, at the single molecule level, the adsorption, lateral diffusion, and lateral partitioning behavior of PLA2, and correlate these directly to the microstructure of the phospholipid monolayers during hydrolysis.
To perform such a study, we developed a novel monolayer trough designed primarily to accommodate high numerical aperture (NA) microscope objectives (see scheme in Fig. 1, setup described in (17)). In comparison to previously published fluorescence studies on doped monolayers (e.g., (3,18)), and labeled PLA2 (19), the combination of the novel Langmuir trough and a wide-field microscope with single molecule detection sensitivity used in this study resulted in a drastic increase in optical resolution, signal/noise ratio, and temporal resolution. As has been shown, single-molecule experiments can reveal phenomena that are hidden from ensemble data and provide new insights into the influence of lipid heterogeneities on the action of lipolytic enzymes (20,21). Phospholipid monolayers were chosen for this study for several reasons. Most importantly, phospholipids are the natural substrate for PLA2, their monolayers at the air-water interface are well characterized in the literature (18,22), and they mimic the natural situation in which the enzymes operate. In addition, they are thermodynamically well-defined lipid systems in which several parameters (surface pressure Π, mean molecular area MMA, temperature T, etc.) can easily be controlled. Furthermore, they are readily visualized using standard fluorescence microscopy methods.
Figure 1.
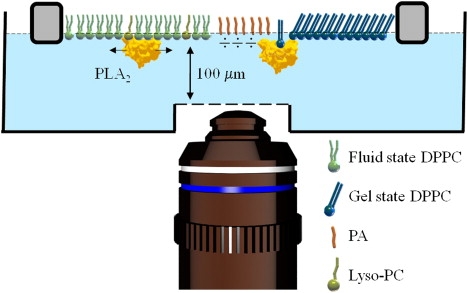
Schematic of the monolayer setup. A DPPC monolayer is compressed into the center of the phase transition region (Π = 8 mN/m, MMA = 65 Å2). At this surface pressure, liquid state lipid molecules (green) coexist with gel state lipid molecules (blue), which form domains. In this coexistence region, L-DPPC monolayers are susceptible to hydrolysis by PLA2-IB (yellow). Hydrolysis leads to formation and accumulation of ionized free palmitic acid (PA, red) and lysophosphatidylcholine (lyso-PC, olive green) in the monolayer. The monolayer trough was custom-designed to accommodate the 200-μm working distance of the high numerical aperture objective (NA = 1.2) and mounted on a home-built epi-fluorescence microscope.
In this study, the action of PLA2-IB on 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) monolayers in the lipid state coexistence region was monitored using two complementary types of fluorescence labeling schemes. In a first set of experiments, L-DPPC monolayers were fluorescently labeled with a lipid fluorophore that partitions exclusively in the fluid regions. In a second set of experiments, only PLA2-IB was marked with a small organic dye, a water-soluble perylene diimide (PDI) (23), which has no influence on enzyme activity (24). When using low concentrations, single enzymes molecules were tracked while diffusing on the monolayer. Both L-DPPC and D-DPPC monolayers were studied. Although the binding affinity of PLA2 for both lipids is similar (25), the enzyme shows no activity for the D-DPPC layer, which therefore served as a nonhydrolyzable surface.
Materials and Methods
Chemicals
L-DPPC and D-DPPC (left- and right-handed enantiomers) were from Avanti Polar Lipids (Cat. No. 850355; Alabaster, AL) and from Sigma-Aldrich (Cat. No. 42566; St. Louis, MO), respectively. Fluorescence label n-(tetramethylrhodamine-6-thiocarbamoyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (TRITC-DHPE) was supplied by Invitrogen (Cat. No. T1391; Carlsbad, CA). Purified porcine pancreas PLA2 (Type IB) was provided by Novozymes (Bagsværd, Denmark). All solvents were spectroscopic grade from Merck (Whitehouse Station, NJ). Milli-Q-water (>18.0 MΩ per 1/2 cm at 25°C) was purified on a desktop system (Millipore, Billerica, MA) and it was used for all steps involving water. Ultra-pure salts for buffer (TRIS pH 8.9, 150 mM NaCl, 5 mM CaCl2) were purchased from Merck. All chemicals were used as received.
Enzyme labeling
The n-hydroxisuccinimide (NHS)-ester of PDI (PDI-NHS) was synthesized and purified as previously described (24). Conjugation of PDI-NHS to PLA2 was performed using standard procedures for protein labeling: PDI-NHS was added in 25-fold excess to an enzyme solution in carbonate buffer (pH 8.0). The solution was then incubated at 4°C for 2 h to produce the dye-labeled enzyme (PLA2-PDI). The PDI-NHS bound itself to the enzyme through the amino groups of the lysine residues. Removal of unreacted dye from the enzyme solution was accomplished by several (≈20) size-exclusion spin filtrations with 5 kDa filters. Successive spin filtrations were performed until the filtrate was free of unreacted dye. The activity of the enzyme was verified on our monolayer setup with no detectable loss of activity. Absorbance spectroscopy showed, on average, three labels per protein. Fluorescence correlation spectroscopy measurements confirmed the presence of a single diffusing species (PLA2-PDI) with D3D = 100 μm2/s (measured relative to rhodamine 6G with known D3D = 300 μm2/s (26)).
Langmuir film preparation
A home-built Langmuir Teflon trough (DuPont, Wilmington, DE) with internal dimensions 150 × 50 mm equipped with two moveable barriers made from Delrin (DuPont) was used for the experiments. The trough was designed for our inverted fluorescence microscopy setup. To accommodate the short working distance of our microscope objective (see below), the glass observation window in the center of the trough was elevated ≈3 mm above the Teflon trough bottom. This made it possible to keep a stable 3–4 mm high subphase in the majority of the trough, while minimizing the height of the subphase directly above the observation window of the required ≈200 μm. The Langmuir trough electronics and control software was from Kibron (Espoo, Finland). A full description of the monolayer trough will be published elsewhere. To reduce surface flow of the monolayer in the observation region, a Teflon ring (Ø 15 mm, height 3 mm) with a slit opening (2 mm) facing one of the barriers was placed on the cover glass in the trough during experiments. A second cover glass was placed on top of the Teflon ring to further reduce air flow. The entire trough was covered with an acrylic case. Monolayers were spread from ≈0.6 mg/mL solution of lipid dissolved in hexane/ethanol 95:5 (v/v). Doped monolayers contained <0.1 mol % TRITC-DHPE.
Addition of enzyme beneath the monolayers
Two different strategies were used for injection of enzyme. In experiments with TRITC-DHPE doped monolayers (Fig. 2), the enzyme was dissolved in the subphase before spreading the monolayer and compression of the monolayer. This ensured a homogenous concentration in the aqueous subphase when the experiment was initiated. For experiments with fluorescently marked PLA2 (PLA2-PDI), the enzyme was injected with a Hamilton syringe that was immersed from behind the monolayer barriers after monolayer compression. The enzyme was injected in proximity to the observation window. This created a gradient in the enzyme concentration near the observation area that made it possible to select areas with suitable surface density of enzyme, i.e., well-separated particles, as required for single particle tracking (SPT).
Figure 2.
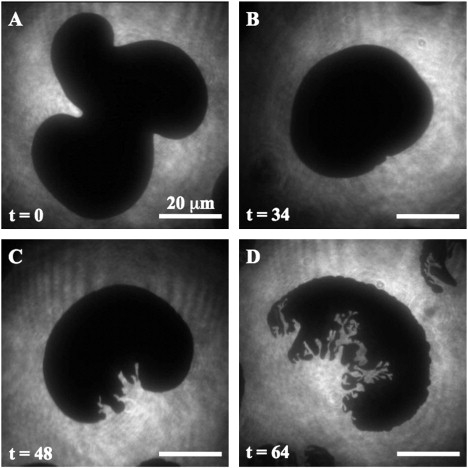
Time evolution of the morphology of a gel domain during PLA2-IB catalyzed hydrolysis at time 0 min (A), 34 min (B), 48 min (C), and 64 min (D) after compression to 65 Å2. Note that the domain in image A is not the same as the domain followed in images B–D, due to a slight drift in the monolayer. The L-DPPC monolayer was doped with the fluorescent lipid analog (TRITC-DHPE).
Wide-field microscopy
Images presented in Fig. 2 were recorded on a charge-coupled device camera (model No. KX85, pixel array: 1300 × 1030, pixel size: 6.7 μm; Apogee, Minneapolis, MN). All other images, including an image series for SPT, were recorded on an electromagnetic charge-coupled device camera (IXON EM+, DU897BV; Andor Technology, Belfast, Northern Ireland), pixel array: 512 × 512, pixel size: 16 μm). All images shown here were recorded using an Olympus 60×, water immersion, NA 1.2, UPLAPO objective (working distance: 0.22 mm). The Apogee camera was used in combination with a 1× camera lens. The Andor camera was used with a 2.5× camera lens (Total magnification: 150×). Samples were excited at 532 nm at a final excitation irradiance at the sample plane of 0.5–2 kW/cm2.
SPT
The determination of single enzyme trajectories was performed using a home-developed routine in MATLAB (The MathWorks, Natick, MA). The enzyme can be located with a precision of ≈100 nm for slowly diffusing enzymes and ≈200 nm for fast diffusing enzymes. The values of the diffusion coefficient for all the different enzyme motions were determined using cumulative distribution functions (CDFs). All trajectories in a given experiment (between 6000 and 16,000 trajectories) were analyzed using these CDFs, where square-displacements at each time interval of all trajectories were calculated and plotted as a histogram. This histogram is a probability function of the square-displacements, from which we are able to extract mean-square displacements (MSDs) at the corresponding time-interval. If, for example, the diffusive motion is heterogeneous and contains two distinct diffusion constants, one will obtain two values of MSD from the CDFs. By estimating the MSD at each time interval, one can construct MSD curves on each diffusion constant. When different regions (i.e., gel or product domains) were present, the trajectories were first classified into separate sets, namely trajectories in gel, fluid, and product domains, respectively, with each set of trajectories analyzed separately. The details of the analysis are described in the literature (20,27). The frame rate was typically 15 images/s, with an exposure time of 20–40 ms depending on the sample.
Results and Discussion
Visualizing enzymatic action on fluorescently labeled monolayers
In this series of experiments, the monolayers were labeled with TRITC-DHPE. Using this fluorophore, the gel L-DPPC domains appeared as dark regions and the fluid regions (L-DPPC + TRITC-DHPE) appeared as bright areas in the fluorescence images. This allowed us to monitor domain structural changes during enzymatic activity.
The first indication of enzymatic action appeared as an accelerated relaxation of the bean-shaped or trilobed L-DPPC domains into circular domains within a time-span of 30 min (Fig. 2, A and B). Relaxations from multilobed shapes are also seen in absence of enzyme, but in that case typically take several hours or days (18,28). The circular gel domains were then degraded from the gel-fluid interface as channels of fluid regions began to spread into the gel domains (Fig. 2 C). Strikingly, the channel formation occurred simultaneously on practically all domains within a given experiment (Fig. S1 in the Supporting Material). This was always observed, even though the lag time before channel formation varied considerably in-between separate but identical experiments (tlag = 40 ± 10 min). This is a strong indication of a thermodynamic control of the process, as simultaneously occurring domain degradation must be controlled by macroscopic properties and not by the microscopic structure of the individual domains. The overall growth pattern of the channels appeared similar on all domains (Fig. S1). Degradation furthermore occurred from only one side of the domains, leaving the opposing side remarkably inert to hydrolysis (Fig. 2, C and D). The channel growth appeared directed, and in many cases followed a straight line over large distances on the molecular level. Taken together, this indicates that channel formation is linked to the internal structure of the L-DPPC gel domain. It seems likely that individual lipids in the gel domains are oriented on a lattice and can only be attacked by the enzyme from one side. The structure of the partly degraded domains, observed here for pancreatic PLA-IB, closely resemble those reported for snake venom PLA2-IIA on a similar system (3,29).
The experiments were performed under constant area conditions to avoid disturbance introduced by possible movement of the barriers. As a consequence, the lateral pressure, Π, typically increased from initially 8 mN/m to finally 17 mN/m (t = 91 min). This was most likely caused by a combination of enzyme adsorption, enzyme penetration into the monolayer, and hydrolysis. Complete pressure-area and pressure-time isotherms associated with Fig. 2 are given in Fig. 3.
Figure 3.
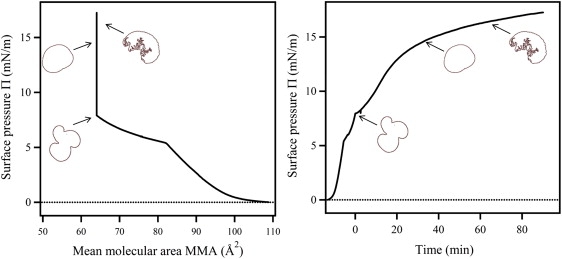
(Left) Pressure-area isotherm from the experiment shown in Fig. 2. Compression was started at a MMA 110 Å2, and the onset of the phase transition region is seen at MMA 82–83 Å2. Compression was stopped at a target MMA of 65 Å2 corresponding to a surface pressure of 8 mN/m. The monolayer was kept a constant area during the enzyme adsorption and hydrolysis process. Domain shapes at corresponding times and pressures during the hydrolysis process are shown for reference. (Right) Pressure-time plot. Compression was started at t = −13 min, the phase coexistence region was reached at t = −6 min, and compression was stopped at MMA 65 Å2 where large trilobed domains were formed (t ≡ 0).
Visualizing individual PLA2-PDI enzymes
In this set of experiments, PLA2-IB was labeled with the highly photostable organic dye PDI (23,24). In this way, the location and mobility of labeled enzyme (PLA2-PDI) could be tracked, analyzed, and linked to different lipid regions in the monolayer. Fig. 4 shows typical images of an undoped L-DPPC monolayer with different amounts of PLA2-PDI added to the aqueous subphase (compare this to Fig. 1). All fluorescence contrast stems from labeled protein associated to the monolayer.
Figure 4.

Wide-field fluorescence images of L-DPPC monolayers compressed to Π = 8 mN/m (22°C) with different concentrations of fluorescently labeled PLA2 enzymes (PLA2-PDI). (A and B) When the enzyme is present in high concentration, it is possible to visualize the domain structure (see text). It is also evident, from the bright regions along the domain interface, that a higher local concentration of PLA2-PDI is found along the fluid/gel boundary. (C) At low concentration, it is possible to discriminate and track single enzyme molecules (fluorescence image after linear deconvolution process). The magnifications show trajectories described by slow diffusing (top) and fast diffusing (bottom) PLA2-PDI molecules. Integration time: 22 ms.
At relatively high concentrations of enzyme (>10−11 M), the fluorescence images reveal the domain-segregated structure of the monolayer (Fig. 4, A and B) due to the different affinities of the enzyme toward regions of different lipid packing (i.e., lipid states). These images directly show that PLA2-PDI interacts preferentially with the fluid regions, as is evident from the bright interface of the domains. In principle, the image contrast should also make it possible to estimate the partition coefficient of PLA2 between the fluid regions and the gel domains. In practice, however, only a very low signal from the enzyme could be observed in the gel regions, and therefore, the enzyme is best characterized as having no affinity toward these dense gel-state lipid domains. The bright regions observed along the domain interface are caused by a higher local PLA2-PDI concentration and/or slower diffusion of the enzyme molecules at the fluid/gel boundary. At the highest concentration used (Fig. 4 A, [PLA2-PDI] ≈ 10−9 M), the entire fluid-gel interface is covered with PLA2-PDI. However, differences in fluorescence intensity witnessed at the liquid/gel interface indicate a nonuniform distribution of labeled PLA2-PDI molecules. Thus, the hydrolysis of the domains from only one side of the domains (Fig. 2 and Fig. S1) cannot be due to different affinities of the enzyme at different sides of the domains. At lower concentration (Fig. 4 B, [PLA2-PDI] ≈ 10−11 M), the enzyme is clearly accumulated at discrete sites on the domain interface (bright spots), where it seems to be virtually immobilized. From the spot size and fluorescence intensity, we speculate that the majority of the spots are aggregates of several enzyme molecules.
To visualize the mobility of enzymes while acting on the phospholipid monolayers, the enzyme concentration was decreased to the picomolar regime. This was needed to ensure that the individual enzymes were well separated so that individual enzyme trajectories could be resolved. For this purpose, image time-series of monolayer regions with low surface density of the enzyme were recorded and analyzed (Fig. 4 C). As a result of the low concentration of enzyme, the monolayer structure could not be inferred from the individual image frames.
Single-particle tracking of PLA2-PDI
To investigate the influence of hydrolysis on the diffusion behavior of PLA2, parallel experiments on L-DPPC and D-DPPC were performed at the single-molecule level. As pointed out previously, PLA2 cannot hydrolyze D-DPPC and is thereby inactive on the D-DPPC substrate monolayers. Nevertheless, PLA2 has the same initial binding affinity for lipid structures of these two enantiomers (19,30,31). Although PLA2 could be inactivated by removal of its cofactor Ca2+ from the buffer, that approach was considered undesirable, as Ca2+ removal might affect the binding affinity of PLA2 to the monolayer (32) and would definitely have influenced the phase behavior and lipid packing of the monolayer structure itself (22).
The advantage, and justification, of SPT experiments and analysis is the ability to detect heterogeneities in the trajectories followed by individual molecules. Single enzyme trajectories show that different PLA2-PDI molecules may present different diffusion coefficients (Fig. 4 C). Moreover, when representative trajectories of single enzymes diffusing on L-DPPC monolayers are laid out, it is possible to discriminate two distinct spatial regions where phospholipase molecules diffuse differently (Fig. 5 A). Although freely diffusing on the largest part of the monolayer in the field of view, on some regions the molecules seem to be immobilized or confined (e.g., top-left corner of the image on Fig. 5 A). Taking into account the morphology that could be outlined at higher enzyme concentrations, we expect these immobilized or slowly diffusing PLA2-PDI molecules to be located at the gel domain boundaries.
Figure 5.
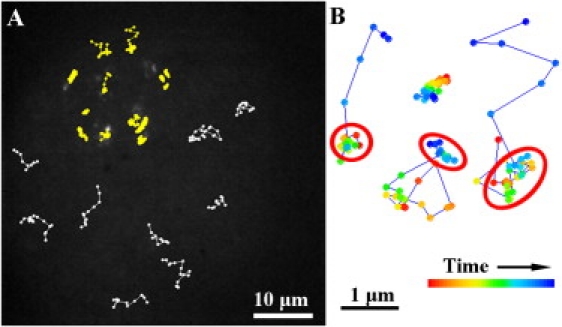
Diffusion behavior of labeled individual PLA2 molecules. (A) Typical trajectories of individual PLA2 molecules diffusing on the fluid region (white) and near the fluid/gel boundary (yellow) as observed in the background image (accumulated over 100 frames). Four of the trajectories colored in yellow are magnified in panel B. In three of the trajectories, it is possible to distinguish the hot spots (indicated by the red circles) where diffusion is slow. The color scale is sequential and does not indicate real time. The first data point is depicted in red and the last one in blue.
A detailed analysis of the trajectories followed by single enzyme molecules located near the fluid/gel boundary in Fig. 5 reveals heterogeneities both between and within the trajectories (Fig. 5 B). Four representative examples of enzymes that showed switching between fast and slow diffusion are depicted in Fig. 5 B. From the results obtained with the fluorescently labeled monolayer, we speculate that the trajectories detected near the gel domain region correspond to PLA2-PDI molecules confined to the channels produced by hydrolysis (compare to Fig. 2, C and D). Such transiently confined enzymes show switching between fast and slow diffusion and vice versa. The periods of slow diffusion are indicated by the red circles in Fig. 5 B. Importantly, this switching diffusion behavior was not detected for enzymes diffusing on the unhydrolyzable D-DPPC monolayer.
The different mobility of the enzyme molecules on the different areas of the L-DPPC monolayer becomes clear when using mobility-localization microscopy (M-LM) images. In all types of localization microscopy, e.g., PALM, STORM, PALMIRA, and S-PALM (33), the position of single molecules within a frame is determined with high accuracy. This is done for all frames in the recorded movie and in the end, a super-resolution image is reconstructed. Here, instead of plotting the position of each molecule in the recorded movie on a final image, we plot the measured displacement of each individual enzyme between two consecutive image frames onto one final image. The images obtained this way map out the relation between the monolayer structure and the mobility of the enzyme. The two regions where PLA2-PDI molecules present different diffusion behavior can now be identified clearly on the L-DPPC monolayer (Fig. 6 A), whereas enzyme motion on the fluid region of the D-DPPC monolayer is found nearly homogeneous (Fig. 6 B).
Figure 6.
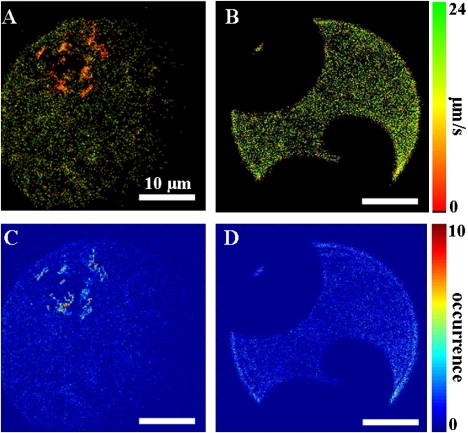
(A) M-LM image constructs the mobility of PLA2-PDI enzymes diffusing on a L-DPPC that is susceptible to hydrolysis. The enzyme diffuses markedly slower in distinct regions (red areas). (B) In the liquid regions of the nonhydrolyzable D-DPPC monolayer, measured enzyme diffusion mobilities are homogeneously distributed. (C and D) H-LM images showing the localization of PLA2-PDI enzymes diffusing on (C) a L-DPPC and (D) a D-DPPC monolayer. Colored scale bars indicate measured diffusion mobilities (in μm/s) and local occurrence of the enzyme. All scale bars in the images are 10 μm. The total number of images was 1000 corresponding to a total time of 68 s (15 frames/s).
In a similar way, the affinity of the enzyme for different regions of the monolayers can be evaluated by constructing histogram-LM images (H-LM (20)). H-LM images are constructed by replotting the detected localization of each individual enzyme from each image frame of a time series of images onto one final histogram image. The H-LM image does not distinguish individual enzymes or contain information on dynamics, but instead provides insight into the likelihood of an enzyme to be found at a particular location. Thus, M-LM and H-LM plots contain different information. The H-LM images for the PLA2-PDI diffusing on the different monolayers are shown in Fig. 6, C and D.
Although not immediately recognizable from the fluorescence images at low enzyme concentration, the gel domain structures are rendered visible both in M-LM and H-LM images (Fig. 6). This is especially true on the D-DPPC monolayer, where the domains are well defined and remain unchanged during time because no hydrolysis can occur. Near the domains, clear differences between the two systems are evident. On the L-DPPC monolayer, the enzyme binds preferentially to localized spots, i.e., hot-spots (shown in yellow and red) located at the gel-fluid boundary region (Fig. 6 A). In contrast, the enzyme is relatively homogeneously dispersed in the fluid region of the D-DPPC layer (Fig. 6 B). It is obvious from Fig. 6, B and D, that neither is there an accumulation of enzyme at the domain boundary of D-DPPC layers nor any measurable change in mobility. When an enzyme hit a domain boundary, it typically did not dissociate but instead continued diffusing in the liquid phase. Interestingly, the hot-spots coincide with the areas where enzyme diffuses slowly and they are limited to L-DPPC monolayers. We can therefore conclude that they are intimately linked to hydrolysis. This indicates that the regions of slow enzyme diffusion on L-DPPC are most likely hydrolysis product domains. It should be added that hydrolysis in the fluid phase is known to be very slow and that hydrolysis mostly happens at the domain boundaries (34). Further, hydrolysis products are known to phase-separate in the solid phase and in the solid-liquid coexistence regime in bilayers (see (35) and the phase diagram therein). We therefore expect that the diffusion in the liquid-expanded phase is not very much affected by product formation.
The values of the diffusion coefficient for the different enzyme motions were determined using cumulative distribution functions (20,27). Within the fluid regions of the L-DPPC monolayer, all the enzymes seemed to diffuse randomly, with slightly different diffusion coefficients. In detail, the trajectories described by the majority of the molecules (87%, 1216 molecules analyzed) exhibited only fast diffusion with D = 3.2 μm2/s, while 13% of the trajectories were found to contain both fast (D = 3.2 μm2/s) and slow periods (D < 0.038 μm2/s). Conversely, near the gel-fluid boundary, 16% of the trajectories (515 molecules analyzed) exhibited slow diffusion (D < 0.031 μm2/s). The majority of trajectories (61%) showed a combination of slow (D < 0.031 μm2/s) and fast (D = 3.2 μm2/s) diffusion steps. The remaining 13% of the molecules showed anomalous diffusion (D = 0.27 μm2/sα, α = 0.24), indicating confinement of the diffusion, probably in channels that are formed in the gel domain (see features in Fig. 2). This behavior is in great contrast to the diffusion behavior found on D-DPPC. In the fluid region on D-DPPC, 90% of the trajectories (2971 molecules analyzed) exhibited normal fast diffusion (D = 4.9 μm2/s). The remaining 10% was heterogeneous, having periods of slow (D < 0.004 μm2/s) and fast diffusion. Because diffusion is faster on D-DPPC (D = 4.9 μm2/s) than on L-DPPC (D = 3.0 μm2/s), it is speculated that some hydrolysis takes place in the fluid region of the L-DPPC monolayer. Slow diffusion constants are only given with upper limits, as they are so slow that they might be influenced by domain or monolayer movement. Photobleaching was not considered as a possible limitation for the determination of the residence times. It has been shown in Peneva et al. (24) that under the conditions used in our work, the lifetime of the label is ∼5 s.
When hydrolysis was allowed to proceed over >1 h, large areas with distinctly slower enzyme diffusion (i.e., localized spots over a large area) were observed within the fluid region of L-DPPC monolayers (Fig. 7, A and B). Because these areas were always detected near the gel domains, we can speculate that they form as the debris of products generated as the gel/fluid boundary diffuses into the fluid region and accumulates. Although the existence of these so-called product domains has been previously reported, their structure and exact composition are largely unknown, but it seems clear that they contain, at least, calcium palmitate salts (22,36,37).
Figure 7.
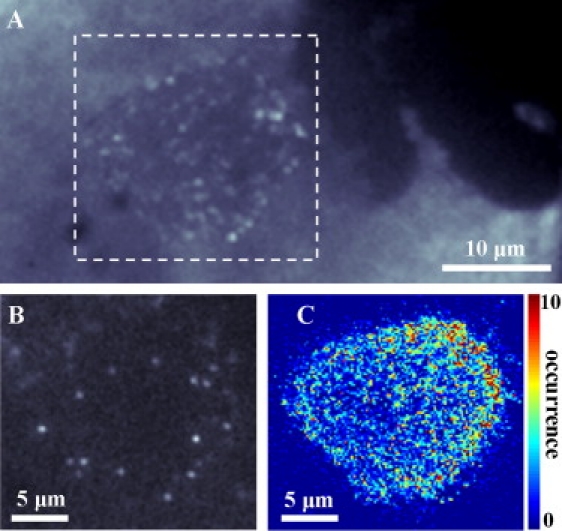
(A) Accumulation over 50 frames of a L-DPPC layer incubated with low enzyme concentration (≈10–12 M) for >60 min. It is possible to discriminate the gel domain (no enzymes, black region in upper-right corner), the fluid region (enzymes diffusing fast leading to a uniform fluorescence) and the product domain (enzymes diffusing slowly or immobilized). The dashed square indicates the product domain region shown in panels B and C. (B) Fluorescence image of the product domain in which single enzyme molecules can be discriminated. Integration time: 30 ms. (C) The H-LM image shows a tendency for the enzyme to preferentially localize near the gel-fluid boundary.
On such product domains, the enzyme molecules (2095 enzymes analyzed) presented diffusion with two characteristic diffusion coefficients: D = 0.16 μm2/s and D < 0.031 μm2/s. Compared to the fluid L-DPPC region, the mobility of PLA2-PDI is reduced by a factor of 20. Approximately one-half of the trajectories showed only the fast component, 1% only the slow, and 48% showed periods of both slow and fast diffusion. The M-LM image (Fig. 8) shows that the diffusion coefficients of PLA2-PDI did not vary systematically within the product domain. Nevertheless, the H-LM shows a preferential localization of the enzyme for the product domain-gel domain interface (Fig. 7 C).
Figure 8.
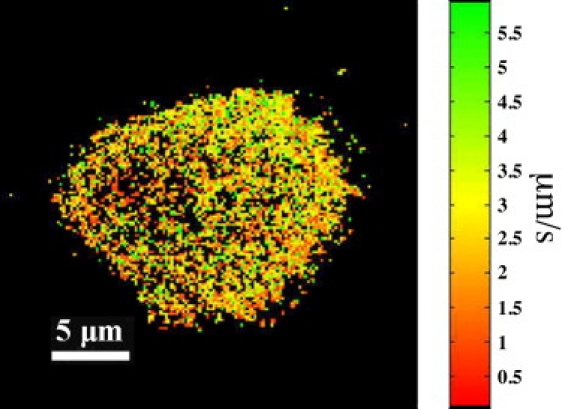
M-LM image of the product-enriched area on L-DPPC (see also Fig. 7). The image shows that the diffusion mobility did not vary systematically within the product-enriched area.
In addition to the diffusion coefficients, the residence time (i.e., average time-length of the trajectories) for PLA2-PDI was determined for the different systems. In all cases, a satisfactory fit was obtained by fitting the obtained distribution of trajectory duration to a two-component exponential decay. All systems contained the same relatively short residence time of 28 ms (corresponding to the exposure time of a single frame), as well as a more long-lived component that varied between 95 and 220 ms. The short residence-time component is attributed to enzymes at the surface that did not bind specifically to the monolayer; e.g., enzyme at the interface with the binding motif (i.e., the i-face) facing away from the monolayer. When within the product domain, the residence time of PLA2-PDI molecules doubled. This further strengthens the hypothesis of a region enriched in negatively charged hydrolysis products where the enzyme binds more strongly. All diffusion coefficients and residence times are summarized in Table 1. The survival time of the fluorescence dye was estimated to be 5 s (24). Therefore, bleaching should not play a major role.
Table 1.
Summary of diffusion coefficients and mean residence times of PLA2-PDI
| Monolayer system |
D (μm2/s) Normal diffusion |
D (μm2/sα) Anomalous diffusion (fast) | Residence time (ms) |
Number of trajectories analyzed | ||
|---|---|---|---|---|---|---|
| Fast components | Slow components | Short components | Long components | |||
| L-DPPC fluid region | 3.2 ± 0.1 | <0.038 | — | 28 | 95 | 1216 |
| L-DPPC near domain | 3.2 ± 0.1 | <0.031 | 0.27 ± 0.01α = 0.24 ± 0.02 | 31 | 220 | 515 |
| L-DPPC product domain | 0.16 ± 0.002 | <0.031 | — | 27 | 190 | 2095 |
| D-DPPC fluid region | 4.9 ± 0.1 | <0.004 | — | 26 | 132 | 2971 |
The typical exposure time of each snapshot is 20–40 ms. Thus, the short residence times correspond to 1–2 snapshots and probably indicate nonbinding events. These events are not used for diffusion analysis. Only events with long residence times were used to obtain the fast and slow diffusion coefficients. The slow diffusion components display values corresponding to virtually immobile enzymes. We only give an upper limit because on this timescale the values may be influenced by movement of domains. The procedure for obtaining the diffusion coefficients and the errors is described in Schütz et al. (27) (compare to Fig. 3 therein, see also text).
Conclusion
Here we studied the action of PLA2 on monolayers under conditions comparable (although not identical) to a bilayer membrane in the melting regime. The primary scope of this study was the direct visualization of activity and diffusion behavior of PLA2 in a heterogeneous lipid environment. This was engaged upon by performing high-resolution and high-sensitivity time-resolved fluorescence imaging and single-particle tracking of PLA2-IB on L-DPPC monolayers during active hydrolysis. By tracking individual enzymes in the different phases of the substrate, differences in their mobility were related to differences in activity toward the different phases. All the experiments reported in the literature so far point to the fact that hydrolysis predominantly takes place at the domain boundaries (4,7,11). Our results corroborate this notion through direct visualization of enzyme activity. The results provide further evidence of a drastic change in the enzyme behavior at the gel-fluid boundary as hydrolysis progresses. The preferential binding of the enzyme to the gel-fluid boundary shown in the H-LM images on L-DPPC monolayer is not seen in absence of hydrolysis on a nonsubstrate D-DPPC monolayer. Moreover, the decreased mobility of PLA2 in the fluid region of L-DPPC monolayers, relative to D-DPPC monolayers, indicates that some hydrolysis may also take place in the fluid region.
The advantage of SPT is clear in the context of this study, because it allows us to distinguish among PLA2's different modes of diffusion in different regions, and to determine the associated diffusion coefficients as well as residence times of the enzyme on the monolayer. In a condensed format, the enzyme was found to diffuse fast in the fluid regions of the L-DPPC monolayer, and slow near domain boundaries where hydrolysis predominantly takes place. The enzyme appears to have very low affinity on gel domains, presumably because the lipid packing is too dense for the enzyme to penetrate the domains (Fig. 9).
Figure 9.
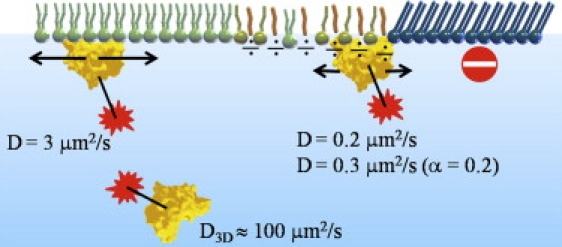
Cartoon representation showing typical modes of diffusion in different environments. For diffusion on the fluid region (lipids in green), the diffusion was generally normal with a diffusion coefficient of 3 μm2/s. Enzymes located near L-DPPC gel domains showed more complex diffusion with, e.g., transient trapping of the enzyme. After extended hydrolysis, areas enriched in hydrolysis product (molecules in olive green and red) showed enzyme diffusion that was significantly slower than on the fluid region. See text for details.
Supporting Material
One figure is available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00200-6.
Supporting Material
Acknowledgments
We thank Allan Svendsen (Novozymes A/S, Denmark) for supplying purified PLA2.
This project was supported by the European Union project BIOSCOPE and the Catholic University of Leuven research fund (grant No. CREA2007). S.R. acknowledges the Portuguese Foundation for Science and Technology for its PhD grant (No. SFRH/BD/27265/2006).
Footnotes
Martin Gudmand performed the experimental work.
Contributor Information
Thomas Bjørnholm, Email: tb@nano.ku.dk.
Thomas Heimburg, Email: theimbu@nbi.dk.
References
- 1.Six D.A., Dennis E.A. The expanding superfamily of phospholipase A2 enzymes: classification and characterization. Biochim. Biophys. Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 2.Saari K.M., Aho V.V., Nevalainen T.J. Group II PLA2 content of tears in normal subjects. Invest. Ophthalmol. Vis. Sci. 2001;42:318–320. [PubMed] [Google Scholar]
- 3.Grainger D.W., Reichert A., Salesse C. An enzyme caught in action: direct imaging of hydrolytic function and domain formation of phospholipase A2 in phosphatidyl choline monolayers. FEBS Lett. 1989;252:73–82. [Google Scholar]
- 4.Hønger T., Jørgensen K., Mouritsen O.G. Systematic relationship between phospholipase A2 activity and dynamic lipid bilayer microheterogeneity. Biochemistry. 1996;35:9003–9006. doi: 10.1021/bi960866a. [DOI] [PubMed] [Google Scholar]
- 5.Burack W.R., Dibble A.R.G., Biltonen R.L. Changes in vesicle morphology induced by lateral phase separation modulate phospholipase A2 activity. Biochemistry. 1997;36:10551–10557. doi: 10.1021/bi970509f. [DOI] [PubMed] [Google Scholar]
- 6.Nielsen L.K., Balashev K., Bjørnholm T. Influence of product phase separation on phospholipase A2 hydrolysis of supported phospholipid bilayers studied by force microscopy. Biophys. J. 2002;83:2617–2624. doi: 10.1016/S0006-3495(02)75272-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Op den Kamp J.A.F., de Gier J., van Deenen L.L.M. Hydrolysis of phosphatidylcholine liposomes by pancreatic phospholipase A2 at the transition temperature. Biochim. Biophys. Acta. 1974;345:253–256. doi: 10.1016/0005-2736(74)90263-6. [DOI] [PubMed] [Google Scholar]
- 8.Heimburg T. Wiley VCH; Berlin, Germany: 2007. Thermal Biophysics of Membranes. [Google Scholar]
- 9.Apitz-Castro R., Jain M.K., De Haas G.H. Origin of the latency phase during the action of phospholipase A2 on unmodified phosphatidylcholine vesicles. Biochim. Biophys. Acta. 1982;688:349–356. doi: 10.1016/0005-2736(82)90346-7. [DOI] [PubMed] [Google Scholar]
- 10.Nielsen L.K., Risbo J., Bjørnholm T. Lag-burst kinetics in phospholipase A2 hydrolysis of DPPC bilayers visualized by atomic force microscopy. Biochim. Biophys. Acta. 1999;1420:266–271. doi: 10.1016/s0005-2736(99)00103-0. [DOI] [PubMed] [Google Scholar]
- 11.Mouritsen O.G., Andersen T.L., Weiss M. Activation of interfacial enzymes at membrane surfaces. J. Phys. Condens. Matter. 2006;18:S1293–S1304. doi: 10.1088/0953-8984/18/28/S12. [DOI] [PubMed] [Google Scholar]
- 12.Leidy C., Linderoth L., Peters G.H. Domain-induced activation of human phospholipase A2 type IIA: local versus global lipid composition. Biophys. J. 2006;90:3165–3175. doi: 10.1529/biophysj.105.070987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verger R., de Haas G.H. Interfacial enzyme kinetics of lipolysis. Annu. Rev. Biophys. Bioeng. 1976;5:77–117. doi: 10.1146/annurev.bb.05.060176.000453. [DOI] [PubMed] [Google Scholar]
- 14.Zhou F., Schulten K. Molecular dynamics study of phospholipase A2 on a membrane surface. Proteins. 1996;25:12–27. doi: 10.1002/(SICI)1097-0134(199605)25:1<12::AID-PROT2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 15.Simonsen A.C. Activation of phospholipase A2 by ternary model membranes. Biophys. J. 2008;94:3966–3975. doi: 10.1529/biophysj.107.114363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiu C.-R., Huang W.-N., Yang T.S. Fluorescence single-molecule study of cobra phospholipase A2 action on a supported gel-phase lipid bilayer. ChemPhysChem. 2009;10:549–558. [Google Scholar]
- 17.Gudmand M., Fidorra M., Heimburg T. Diffusion and partitioning of fluorescent lipid probes in phospholipid monolayers. Biophys. J. 2009;96:4598–4609. doi: 10.1016/j.bpj.2009.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McConlogue C.W., Vanderlick T.K. A close look at domain formation in DPPC monolayers. Langmuir. 1997;13:7158–7164. [Google Scholar]
- 19.Dahmen-Levison U., Brezesinski G., Möhwald H. Specific adsorption of PLA2 at monolayers. Thin Solid Films. 1998;327–329:616–620. [Google Scholar]
- 20.Rocha S., Hutchison J.A., Uji-i H. Linking phospholipase mobility to activity by single-molecule wide-field microscopy. ChemPhysChem. 2009;10:151–161. doi: 10.1002/cphc.200800537. [DOI] [PubMed] [Google Scholar]
- 21.Sonesson A.W., Elofsson U.M., Brismar H. Tracking single lipase molecules on a trimyristin substrate surface using quantum dots. Langmuir. 2007;23:8352–8356. doi: 10.1021/la700918r. [DOI] [PubMed] [Google Scholar]
- 22.Maloney K.M., Grainger D.W. Phase separated anionic domains in ternary mixed lipid monolayers at the air-water interface. Chem. Phys. Lipids. 1993;65:31–42. doi: 10.1016/0009-3084(93)90079-i. [DOI] [PubMed] [Google Scholar]
- 23.Margineanu A., Hofkens J., De Schryver F.C. Photophysics of a water-soluble rylene dye: comparison with other fluorescent molecules for biological applications. J. Phys. Chem. B. 2004;108:12242–12251. [Google Scholar]
- 24.Peneva K., Mihov G., Muellen K. Water-soluble monofunctional perylene and terrylene dyes: powerful labels for single-enzyme tracking. Angew. Chem. Int. Ed. 2008;47:3372–3375. doi: 10.1002/anie.200705409. [DOI] [PubMed] [Google Scholar]
- 25.Pattus F., Slotboom A.J., de Haas G.H. Regulation of the interaction of pancreatic phospholipase A2 with lipid-water interfaces by Ca2+ ions: a monolayer study. Biochemistry. 1979;18:2698–2702. doi: 10.1021/bi00580a002. [DOI] [PubMed] [Google Scholar]
- 26.Webb W.W. Fluorescence correlation spectroscopy. Bull. Am. Phys. Soc. 1974;19:197. [Google Scholar]
- 27.Schütz G.J., Schindler H., Schmidt T. Single-molecule microscopy on model membranes reveals anomalous diffusion. Biophys. J. 1997;73:1073–1080. doi: 10.1016/S0006-3495(97)78139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klopfer K.J., Vanderlick T.K. Isotherms of dipalmitoylphosphatidylcholine (DPPC) monolayers: features revealed and features obscured. J. Coll. Interf. Sci. 1996;182:220–229. [Google Scholar]
- 29.Grainger D.W., Reichert A., Salesse C. Hydrolytic action of phospholipase A2 in monolayers in the phase transition region: direct observation of enzyme domain formation using fluorescence microscopy. Biochim. Biophys. Acta. 1990;1023:365–379. doi: 10.1016/0005-2736(90)90128-b. [DOI] [PubMed] [Google Scholar]
- 30.Li J.B., Chen Z.J., Möhwald H. Dynamic observations of the hydrolysis of a DPPC monolayer at the air/water interface catalyzed by phospholipase A2. Angew. Chem. Int. Ed. 2000;39:3059–3062. doi: 10.1002/1521-3773(20000901)39:17<3059::aid-anie3059>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 31.Bonsen P.P., de Haas G.H., van Deenen L.L. Studies on phospholipase A and its zymogen from porcine pancreas. IV. The influence of chemical modification of the lecithin structure on substrate properties. Biochim. Biophys. Acta. 1972;270:364–382. doi: 10.1016/0005-2760(72)90200-7. [DOI] [PubMed] [Google Scholar]
- 32.Pattus F., Slotboom A.J., de Haas G.H. Regulation of phospholipase A2 activity by the lipid-water interface: a monolayer approach. Biochemistry. 1979;18:2691–2697. doi: 10.1021/bi00580a001. [DOI] [PubMed] [Google Scholar]
- 33.Flors C., Hotta J., Hofkens J. A stroboscopic approach for fast photoactivation-localization microscopy with Dronpa mutants. J. Am. Chem. Soc. 2007;129:13970–13977. doi: 10.1021/ja074704l. [DOI] [PubMed] [Google Scholar]
- 34.Biltonen R.L. A statistical-thermodynamic view of cooperative structural changes in phospholipid bilayer membranes: their potential role in biological function. J. Chem. Thermodyn. 1990;220:1–19. [Google Scholar]
- 35.Burack W.R., Yuan Q., Biltonen R.L. Role of lateral phase separation in the modulation of phospholipase A2 activity. Biochemistry. 1993;32:583–589. doi: 10.1021/bi00053a025. [DOI] [PubMed] [Google Scholar]
- 36.Maloney K.M., Grandbois M., Roberts M.F. Phospholipase A2 domain formation in hydrolyzed asymmetric phospholipid monolayers at the air/water interface. Biochim. Biophys. Acta. 1995;1235:395–405. doi: 10.1016/0005-2736(95)80029-f. [DOI] [PubMed] [Google Scholar]
- 37.Grandbois M., Desbat B., Salesse C. Monitoring of phospholipid monolayer hydrolysis by phospholipase A2 by use of polarization-modulated Fourier transform infrared spectroscopy. Biophys. Chem. 2000;88:127–135. doi: 10.1016/s0301-4622(00)00204-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.