

Abstract
A set of bivalent mannose 6-phosphonate “molecular rulers” has been synthesized to examine ligand binding to the M6P/IGF2R. The set is estimated to span a P-P distance range of 16–26 Å (MMFF energy minimization on the hydrated phosphonates). Key synthetic transformations include sugar triflate displacement for phosphonate installation and Grubbs I cross-metathesis to achieve bivalency. Relative binding affinities were tested by radioligand displacement assays versus PMP-BSA (pentamannose phosphate-bovine serum albumin). These compounds exhibit slightly higher binding affinities for the receptor (IC50’s = 3.7–5 μM) than the parent, monomeric mannose 6-phosphonate ligand and M6P itself (IC50 = 11.5 ± 2.5 μM). These results suggest that the use of an α-configured anomeric alkane tether is acceptable, as no significant thermodynamic penalty is apparently paid with this design. On the other hand, the modest gains in binding affinity observed suggest that this ligand set has not yet found true bivalent interaction with the M6P/IGF2R (i.e. binding to two distinct M6P-binding pockets).
The mannose 6-phosphate/insulin-like growth factor II receptor (M6P/IGF2R) is a type I transmembrane glycoprotein that cycles through the Golgi, endosomes, and the plasma membrane to carry out its role in the transport of lysosomal enzymes to their cellular destination.1 The receptor also functions in the binding, uptake, and degradation of the mitogen, insulin-like growth factor II (IGF-II) and facilitates activation of the growth inhibitor, transforming growth factor-β. The ability of the M6P/IGF2R to inhibit cell proliferation, or stimulate apoptosis, by these mechanisms has implicated the receptor as a tumor suppressor. The IGF-II binding activity of the M6P/IGF2R is mainly responsible for its growth suppressor function. Many cancers become growth factor-independent by high-level expression of IGF-II, which not only binds to the M6P/IGF2R, but also to the IGF1R. The high affinity interaction of IGF-II with the IGF1R leads to activation of IGF1R signaling pathways that promote cell division and survival.2
The extracellular portion of the M6P/IGF2R contains 15 homologous repeat domains of ~147 amino acid residues each. There are two M6P binding sites located in domains 3 and 9, and there is one IGF-II binding site in domain 11.3 Binding of high-affinity, M6P-based ligands and rapid internalization of extracellular ligands, such as IGF-II, are aided by the M6P/IGF2R’s ability to dimerize.4,5 York et al. demonstrated that β-glucuronidase (hGUS), a homotetrameric lysosomal enzyme bearing multiple M6P moieties, stabilized the receptor’s dimeric structure by cross-bridging the M6P binding sites on two adjacent subunits.5 These data support a dimeric model for binding of bivalent M6P-based ligands by the M6P/IGF2R (Figure 1). Importantly, they also observed that hGUS binding increased the rate of internalization of the receptor and consequently stimulated the degradation of any passenger ligands, including IGF-II, by 3- to 4-fold. The long-term goal of the present work is to exploit this unique property of bivalent M6P ligands as a potential strategy for therapeutic intervention in IGF-II-dependent cancer.
Figure 1.
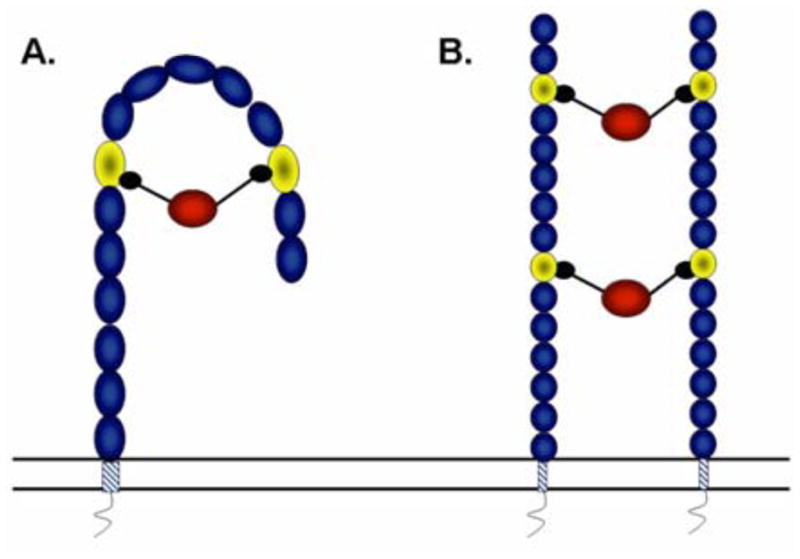
M6P ligand binding to the M6P/IGF2R: The alternative “hook and ladder” models: A. One monomeric unit of the M6P/IGF2R consisting of the 15 extracellular repeating domains, transmembrane domain, and a short cytoplasmic tail. The M6P/IGF2R is depicted as forming a hook-like structure when a ligand bearing two M6P groups binds to domains 3 and 9 (lighter shaded ovals). B. Two monomeric units of the M6P/IGF2R are connected through binding by ligands that interact with either domain 3 or 9 of two individual monomeric units to form a dimeric ladder-like structure.
Multivalent interactions between receptors and their ligands,6 which are common in biology, involve a multistep mechanism in which most of the entropic cost is paid by the initial binding event and subsequent contacts contribute a favorable enthalpy without further sacrifice of rotational and translational entropy.7 The resultant high binding affinity in these interactions is due to a reduced rate of ligand-receptor dissociation. This type of interaction occurs in carbohydrate binding to lectins and is particularly important in the binding of M6P-bearing oligosaccharides by P-type lectins such as the M6P/IGF2R.
Tong et al. demonstrated that there are two M6P binding sites per monomeric unit of the M6P/IGF2R.8 Some native glycoprotein ligands and model compounds (e.g. PMP-BSA) display up to 100- to 1000-fold lower dissociation constants, i.e., higher affinities, than ligands bearing a single phosphorylated mannoside. Given that two M6P-binding pockets are available per receptor in the monomeric binding model and four per receptor in the dimeric model, bi- or multivalency may account for this effect. This could result from simultaneous contact with two M6P groups on two distinct oligosaccharides. Alternatively, the pioneering work of Varki and Kornfeld suggested that such high affinity bivalent binding might also be achieved with a single N-linked oligosaccharide phosphorylated on the two ultimate mannose residues, at the first and third antennae (Figure 2).9 This high affinity could arise either from intramolecular contact between a single receptor molecule and the two phosphate groups on the ligand or by intermolecular cooperation between two subunits within a dimeric receptor structure, as depicted in Figure 1.
Figure 2.

Hindsgaul’s model biantennary ligands
Later work by the Hindsgaul group demonstrated that the linkage between the ultimate Man and penultimate Man on the phosphorylated branch is important, as an α-1,2-glycosidic linkage results in a higher binding affinity to the receptor than an α-1,3-linkage (Figure 2).10 A series of synthetic multivalent ligands for the M6P/IGF2R was prepared by Bock and coworkers, using a glycopeptide design.11 The best of these compounds bore two mannose disaccharides capped with phosphate connected by a core peptide of 3 to 5 amino acids. A tripeptide version of this compound bound the M6P/IGF2R with high affinity, which led to the hypothesis of a bivalent M6P-based mechanism.5, 11 However, upon closer inspection, it appears that the exceptional binding affinity of this compound was attributable to an anthranoyl group present on the lysine ε-amino group within the core peptide (Figure 3). This modification increased the affinity by ~200-fold relative to the same compound with an unmodified peptide,11 presumably through interaction with a hydrophobic patch on the receptor proximal to the M6P binding site. Considering that the high affinity of this compound did not arise from a bivalent M6P-based binding mechanism, it is not surprising that York et al. found that the compound failed to stabilize the receptor’s dimeric structure or to stimulate its rapid internalization.5 In summary, there is currently no evidence in the literature of a small synthetic compound capable of bivalent binding to the M6P/IGF2R by a M6P-based mechanism.
Figure 3.

Bock’s tripeptide bis-M6P-bearing ligands
Thus, the goal of this work is to develop high affinity bivalent M6P-based ligands that accelerate disposal of IGF-II as a passenger ligand directly in tumors, by cross-bridging the M6P/IGF2R thereby enhancing its ability to internalize IGF-II. In our previous work, we discovered that the phosphonate is an excellent surrogate for phosphate to promote equivalent interaction with the M6P binding domains of the M6P/IGF2R12,13 The phosphonate has the advantage of resistance to hydrolysis with the potential for improved pharmacokinetics and efficacy in vivo. In light of the aforementioned studies, we sought to improve affinity by building bivalency into such ligands.
We have recently demonstrated proof of principle for a Ru-mediated cross-metathesis (CM) route to joining two M6P surrogates, of both the malonate and phosphonate varieties with a hydrocarbon tether.12 Herein, we describe the exploitation of this methodology to synthesize a series of bis-M6P-phosphonates, with incrementally increasing tether lengths as a sort of “molecular ruler” set to probe for such a bivalent interaction with the receptor. To systematically increase tether length in two carbon increments, the initial mannosidation reaction was performed with a series of terminally unsaturated alcohols bearing 4–7 carbons [from 3-buten-1-ol through 6-hepten-1-ol (Scheme 1).
Scheme 1.

Reagents and conditions: (a) NaH, BnBr, DMF, 0 °C to rt (90%); (b) AcOH, Ac2O, H2SO4, 0 °C (82%); (c) 2 TMSOTf, 3-buten-1-ol, rt (86%); 3 TMSOTf, 4-penten-1-ol, rt (70%); 4 TMSOTf, 5-hexen-1-ol, rt (63%); 5 TMSOTf, 6-hepten-1-ol, rt (62%); (d) Grubbs I catalyst, DCM, 40 °C: 6 (81%); 7 (78%); 8 (69%); 9 (70%); (e) Sodium methoxide, methanol, rt (quantitative); (f) 2,6-di-tert-butyl-4-methylpyridine, Tf2O, DCM, −40 °C; (g) n-BuLi, dibenzyl methyl-phosphonate, THF, −78 °C (two step yields): 14 (67%); 15 (59%); 16 (33%); 17 (18%); (h) H2 (balloon pressure), Pd/C, rt; (i) NH4HCO3 (50 mM) aqueous solution, rt (two step yields): 18 (66%); 19 (71%); 20 (68%); 21 (62%).
Initial studies pointed to the need for a modified glycosylation protocol. The previous work employed HCl gas-mediated glycosylation for allyl alcohol itself, but this approach gave low yields, in the present work, when applied to longer chain alcohols. Instead, it was found that TMSOTf-mediated, Vorbrüggen-type glycosylation, using an α-mannosyl acetate glycosyl donor was quite an efficient reaction. Alkene cross metathesis14 and triflate displacement15 then followed as the key steps, as before, in constructing these compounds. Following a final alkene hydrogenation/global debenzylation step, the free tethered sugar phosphonates were obtained. Pleasingly, even with the longest hydrocarbon tether lengths studied here, no solubility issues were encountered in preparing stock solutions up to 200 mM in a HEPES-saline buffer, pH 7.4.
Relative binding affinities to M6P/IGF2R were determined by displacement assay using radiolabeled PMP-BSA as the tracer in the presence of increasing concentrations of each of the synthetic ligands (Table 1). All the compounds in this new series showed IC50 values in the micromolar range with ~2-fold increase in RBA compared to M6P alone. This small increase in RBA likely results from the availability of 2 moles of M6P per mole of ligand providing a 2-fold increase in the effective competitor concentration, as opposed to any effect of tether length. Thus, we conclude that these synthetic compounds are binding the M6P/IGF2R in a monovalent manner. Moreover, these results imply that the two M6-phosphonate moieties are binding essentially independently, and with no apparent thermodynamic or conformational penalty paid for the linker (possible issues include: position of attachment, hydrophobicity, trajectory, etc.)
Table 1.
Relative M6P/IGF2R binding affinities
| Ligand | IC50(n) μMa | RBAb | Mr | Lgthc |
|---|---|---|---|---|
| M6P | 11.5 ± 2.51 (4) | 1.0 | 340 | NA |
| G6P | >10 (4) | NA | 282 | NA |
| 18 (6C) | 4.76 ± 2.50 (4) | 2.63 ± 0.74 | 666 | 16.2–19.5 Å |
| 19 (8C) | 5.03 ± 1.34 (4) | 2.39 ± 0.83 | 694 | 19.2–20.9 Å |
| 20 (10C) | 4.44 ± 1.40 (4) | 2.65 ± 0.52 | 722 | 19.6–22.7 Å |
| 21 (12C) | 3.70 ± 0.56 (4) | 3.02 ± 0.41 | 750 | 24.6–26.0 Å |
IC50’s for competitive displacement of radiolabeled PMP-BSA from the receptor (n = no. of trials, see SI for details); G6P = glucose 6-phosphate
RBA = relative binding affinity, normalized to free M6P;
Length = P-P distance, as estimated by molecular mechanics minimization (MMFF). For each compound, minimizations were run from five different, chain-extended starting conformers. The P-P distances given represent the ranges seen for the set of low energy, chain-extended conformers found.
Dahms and coworkers performed very preliminary modeling studies of the whole receptor, based on the crystal structure of domains 1–3 using topographical information based on the amino acid sequence of each domain.16 Using this approach, they estimated the intramolecular distance of closest approach between the domain 3 and domain 9 M6P binding sites to be ~45 Å. In contrast, they estimated the interphosphate distance between the M6P caps of a bis-phosphorylated oligosaccharide to be ~30 Å. For comparison, we conducted modeling studies of our bis-phosphonate ligands to determine if they could span these distances. Using Spartan 04, we built the model structures of the four compounds and added a cluster of six waters around each phosphonate. In clustering these waters, water-proton/phosphonate oxygen distances were set at 2.5 Å (hydrogen bonding distance). From energy minimization by molecular mechanics methods (Spartan 04) using the Merck Molecular Force Field (MMFF),17 one obtains an estimate of the distance between the two phosphorus atoms in each synthetic bis-phosphonate (Table 1). The longest ligand could have a maximum span of ~26 Å (Figure 4). Based on these estimates, our compounds may still be too short to bind in a bivalent manner, regardless of which receptor binding model is correct.
Figure 4.

One of several low energy chain-extended conformers found by an MMFF molecular mechanics minimization on the hydrated 12-carbon-spaced ligand. See Table 1 for the P,P-distance range found for the set of such conformers.
In addition to the importance of tether length, the structure of the tether is critical to development of a high-affinity bivalent ligand. The adventitious binding properties of the Bock compound suggest that additional binding energy may be achieved by adjusting hydrophobicity, charge and/or π-surface of the tether.11 Moreover, the success enjoyed by the groups of Bock and Hindsgaul, respectively, in attaining 2–3 orders of magnitude improved receptor binding over M6P, suggests that peptide- or carbohydrate-based linkers may be advantageous. Both such tethers present H-bond donor/acceptor functionality across the M6P-M6P span. They also confer more rigidity than a simply sp3-hybridized alkane tether. Thus, each peptide bond really represents a degree of pseudo-unsaturation (planarity) with an expected bias toward a transoid amide geometry.
In conclusion, this study introduces the design and successful synthesis of the first array of bis-M6-phosphonate-presenting “molecular rulers” to measure distances between M6P-binding pockets at MPR’s, and to distinguish between intramolecular and intermolecular modes of bivalent binding. Although the highest M6P/IGF2R binding affinity seen in the ligand set is in the micromolar range [IC50 ~ 4 μM], no solubility problems or tether penalty issues were encountered. Moreover, the replacement of the M6P ester with a hydrolytically stable phosphonate surrogate persists as an effective design, across the entire set, and reinforces the notion that phosphatase resistance can be incorporated into such small molecule probes. Completion of these studies will require that we find a high-affinity ligand that stabilizes the dimeric structure of the receptor and thereby promotes rapid internalization of IGF-II in a cellular model. Ultimately, a new compound that exhibits all these properties would potentially be testable in an animal model for inhibition for IGF-II-driven tumor growth.
In closing, we note that the combination of cross-metathesis to build the bivalent sugar scaffold, and bis-triflate displacement to introduce the phosphate-surrogate late in the synthesis, is a powerful approach. This strategy is likely amenable to the introduction of other phosphate-mimicking functionality in the endgame, and more generally, is likely extendible to the study of other multivalent ligand-protein interactions.
Supplementary Material
Acknowledgments
The authors are indebted to the American Heart Association (GIA-060014Z to DBB), the NIH (CA-91885 to RGM) and to the University of Nebraska Biomedical Research Retreat 2006 Collaboration Seed Grant Program (jointly to RGM and DBB) for funding. CMC was supported by the Nebraska Center for Cellular Signaling (NIH grant P20 RR18759), UNMC Graduate Studies assistantship, the Dr. Fred W. Upson grant-in-aid award through UNMC, and the UNMC Faculty Women’s Club Scholarship. We acknowledge the NSF (CHE-0091975, MRI-0079750) and NIH (SIG-1-510-RR-06307) for NMR instrumentation, and the NIH (RR016544) for facilities renovation.
Footnotes
Experimental procedures, spectral data, copies of NMR spectra and details of the radioligand displacement assays can be found in the online version at ____.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Scott CD, Firth SM. Horm Metab Res. 2004;36:261. doi: 10.1055/s-2004-814477. [DOI] [PubMed] [Google Scholar]; (b) Ghosh P, Dahms NM, Kornfeld S. Nat Rev Mol Cell Biol. 2003;4:202. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]; (c) Dahms NM, Hancock MK. Biochim Biophys Acta. 2002;1572:317. doi: 10.1016/s0304-4165(02)00317-3. [DOI] [PubMed] [Google Scholar]
- 2.(a) Foulstone E, Prince S, Zaccheo O, Burns JL, Harper J, Jacobs C, Church D, Hassan AB. J Pathol. 2005;205:145. doi: 10.1002/path.1712. [DOI] [PubMed] [Google Scholar]; (b) Pollak MN, Schernhammer ES, Hankinson SE. Nat Rev Cancer. 2004;4:505. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- 3.(a) Hancock MK, Haskins DJ, Sun G, Dahms NM. J Biol Chem. 2002;277:11255. doi: 10.1074/jbc.M109855200. [DOI] [PubMed] [Google Scholar]; (b) Schmidt B, Kiecke-Siemsen C, Waheed A, Braulke T, von Figura K. J Biol Chem. 1995;270:14975. doi: 10.1074/jbc.270.25.14975. [DOI] [PubMed] [Google Scholar]; (c) Garmroudi F, MacDonald RG. J Biol Chem. 1994;269:26944. [PubMed] [Google Scholar]; (d) Dahms NM, Wick DA, Brzycki-Wessell MA. J Biol Chem. 1994;269:3802. [PubMed] [Google Scholar]; (e) Dahms NM, Rose PA, Molkentin JD, Zhang Y, Brzycki MA. J Biol Chem. 1993;268:5457. [PubMed] [Google Scholar]
- 4.(a) Byrd JC, Park JHY, Schaffer BS, Garmroudi F, MacDonald RG. J Biol Chem. 2000;275:18647. doi: 10.1074/jbc.M001273200. [DOI] [PubMed] [Google Scholar]; (b) Byrd JC, MacDonald RG. J Biol Chem. 2000;275:18638. doi: 10.1074/jbc.M000010200. [DOI] [PubMed] [Google Scholar]
- 5.York SJ, Arneson LS, Gregory WT, Dahms NM, Kornfeld S. J Biol Chem. 1999;274:1164. doi: 10.1074/jbc.274.2.1164. [DOI] [PubMed] [Google Scholar]
- 6.(a) Puffer EB, Pontrello JK, Hollenbeck JJ, Kink JA, Kiessling LL. ACS Chem Biol. 2007;2:252. doi: 10.1021/cb600489g. [DOI] [PubMed] [Google Scholar]; (b) Gordon EJ, Gestwicki JE, Strong LE, Kiessling LL. Chem Biol. 2000;7:9. doi: 10.1016/s1074-5521(00)00060-0. [DOI] [PubMed] [Google Scholar]; (c) Strong LE, Kiessling LL. J Am Chem Soc. 1999;121:6193. [Google Scholar]
- 7.Mammen M, Chio SK, Whitesides GM. Angew Chem Int Ed. 1998;37:2755. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 8.Tong PY, Gregory W, Kornfeld S. J Biol Chem. 1989;264:7962. [PubMed] [Google Scholar]
- 9.Varki A, Kornfeld S. J Biol Chem. 1983;258:2808. [PubMed] [Google Scholar]
- 10.Distler JJ, Guo JF, Jourdian GW, Srivastava OP, Hindsgaul O. J Biol Chem. 1991;266:21687. [PubMed] [Google Scholar]
- 11.Franzyk H, Christensen MK, Jorgensen RM, Meldal M, Cordes H, Mouritsen S, Bock K. Bioorg Med Chem. 1997;5:21. doi: 10.1016/s0968-0896(96)00194-0. [DOI] [PubMed] [Google Scholar]
- 12.Berkowitz DB, Maiti G, Charette BD, Dreis CD, MacDonald RG. Org Lett. 2004;6:4921. doi: 10.1021/ol0479444. [DOI] [PubMed] [Google Scholar]
- 13.(a) Jeanjean A, Garcia M, Leydet A, Montero JL, Morere A. Bioorg Med Chem. 2006;14:3575. doi: 10.1016/j.bmc.2006.01.024. [DOI] [PubMed] [Google Scholar]; (b) Vidal S, Garcia M, Montero JL, Morere A. Bioorg Med Chem. 2002;10:4051. doi: 10.1016/s0968-0896(02)00264-x. [DOI] [PubMed] [Google Scholar]; (c) Vidal S, Vidil C, Morere A, Garcia M, Montero JL. Eur J Org Chem. 2000:3433. [Google Scholar]
- 14.Chatterjee AK, Morgan JP, Scholl M, Grubbs RH. J Am Chem Soc. 2000;122:3783. [Google Scholar]
- 15.(a) Berkowitz DB, Bose M, Asher NG. Org Lett. 2001;3:2009. doi: 10.1021/ol015983z. [DOI] [PubMed] [Google Scholar]; (b) Berkowitz DB, Bhuniya D, Peris G. Tet Lett. 1999;40:1869. [Google Scholar]; (c) Berkowitz DB, Sloss DG. J Org Chem. 1995;60:7047. [Google Scholar]; (d) Shen Q, Sloss DG, Berkowitz DB. Syn Commun. 1994;24:1519. [Google Scholar]; (e) Berkowitz DB, Eggen M, Shen Q, Sloss DG. J Org Chem. 1993;58:6174. [Google Scholar]
- 16.Olson LJ, Yammani RD, Dahms NM, Kim JJP. EMBO J. 2004;23:2019. doi: 10.1038/sj.emboj.7600215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halgren TA. J Am Chem Soc. 1992;114:7827. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.