

Abstract
Traditional small-molecule fluorophores are always fluorescent. That attribute can obscure valuable information in biological experiments. Here, we report on a versatile “latent” fluorophore that overcomes this limitation. At the core of the latent fluorophore is a derivative of rhodamine in which one nitrogen is modified as a urea. That modification enables rhodamine to retain half of its fluorescence while facilitating conjugation to a target molecule. The other nitrogen of rhodamine is modified with a “trimethyl lock”, which enables fluorescence to be unmasked fully by a single user-designated chemical reaction. An esterase-reactive latent fluorophore was synthesized in high yield and attached covalently to a cationic protein. The resulting conjugate was not fluorescent in the absence of esterases. The enzymatic activity of esterases in endocytic vesicles and the cytosol educed fluorescence, enabling the time-lapse imaging of endocytosis into live human cells and thus providing unprecedented spatiotemporal resolution of this process. The modular design of this “fluorogenic label” enables the facile synthesis of an ensemble of small-molecule probes for the illumination of numerous biochemical and cell biological processes.
Introduction
Fluorescent molecules are critical tools in the study of biochemical and cell biological processes (1). In many studies, however, only few of the fluorescent molecules experience a phenomenon of interest. Because traditional fluorophores, such as rhodamine and fluorescein, are always fluorescent, bulk fluorescence can obscure valuable information. To overcome this limitation, molecules can be designed such that a chemical reaction elicits a change in their fluorescence. Such “latent” fluorophores are at the core of common methods, including the enzyme linked immunosorbent assay (ELISA), high-throughput screening of enzyme inhibitors, detection of reporter genes, and evaluation of cell viability (1). We reasoned that the use of a latent fluorophore as a “fluorogenic label” could overcome limitations of traditional fluorescent labels and thereby improve the spatial and temporal resolution of bioimaging.
Recently, our laboratory reported on a new class of latent fluorophores based on the “trimethyl lock” (2, 3). The rapid lactonization (4, 5) of the trimethyl lock had been exploited previously to prepare stable pro-drugs that were unmasked by an enzyme-catalyzed reaction (6, 7). We first used the trimethyl lock to shroud the fluorescence of a xanthene dye, rhodamine 110 (Rh110) (2), and then an oxazine dye, cresyl violet (3). This approach afforded highly stable bis(trimethyl lock) “pro-fluorophores” that were labile to esterase catalysis in vitro and in cellulo.
Our bis(trimethyl lock) pro-fluorophores had two problematic attributes. First, two chemical reactions were necessary to unveil the vast majority of their fluorescence, decreasing the rate of fluorescence manifestation and limiting the linear range of assays (8, 9). Analogous fluorogenic protease substrates based on a rhodamine diamide display complex hydrolysis kinetics (10–12), as we observed with our bis(trimethyl lock) pro-fluorophores (2, 3). The second problematic attribute was the absence of a handle for target-molecule conjugation. Such a handle is available in derivatives, such as 5- or 6-carboxyrhodamine, that are accessible only from low-yielding synthetic routes.
We suspected that we could solve both problems by capping one of the amino groups of Rh110. The capping of rhodamine dyes with an amide (13–15), carbamate (16), or urea (17) can preserve much of their fluorescence. We were especially intrigued by the attributes of urea–rhodamine, which according to recent reports in the scientific (17) and patent (18, 19) literature appears to retain significant fluorescence intensity relative to Rh110.
Here, we report on a versatile fluorogenic label for biomolecular imaging. First, we describe the synthesis of a complete set of ureated and amidated derivatives of Rh110, as well as a characterization of their fluorescent properties. Then, we show that imposing our trimethyl lock strategy upon a urea–rhodamine yields a stable latent fluorophore with a high rate of enzymatic hydrolysis. Finally, we demonstrate the power of our modular approach by using the urea moiety as a handle for protein conjugation and subsequent continuous imaging of endocytosis by live human cells.
RESULTS AND DISCUSSION
Synthesis of Model Compounds
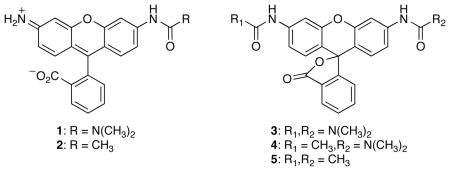
To gain a comprehensive understanding of the urea and amide derivatives of rhodamine, we undertook the synthesis of compounds 1–5 (Table 1). Rhodamine itself and these five derivatives encompass the ensemble of possible ureated and amidated derivatives. We were especially interested in those properties of 1–5 with biological implications, such as the extinction coefficient and quantum yield in aqueous solution. Previous reports (20, 10, 11, 18, 19, 17) of similar derivatives did not provide a complete listing of relevant fluorescent characteristics.
Table 1.
Spectroscopic Properties of Rh110 and Its Derivatives
 | |||||
|---|---|---|---|---|---|
| Dye | λmax (nm) | ε (M−1cm−1) | λem (nm) | Φ | ε × Φ (rel) |
| Rh110 | 496 | 74,000 | 517 | 0.92 | 100% |
| 1 | 492 | 48,600 | 518 | 0.49 | 35% |
| 2 | 489 | 30,200 | 522 | 0.28 | 12% |
| 3 | 482 | 3300 | 517 | 0.01 | 0.05% |
| 4 | 475 | 400 | — | — | — |
| 5 | ~469 | ≤200 | — | — | — |
Installation of the urea moiety to produce urea 1 proved to be surprisingly difficult. In our hands, the reported conditions (17) involving the reaction of Rh110 with a carbamoyl chloride using Hünig’s base gave an intractable mixture of products. In contrast, we found that Rh110 was deprotonated effectively with NaH, and that the resulting anion reacted with dimethylcarbamyl chloride to yield the desired urea 1. This deprotonation strategy also proved useful for the synthesis of amide 2 and diurea 3. The additional acetamide group in urea–amide 4 and diamide 5 were installed by reaction with acetyl chloride in the presence of a base.
Fluorescence Properties
The absorbance and fluorescence spectra of Rh110 and each derivative are shown in Figure 1. The corresponding values of λmax, extinction coefficient at λmax (ε), λem, and quantum yield (Φ) are listed in Table 1. We determined the relative fluorescence intensity of these compounds by calculating the product of extinction coefficient and quantum yield and then normalizing these values to those of Rh110. In our measurement, urea 1 retained 35% of the fluorescence intensity of Rh110 with a quantum yield value of 0.49. Amide 2 is only 12% as fluorescent as Rh110, which is consistent with earlier reports (10, 11). The fluorescence of the bis-substituted dyes was largely quenched in aqueous solution. Diurea 3 did, however, possess significant absorbance and fluorescence compared to the urea–amide 4 or diamide 5. These latter two rhodamine derivatives are essentially nonfluorescent.
Figure 1.

Spectra of Rh110 and its derivatives. a) Absorption spectra of Rh110 and derivatives 1–5 (7.5 μM). b) Fluorescent emission spectra of Rh110 and 1–3 (λex = 450 nm, not to scale).
We also determined the pH-dependence of the fluorescence of urea 1 and amide 2. The fluorescence of Rh110 is relatively insensitive to pH values between 4 and 10 (1). This property is beneficial in biological assays, where unknown variations in pH can hamper quantitative measurements. Like Rh110, urea 1 and amide 2 show no significant spectral change between pH values of 4 and 10 (see: Figure S1 in the Supporting Information).
Substituent effects on the fluorescent properties of rhodamine dyes are challenging to predict or interpret due to the complexity of the rhodamine system (21). In solution, rhodamine derivatives exist in equilibrium between a zwitterion that absorbs visible light and is fluorescent and a lactone that is colorless and nonfluorescent. Substitution on nitrogen can affect both this open closed equilibrium and the spectral characteristics of the fluorescent zwitterions (22, 23). We suspected that the differences in optical properties seen in compounds 1–5 could be rationalized, in part, through examination of the electron-donation capability of the different nitrogen substituents. According to this reasoning, weakly donating substituents would favor the colorless lactone as well as decrease the intrinsic absorptivity of the zwitterions and, hence, the extinction coefficient. Weakly donating substituents could also reduce the quantum yield by decreasing the C–N bond-order and thereby enhancing nonradiative decay of the excited state through vibrational relaxation processes (24, 25).
We explored the relationship between the values of extinction coefficient and quantum yield and the Hammett σp substituent constants (26). An unprotonated amino group is a good electron donor (σp = −0.66), whereas an amide group is a relatively poor donor (σp = 0.00), due to amidic resonance. A urea group is peculiar—its carbonyl group is cross-conjugated and both of its nitrogens participate in amidic resonance. This cross-conjugation attenuates its electron-donating ability, as reflected in an intermediate Hammett constant (σp = −0.26). A plot of both extinction coefficient and quantum yield versus σp substituent constant for Rh110 and monosubstituted rhodamines 1 and 2 are shown in Figure 2. The correlation indicates that both spectral properties are affected by electron donation from the nitrogens. A similar trend in quantum yields has been observed in substituted phenoxazinone dyes (27).
Figure 2.

Hammett plot of extinction coefficient (○) and quantum yield (●) versus σp for Rh110, urea 1, and amide 2.
The moderate electron-donating character of the urea moiety provides an explanation for the advantageous properties of urea 1. Substitution with the cross-conjugated urea suppresses the fluorescence intensity of urea 1 relative to Rh110. This decrease is not, however, as severe as seen in amide 2, due to the greater electron-donating properties of the urea moiety. Still, the attenuated electron-donation allows complete suppression of fluorescence upon amidation of the remaining nitrogen in urea–amide 4. Finally, the effect of electron-rich substituents on the rhodamine system are apparent again in the fluorescence of diurea 3 being greater than that of diamide 5.
Synthesis of Urea–Rhodamine Trimethyl Lock
Having affirmed the desirable properties of urea–rhodamine, we next sought to apply our trimethyl lock strategy to this dye. The synthetic route to the fluorogenic substrate, which employs rhodamine morpholino-urea 6 (17), is shown in Scheme 1. Again, we found that the use of Hünig’s base in the synthesis afforded a mixture of products. Deprotonation of Rh110 with NaH followed by dropwise addition of 4-morpholinecarbonyl chloride furnished rhodamine morpholino-urea 6. This compound exhibited similar fluorescent characteristics to urea 1 (Table 1), having an extinction coefficient of 51,700 M−1cm−1 and quantum yield of 0.44. Carbodiimide coupling of rhodamine morpholino-urea 6 with acid 7 (28) afforded the desired pro-fluorophore 8.
Scheme 1.

Synthetic Route to Pro-Fluorophore 8
Chemical Stability
Pro-fluorophore 8 must be stable in aqueous solution to be useful in biological assays. Such stability can be problematic for hydrolase substrates, as spontaneous hydrolysis can compete effectively with enzymatic activity and raise background levels. As shown in Figure 3, pro-fluorophore 8 showed remarkable stability in both phosphate-buffered saline (PBS) and Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% v/v fetal bovine serum (FBS). In contrast, fluorescein diacetate, which is a widely used esterase substrate (29), suffered relatively rapid hydrolysis in both solutions. This dramatic increase in stability arises from the large difference in pKa values between the conjugate acids of the two leaving groups. Specifically, fluorescein (pKa 6.32 (30)) is a much better leaving group than is the electron-rich trimethyl-lock phenol (o-methylphenol has pKa 10.28 (31)).
Figure 3.

Stability of pro-fluorophore 8 and fluorescein diacetate in aqueous solution. a) Time course for the generation of fluorescence (λex 496 nm, λem 520 nm) of pro-fluorophore 8 (25 nM) and fluorescein diacetate (25 nM) in PBS. b) Time course of the generation of fluorescence (λex 496 nm, λem 520 nm) of pro-fluorophore 8 (25 nM) and fluorescein diacetate (25 nM) in DMEM containing FBS (10% v/v).
Enzymatic Reactivity
An objective in the design of pro-fluorophore 8 was to improve its reactivity as an esterase substrate relative to the original bis(trimethyl lock) rhodamine substrate. The appearance of fluorescence upon reaction of porcine liver esterase (PLE) with pro-fluorophore 8 was indicative of single-hit kinetics (Figure 4). The kinetic constants were calculated to be kcat/KM = 8.2 × 105 M−1s−1 and KM = 0.10 μM. Comparison with the apparent kinetic constants from the original bis(trimethyl lock) rhodamine substrate (2) (kcat/KM = 1.9 × 103 M−1s−1 and KM = 0.47 μM) shows a 430-fold increase in kcat/KM value. A more appropriate comparison takes into account the expected 65% decrease in fluorescence of urea 6 (Table 1), which is the hydrolysis product of pro-fluorophore 8, relative to Rh110. After this adjustment, latent fluorophore performance is still enhanced by 150-fold.
Figure 4.

Kinetic traces (λex 496 nm, λem 520 nm) and Michaelis–Menten plot (inset) of a serial dilution of pro-fluorophore 8 (0.5 μM → 2 nM) with PLE (2.5 μg/mL).
The substantial increase in catalytic efficiency is likely due to the change from the double-hit kinetics observed for the bis-substituted substrate to the single-hit kinetics of pro-fluorophore 8. Hydrolysis of the bis-substituted fluorogenic substrate progresses from diamide to free Rh110 via a monoamide intermediate, with the unmasking of the second amino group producing the majority (<90%) of the fluorescence (10, 11). In contrast, the urea–rhodamine substrate requires only a single cleavage event for the complete manifestation of fluorescence.
Cellular Imaging
Having established the high chemical stability and enzymatic reactivity of pro-fluorophore 8, we next evaluated the behavior of this compound in live human cells. Pro-fluorophore 8 was incubated with HeLa cells and imaged using confocal fluorescence microscopy. As shown in Figure 5A, the substrate was activated in cellulo by endogenous esterases to produce diffuse green cytosolic staining. Importantly, the high chemical stability of the fluorogenic probe allowed for its imaging in the cytosol without an intermediate washing step. Counter-staining with LysoTracker Red showed significant but incomplete colocalization, suggesting that after hydrolysis, a portion of the free urea rhodamine localized in acidic vesicles (yellow color in Figure 5B). To ensure that the fluorescence increase was due to trimethyl lock activation and not hydrolysis of the urea moiety, we incubated HeLa cells with the relatively non-fluorescent diurea-rhodamine 5. In these experiments we observed virtually no intracellular fluorescence (see: Figure S2 in the Supporting Information).
Figure 5.
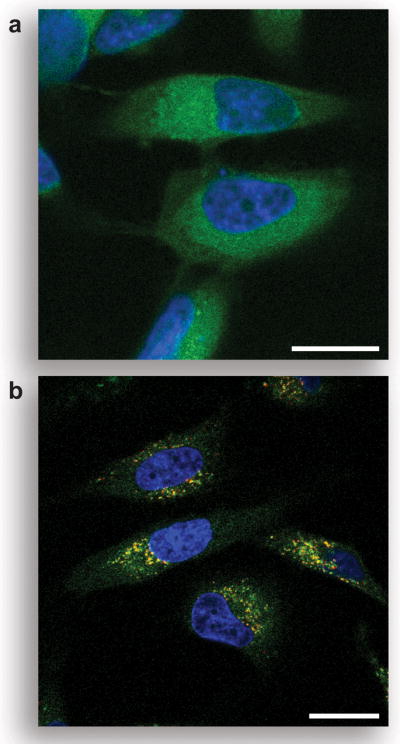
Unmasking of pro-fluorophore 8 in live human cells. a) Unwashed HeLa cells incubated for 1 h with pro-fluorophore 8 (10 μM) at 37 °C in DMEM and counter-stained with Hoechst 33342. b) Washed HeLa cells incubated for 1 h with pro-fluorophore 8 (10 μM) at 37 °C in DMEM and counter-stained with Hoechst 33342 and LysoTracker Red (5% v/v CO2(g), 100% humidity). Scale bar: 20 μm.
Fluorogenic Label
The high chemical stability and rapid in cellulo unmasking of pro-fluorophore 8 prompted us to develop a derivative for bioconjugation. We reasoned that such a fluorogenic label would be stable enough to survive conjugation and purification protocols while still providing a strong signal for continuous biological experiments. It is noteworthy that simple fluorescein diesters have found only limited use as fluorogenic labels (32–35), as fluorescein diesters suffer from low chemical stability in aqueous solution (Figure 3).
Developing pro-fluorophore 8 into a fluorogenic label requires the installation of a functional group with selective reactivity. We chose to install the maleimide functionality (36, 37), which react rapidly with thiol groups (38). The resulting conjugates are stable (39), even after the slow hydrolysis of the nascent sulfosuccinimidyl ring (40).
Traditionally, reactive groups are attached to the pendant carboxyphenyl ring of rhodamine and fluorescein dyes (1). Synthesis of these compounds requires difficult chromatographic steps to obtain isomerically pure compounds (41). We envisioned a facile and economical alternative involving the attachment of a maleimide derivative via the desirable urea functionality. Although uncommon, bioconjugation via the amino groups of rhodamines has been used previously (42, 14, 43). This strategy allows for the use of commercially available (and relatively inexpensive) Rh110 as the starting material for the synthesis of maleimidourea–rhodamine trimethyl lock 13, as shown in Scheme 2. Desymmetrization of Rh110 was accomplished by its deprotonation with NaH and reaction with Boc2O to give t-Boc–rhodamine 9. An isocyanate was generated in situ from maleimide 10 by a Curtius rearrangement (44, 45), and that isocyanate was reacted with t-Boc–rhodamine 9 to generate a urea (46, 47). Deprotection of maleimidourea–rhodamine–t-Boc 11 with TFA afforded fluorescent urea–rhodamine 12. Condensation with 7 using EDC gave thiol-reactive fluorogenic label 13.
Scheme 2.

Synthetic Route to Fluorogenic Label 13
Bioconjugation
To test the utility of fluorogenic label 13 in a biological experiment, we attached it to a thiol-containing variant of bovine pancreatic ribonuclease (RNase A (48)). RNase A is a cationic protein that is internalized by mammalian cells via endocytosis (49). This internalization is critical to the action of cytotoxic RNase A variants and homologs (50). Fluorogenic label 13 reacted cleanly with the A19C variant of RNase A to give a mono-substituted conjugate as determined by MALDI mass spectrometry. This protein conjugate was stable to purification by cation-exchange chromatography at pH 5.0 and showed a 1200-fold increase in fluorescence upon incubation with PLE (data not shown).
At physiological pH, the protein conjugate was less stable than unconjugated pro-fluorophore 8. Spontaneous hydrolysis of the acetate ester was slow but significant in PBS, presumably because conjugation to the protein places the probe in close proximity to nucleophilic functional groups of the protein. Storage at pH 5.0 did, however, extend the stability of the conjugate, allowing multiple experiments to be performed with one preparation.
Cellular Imaging with a Bioconjugate
Fluorescently labeled biomolecules have been used to image endocytotic events (51). We sought to determine the efficacy of our fluorogenic label approach by comparing endocytosis of HeLa cells incubated with Oregon Green-labeled RNase A (49) to that of cells incubated with the protein conjugated with fluorogenic label 13. As shown in Figure 6A, the Oregon Green conjugate showed intense extracellular background signal that obscures the fluorescence from endocytosed material. This background could be eliminated only with many vigorous washing steps (Figure 6B). In contrast, the pro-fluorophore conjugate allowed imaging without intermediate washing steps. As shown in Figure 6C, unwashed HeLa cells incubated with the RNase A conjugate have bright, punctate staining, indicative of the conjugate being localized in small vesicles. Counterstaining with LysoTracker Red shows a large degree of colocalization (Figure 6D), suggesting that the latent conjugate is internalized via endocytosis and activated by endosomal or lysosomal esterases (52–54). Images with the protein conjugate (Figure 6C) are less diffuse and more punctate that are images with free pro-fluorophore 8 (Figure 5), which has much more ready access to the cytosol. To ensure that the signal in Figure 6C is due to unmasked fluorophore attached to RNase A, we fixed cells incubated with our latent conjugate and counterstained them with a primary antibody to RNase A and a secondary antibody labeled with AlexaFluor 594. In a fluorescence microscopy image, we observed a significant overlap of the green and red fluorescent signals to produce a yellow signal, indicating that the unmasked RNase A conjugate is largely intact (see: Figure S3 in the Supporting Information).
Figure 6.

Live-cell imaging experiments with protein conjugates. a) Unwashed HeLa cells incubated for 1 h with Oregon Green–RNase A conjugate (10 μM) at 37 °C in DPBS and counter-stained with Hoechst 33342. b) Washed HeLa cells incubated for 1 h with Oregon Green–RNase A conjugate (10 μM) at 37 °C in DPBS and counter-stained with Hoechst 33342. c) Unwashed HeLa cells incubated for 1 h with fluorogenic label 13–RNase A conjugate (10 μM) at 37 °C in DPBS and counter-stained with Hoechst 33342. d) Washed HeLa cells incubated for 1 h with fluorogenic label 13–RNase A conjugate (10 μM) at 37 °C in DPBS and counter-stained with Hoechst 33342 and LysoTracker Red (5% v/v CO2(g), 100% humidity). Scale bar: 20 μm.
The high chemical stability and low background fluorescence of the fluorogenic label conjugate allowed for the time-lapse imaging of its endocytosis. Cells were incubated with the fluorogenic label 13–RNase A conjugate at room temperature, and images were recorded without washing during the next 90 min. The compilation of these images into a movie revealed that internalization of the conjugate occurred continuously and that vesicular fluorescence increased monotonically (see: Movie in the Supporting Information).
Envoi
We have demonstrated how a common fluorophore, Rh110, can be elaborated into a powerful new tool for biochemistry and cell biology. The use of a trimethyl lock provides a latent fluorophore with high chemical stability while maintaining enzymatic reactivity (2, 3). The use of a urea group (rather than a second trimethyl lock) improves enzymatic reactivity markedly while preserving desirable fluorescence properties, as in pro-fluorophore 8. The elaboration of the urea to include an electrophile outfits the latent fluorophore for conjugation, as in fluorogenic label 13. Conjugation of this fluorogenic label to a target molecule enables, for example, the continuous imaging of the endocytosis of a target molecule by live human cells.
We note that the urea–rhodamine–trimethyl lock probe is modular, and hence can be tailored to suit a variety of applications (Scheme 3). For example, alteration of the bioconjugative group on the urea moiety could be used to change conjugation chemistry, enhance cellular internalization, or target a conjugate to a specific subcellular location. Modification of the enzyme-reactive group on the trimethyl lock could enable the detection of a conjugate in a particular organelle. The use of fluorogenic labels could even transcend cultured cells, allowing for continuous imaging in tissues or in vivo. These applications would be facilitated by extant comprehensive inventories of the enzymes in numerous organs and organelles (55, 56). Accordingly, the fluorogenic label strategy could enable the development of specific probes for biological experiments of ever-increasing sophistication.
Scheme 3.

Modules in Fluorogenic Label 13
METHODS
General Spectroscopic Methods
HEPES (2[4-(2-hydroxyethyl)-1-piperazinyl]ethanesulfonic acid) was from Research Products International. Fluorescein (reference standard grade) was from Molecular Probes. Other reagents were from Sigma–Aldrich or Fisher Scientific. Phosphate-buffered saline, pH 7.4 (PBS) contained (in 1.00 liter) KCl (0.20 g), KH2PO4 (0.20 g), NaCl (8.0 g), and Na2HPO4·7H2O (2.16 g). All measurements were recorded at ambient temperature (23 ± 2 °C) and buffers were not degassed prior to measurements. Compounds were prepared as stock solutions in DMSO and diluted such that the DMSO concentration did not exceed 1% v/v. Porcine liver esterase (PLE, MW = 163 kDa (57)) was obtained from Sigma Chemical (product number E2884) as a suspension in 3.2 M (NH4)2SO4, and was diluted to appropriate concentrations in PBS before use. In pH-dependency studies, the pH of PBS was adjusted by additions of 1.0 M HCl or 1.0 M NaOH and measured using a Beckmann glass electrode that was calibrated prior to each use. Graphs were manipulated and parameters were calculated with Microsoft Excel 2003 and GraphPad Prism 4.
Ultraviolet–Visible and Fluorescence Spectroscopy
Absorption spectra were recorded in 1-cm path length cuvettes having a volume of 1.0 or 3.5 mL on a Cary Model 50 spectrometer from Varian. The extinction coefficients were measured in 10 mM HEPES–NaOH buffer, pH 7.5. Fluorometric measurements were made using fluorescence grade quartz or glass cuvettes from Starna Cells and a QuantaMaster1 photon-counting spectrofluorometer from Photon Technology International equipped with sample stirring. The quantum yields of Rh110 and compounds 1–5 were measured with dilute samples (A ≤ 0.1) in 10 mM HEPES–NaOH buffer, pH 7.5. These values were obtained by the comparison of the integrated area of the emission spectrum of the samples with that of fluorescein in 0.1 M NaOH, which has a quantum efficiency of 0.95 (58). The concentration of the fluorescein reference was adjusted to match the absorbance of the test sample at the excitation wavelength. Under these conditions, quantum yields were calculated by using eq 1.
| (1) |
Protein Purification and Labeling
The TNB-protected A19C variant of RNase A and the Oregon Green-labeled RNase A conjugate were prepared as described previously (49). The TNB-protected protein was deprotected with a three-fold molar excess of dithiothreitol (DTT) and desalted by chromatography using a HiTrap Desalting column (Amersham). The protein conjugate then was prepared by reaction with ten-fold molar excess of thiol-reactive maleimide 13 for 16 h at 4 °C. Purification by chromatography using a HiTrap HP SP column (Amersham) afforded the desired conjugate (MS (MALDI): m/z 14,468 (expected: 14,475)). Protein concentration was determined by using a bicinchoninic acid (BCA) assay kit from Pierce with wild-type RNase A as a standard.
Cell Preparation
HeLa cells were plated on Nunc Lab-Tek II 8-well Chamber Coverglass (Fisher Scientific) and grown to 70–80% confluence at 37 °C in DMEM (Invitrogen) containing FBS (10% v/v). For static imaging, cells were first washed with Dulbecco’s phosphate-buffered saline (DPBS, Invitrogen). Cells were then incubated with pro-fluorophore 8 (10 μM), RNase A conjugated to maleimide 13 (10 μM), or Oregon Green-labeled RNase A (10 μM) for 1 h at 37 °C prior to imaging. Nuclear staining was accomplished by addition of Hoechst 33342 (2 μg/mL) for the final 5 min of incubation. Lysosomal staining involved washing the cells with DPBS followed by incubation with 100 nM LysoTracker Red (Molecular Probes) in DPBS for 1 min at ambient temperature. For dynamic imaging, cells were incubated with Hoechst 33342 (2 μg/mL) for 5 min at 37 oC, and then washed twice with DPBS. Pro-fluorophore 13–RNase A conjugate (10 μM) was added to the cells at ambient temperature (23 ± 2 °C). Imaging of endocytosis started within 1 min after the addition of the conjugate.
Cell Imaging
Cells were imaged on a Nikon Eclipse TE2000-U confocal microscope equipped with a Zeiss AxioCam digital camera, unless indicated otherwise. Excitation at 408 nm was provided by a blue-diode laser, and emission light was passed though a filter centered at 450 nm with a 35-nm band-pass. Excitation at 488 nm was provided by an argon-ion laser and emission light was passed through a filter centered at 515 nm with a 40-nm band-pass. Excitation at 543 nm was provided by a HeNe laser, and emission light was passed through a filter centered at 605 nm with a 75-nm band-pass. For time-lapse imaging, 1 image/min was recorded during the first 30 min of incubation, 2 images/min were recorded during the next 10 min, and 5 images/min were recorded during the last 50 min. The resulting movie condenses these 300 images recorded over 90 min into 40 s. Brightfield images indicated that the cells were alive and appeared to have normal physiology, both before and after the time-lapse imaging.
Supplementary Material
Acknowledgments
We are grateful to K. A. Dickson for preliminary bioimaging experiments and S. S. Chandran, Z. Diwu, and M. B. Soellner for contributive discussions. L.D.L was supported by Biotechnology Training grant 08349 (NIH). This work was supported by Grant CA73808 (NIH). The University of Wisconsin Madison Biophysics Instrumentation Facility was established with grants BIR-9512577 (NSF) and RR13790 (NIH). NMRFAM was supported by grant P41RR02301 (NIH).
Footnotes
Supporting Information Available: All synthetic methods; additional data on the pH sensitivity of Rh110, urea 1, and amide 2; image of HeLa cells incubated with diurea 3; image of HeLa cells incubated with the fluorogenic label 13–RNase A conjugate and counter-stained with anti-RNase A antibodies; and a QuickTime movie of the endocytosis of the fluorogenic label 13–RNase A conjugate into living HeLa cells. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Haugland RP, Spence MTZ, Johnson ID, Basey A. The Handbook: A Guide to Fluorescent Probes and Labeling Technologies. 10. Molecular Probes; Eugene, OR: 2005. [Google Scholar]
- 2.Chandran SS, Dickson KA, Raines RT. Latent fluorophore based on the trimethyl lock. J Am Chem Soc. 2005;127:1652–1653. doi: 10.1021/ja043736v. [DOI] [PubMed] [Google Scholar]
- 3.Lavis LD, Chao T-Y, Raines RT. Latent blue and red fluorophores based on the trimethyl lock. ChemBioChem. 2006;7:xxx–xxx. doi: 10.1002/cbic.200500559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borchardt RT, Cohen LA. Stereopopulation control. II. Rate enhancement of intramolecular nucleophilic displacement. J Am Chem Soc. 1972;94:9166–9174. doi: 10.1021/ja00781a030. [DOI] [PubMed] [Google Scholar]
- 5.Milstein S, Cohen LA. Stereopopulation control. I. Rate enhancement in the lactonizations of o-hydroxyhydrocinnamic acids. J Am Chem Soc. 1972;94:9158–9165. doi: 10.1021/ja00781a029. [DOI] [PubMed] [Google Scholar]
- 6.Shan D, Nicolaou MG, Borchardt RT, Wang B. Prodrug strategies based on intramolecular cyclization reactions. J Pharm Sci. 1997;86:765–767. doi: 10.1021/js970069d. [DOI] [PubMed] [Google Scholar]
- 7.Testa B, Mayer JM. Hydrolysis in Drug and Prodrug Metabolism: Chemistry, Biochemistry, and Enzymology. Verlag Helvetica Chimica Acta; Zürich, Switzerland: 2003. [Google Scholar]
- 8.Fiering SN, Roederer M, Nolan GP, Micklem DR, Parks DR, Herzenberg LA. Improved FACS-Gal: Flow cytometric analysis and sorting of viable eukaryotic cells expressing reporter gene constructs. Cytometry. 1991;12:291–301. doi: 10.1002/cyto.990120402. [DOI] [PubMed] [Google Scholar]
- 9.Urano Y, Kamiya M, Kanda K, Ueno T, Hirose K, Nagano T. Evolution of fluorescein as a platform for finely tunable fluorescence probes. J Am Chem Soc. 2005;127:4888–4894. doi: 10.1021/ja043919h. [DOI] [PubMed] [Google Scholar]
- 10.Leytus SP, Melhado LL, Mangel WF. Rhodamine-based compounds as fluorogenic substrates for serine proteinases. Biochem J. 1983;209:299–307. doi: 10.1042/bj2090299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leytus SP, Patterson WL, Mangel WF. New class of sensitive and selective fluorogenic substrates for serine proteinases. Amino acid and dipeptide derivatives of rhodamine. Biochem J. 1983;215:253–260. doi: 10.1042/bj2150253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Bhalgat M, Zhang C, Diwu Z, Hoyland B, Klaubert DH. Fluorescent molecular probes V: A sensitive caspase-3 substrate for fluorometric assays. Bioorg Med Chem Lett. 1999;9:3231–3236. doi: 10.1016/s0960-894x(99)00566-1. [DOI] [PubMed] [Google Scholar]
- 13.Guzikowski AP, Naleway JJ, Shipp CT, Schutte RC. Synthesis of a macrocyclic rhodamine 110 enzyme substrate as an intracellular probe for caspase 3 activity. Tetrahedron Lett. 2000;41:4733–4735. [Google Scholar]
- 14.Lorey S, Faust J, Mrestani-Klaus C, Kaehne T, Ansorge S, Neubert K, Buehling F. Transcellular proteolysis demonstrated by novel cell surface-associated substrates of dipeptidyl peptidase IV (CD26) J Biol Chem. 2002;277:33170–33177. doi: 10.1074/jbc.M200798200. [DOI] [PubMed] [Google Scholar]
- 15.Zhang HZ, Kasibhatla S, Guastella J, Tseng B, Drewe J, Cai SX. N-Ac-DEVD-N′-(polyfluorobenzoyl)-R110: Novel cell-permeable fluorogenic caspase substrates for the detection of caspase activity and apoptosis. Bioconjugate Chem. 2003;14:458–463. doi: 10.1021/bc0256188. [DOI] [PubMed] [Google Scholar]
- 16.Cai SX, Zhang HZ, Guastella J, Drewe J, Yang W, Weber E. Design and synthesis of rhodamine 110 derivative and caspase-3 substrate for enzyme and cell-based fluorescent assay. Bioorg Med Chem Lett. 2001;11:39–42. doi: 10.1016/s0960-894x(00)00590-4. [DOI] [PubMed] [Google Scholar]
- 17.Wang ZQ, Liao J, Diwu Z. N-DEVD-N′-morpholinecarbonyl-rhodamine 110: Novel caspase-3 fluorogenic substrates for cell-based apoptosis assay. Bioorg Med Chem Lett. 2005;15:2335–2338. doi: 10.1016/j.bmcl.2005.02.081. [DOI] [PubMed] [Google Scholar]
- 18.Zhang H-Z, Cai SX, Drewe JA, Yang W. Novel fluorescence dyes and their applications for whole cell fluorescence screening assays for caspases, peptidases, proteases and other enzymes and the use thereof. 2000004914 WO Patent Application. 2000
- 19.Diwu Z, Liao J, Wang Z. Preparation of rhodamine peptide derivatives as luminogenic protease substrates. 2003099780 WO Patent Application. 2003
- 20.Ioffe IS, Otten VF. Rhodamine dyes and related compounds. XII. Diacetyl derivatives of rhodamine and rhodol; structure of colorless forms of fluoran dyes. Zh Obshch Khim. 1965;1:336–339. [Google Scholar]
- 21.López Arbeloa F, López Arbeloa T, Tapia Estevez MJ, López Arbeloa I. Photophysics of rhodamines: Molecular structure and solvent effects. J Phys Chem. 1991;95:2203–2208. [Google Scholar]
- 22.Ioffe IS, Otten VF. Rhodamine dyes and related compounds. XIV. Mutual conversions of colorless and colored forms of rhodamine and rhodol. Zh Obshch Khim. 1965;1:343–346. [Google Scholar]
- 23.López Arbeloa F, Urrecha Aguirresacona I, López Arbeloa I. Influence of the molecular structure and the nature of the solvent on the absorption and fluorescence characteristics of rhodamines. Chem Phys. 1989;130:371–378. [Google Scholar]
- 24.Vogel M, Rettig W, Sens R, Drexhage KH. Structural relaxation of rhodamine dyes with different N-substitution patterns—A study of fluorescence decay times and quantum yields. Chem Phys Lett. 1988;147:452–460. [Google Scholar]
- 25.López Arbeloa T, López Arbeloa F, Hernández Bartolomé P, López Arbeloa I. On the mechanism of radiationless deactivation of rhodamines. Chem Phys. 1992;160:123–130. [Google Scholar]
- 26.Hansch C, Leo A, Taft RW. A survey of Hammett substituent constants and resonance and field parameters. Chem Rev. 1991;91:165–195. [Google Scholar]
- 27.Descalzo AB, Rurack K, Weisshoff H, Martinex-Manez R, Marcos MD, Amoros P, Hoffmann K, Soto J. Rational design of a chromo- and fluorogenic hybrid chemosensor material for the detection of long-chain carboxylates. J Am Chem Soc. 2005;127:184–200. doi: 10.1021/ja045683n. [DOI] [PubMed] [Google Scholar]
- 28.Amsberry KL, Gerstenberger AE, Borchardt RT. Amine prodrugs which utilize hydroxy amide lactonization. II. A potential esterase-sensitive amide prodrug. Pharm Res. 1991;8:455–461. doi: 10.1023/a:1015890809507. [DOI] [PubMed] [Google Scholar]
- 29.Rotman B, Papermaster BW. Membrane properties of living mammalian cells as studied by enzymatic hydrolysis of fluorogenic esters. Proc Natl Acad Sci USA. 1966;55:134–141. doi: 10.1073/pnas.55.1.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldberg JM, Baldwin RL. Kinetic mechanism of a partial folding reaction. 1 Properties of the reaction and effects of denaturants. Biochemistry. 1998;37:2546–2555. doi: 10.1021/bi972402y. [DOI] [PubMed] [Google Scholar]
- 31.Fickling MM, Fischer A, Mann BR, Packer J, Vaughan J. Hammett substituent constants for electron-withdrawing substituents: Dissociation of phenols, anilinium ions and dimethylanilinium ions. J Am Chem Soc. 1959;81:4226–4230. [Google Scholar]
- 32.Laurent A, Debart F, Lamb N, Rayner B. Esterase-triggered fluorescence of fluorogenic oligonucleotides. Bioconjugate Chem. 1997;8:856–861. doi: 10.1021/bc970168i. [DOI] [PubMed] [Google Scholar]
- 33.Bergsdorf C, Beyer C, Umansky V, Werr M, Sapp M. Highly efficient transport of carboxyfluorescein diacetate succinimidyl ester into COS7 cells using human papillomavirus-like particles. FEBS Lett. 2003;536:120–124. doi: 10.1016/s0014-5793(03)00039-5. [DOI] [PubMed] [Google Scholar]
- 34.Drobni P, Mistry N, McMillan N, Evander M. Carboxy-fluorescein diacetate, succinimidyl ester labeled papillomavirus virus-like particles fluoresce after internalization and interact with heparan sulfate for binding and entry. Virology. 2003;310:163–172. doi: 10.1016/s0042-6822(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 35.Kamal A, Ramulu P, Srinivas O, Ramesh G, Kumar PP. Synthesis of C8-linked pyrrolo[2,1-c][1,4]benzodiazepine–benzimidazole conjugates with remarkable DNA-binding affinity. Bioorg Med Chem Lett. 2004;14:4791–4794. doi: 10.1016/j.bmcl.2004.06.069. [DOI] [PubMed] [Google Scholar]
- 36.Ji TH. Bifunctional reagents. Methods Enzymol. 1983;91:580–609. doi: 10.1016/s0076-6879(83)91053-4. [DOI] [PubMed] [Google Scholar]
- 37.Aslam M, Dent A. Bioconjugation: Protein Coupling Techniques for the Biomedical Sciences. Macmillan Reference; London: 1998. [Google Scholar]
- 38.Bednar RA. Reactivity and pH dependence of thiol conjugation to N-ethylmaleimide: Detection of a conformational change in chalcone isomerase. Biochemistry. 1990;29:3684–3690. doi: 10.1021/bi00467a014. [DOI] [PubMed] [Google Scholar]
- 39.Yoshitake S, Yamada Y, Ishikawa E, Masseyeff R. Conjugation of glucose-oxidase from Aspergillus niger and rabbit antibodies using N-hydroxysuccinimide ester of N-(4-carboxycyclohexylmethyl)-maleimide. Eur J Biochem. 1979;101:395–399. doi: 10.1111/j.1432-1033.1979.tb19731.x. [DOI] [PubMed] [Google Scholar]
- 40.Ishii Y, Lehrer SS. Effects of the state of succinimido-ring on the fluorescence and structural properties of pyrene maleimide-labeled alpha-alpha-tropomyosin. Biophys J. 1986;50:75–80. doi: 10.1016/S0006-3495(86)83440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiao GS, Han JW, Burgess K. Syntheses of regioisomerically pure 5- and 6-halogenated fluoresceins. J Org Chem. 2003;68:8264–8267. doi: 10.1021/jo034724f. [DOI] [PubMed] [Google Scholar]
- 42.Corrie JET, Craik JS, Munasinghe VRN. A homobifunctional rhodamine for labeling proteins with defined orientations of a fluorophore. Bioconjugate Chem. 1998;9:160–167. doi: 10.1021/bc970174e. [DOI] [PubMed] [Google Scholar]
- 43.Meunier F, Wilkinson KJ. Nonperturbing fluorescent labeling of polysaccharides. Biomacromolecules. 2002;3:857–864. doi: 10.1021/bm0255241. [DOI] [PubMed] [Google Scholar]
- 44.Curtius T. Ueber Stickstoffwasserstoffsäure (Azoimid) N3H. Ber Dtsch Chem Ges. 1890;23:3023–3033. [Google Scholar]
- 45.Curtius T. Hydrazide und Azide organischer Säuren. J Prakt Chem. 1894;50:275–294. [Google Scholar]
- 46.Scriven EFV, Turnbull K. Azides: Their preparation and synthetic uses. Chem Rev. 1988;88:297–368. [Google Scholar]
- 47.Bräse S, Gil C, Knepper K, Zimmermann V. Organic azides: An exploding diversity of a unique class of compounds. Angew Chem Int Ed Engl. 2005;44:5188–5240. doi: 10.1002/anie.200400657. [DOI] [PubMed] [Google Scholar]
- 48.Raines RT. Ribonuclease A. Chem Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 49.Haigis MC, Raines RT. Secretory ribonucleases are internalized by a dynamin-independent endocytic pathway. J Cell Sci. 2003;116:313–324. doi: 10.1242/jcs.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haigis MC, Kurten EL, Raines RT. Ribonuclease inhibitor as an intracellular sentry. Nucleic Acids Res. 2003;31:1024–1032. doi: 10.1093/nar/gkg163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Watson P, Jones AT, Stephens DJ. Intracellular trafficking pathways and drug delivery: Fluorescence imaging of living and fixed cells. Adv Drug Deliv Rev. 2005;57:1024–1032. doi: 10.1016/j.addr.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Leinweber FJ. Possible physiological roles of carboxylic ester hydrolases. Drug Metab Rev. 1987;18:379–439. doi: 10.3109/03602538708994129. [DOI] [PubMed] [Google Scholar]
- 53.Runquist EA, Havel RJ. Acid hydrolases in early and late endosome fractions from rat liver. J Biol Chem. 1991;266:22557–22563. [PubMed] [Google Scholar]
- 54.Hornick CA, Thouron C, Delamatre JG, Huang J. Triacylglycerol hydrolysis in isolated hepatic endosomes. J Biol Chem. 1992;267:3396–3401. [PubMed] [Google Scholar]
- 55.Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, Scott MS, Gramolini AO, Morris Q, Hallett MT, Rossant J, Hughes TR, Frey B, Emili A. Global survey of organ and organelle protein expression in mouse: Combined proteomic and transcriptomic profiling. Cell. 2006;125:173–186. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 56.Foster LJ, de Hoog CL, Zhang Y, Zhang Y, Xie X, Mootha VK, Mann M. A mammalian organelle map by protein correlation profiling. Cell. 2006;125:187–199. doi: 10.1016/j.cell.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 57.Horgan DJ, Dunstone JR, Stoops JK, Webb EC, Zerner B. Carboxylesterases (EC 3.1.1). The molecular weight and equivalent weight of pig liver carboxylesterase. Biochemistry. 1969;8:2006–2013. doi: 10.1021/bi00833a034. [DOI] [PubMed] [Google Scholar]
- 58.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Kluwer Academic/Plenum; New York: 1999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.