

Abstract
Background
ASCOT-BPLA study demonstrates that in hypertensive subjects, atenolol+bendroflumethiazide therapy is associated with higher incidence of adverse cardiovascular outcomes and developing diabetes than an amlodipine+perindopril regimen. This is not explained by changes in blood pressure alone. We hypothesized that distinct vascular and metabolic effects of anti-hypertensive drugs may explain these differential effects.
Methods
Either placebo or one class of anti-hypertensive drug (atenolol 100 mg, amlodipine 10 mg, hydrochlorothiazide 50 mg, ramipril 10 mg, or candesartan 16 mg) was given daily during 8 weeks to 31 patients in each of 6 arms of a randomized, single-blind, placebo-controlled, parallel study.
Results
Atenolol, amlodipine, and candesartan therapies significantly reduced systolic blood pressure when compared with ramipril (P<0.05 by ANOVA). Atenolol and thiazide therapies increased triglycerides levels greater than ramipril or candesartan (P=0.005 by ANOVA). Amlodipine significantly increased HDL cholesterol levels greater than atenolol (P=0.011 by ANOVA). Ramipril and candesartan therapies improved FMD and increased adiponectin levels and insulin sensitivity to a greater extent than atenolol or thiazide therapies (P<0.001 and P<0.015 by ANOVA). Amlodipine therapy increased adiponectin levels greater than atenolol therapy (P<0.05 by ANOVA). Ramipril, candesartan, and amlodipine therapies significantly decreased leptin levels to a greater extent when compared with atenolol or thiazide therapies (P<0.001 by ANOVA). Amlodipine therapies significantly decreased resistin levels greater than ramipril or candesartan therapies (P=0.001 by ANOVA).
Conclusions
We observed differential effects of anti-hypertensive drugs on endothelial dysfunction and plasma adipocytokines.
Keywords: Anti-hypertensive drugs, Endothelial function, Adipocytokines, Hypertension, Insulin resistance
1. Introduction
Hypertension and coronary heart disease are cardiovascular diseases characterized by endothelial dysfunction that frequently cluster with disorders of metabolic homeostasis including obesity and type 2 diabetes that are characterized by insulin resistance [1-3]. These co-morbidities may be explained, in part, by reciprocal relationships between endothelial dysfunction and insulin resistance [1,2]. The Nurses’ Health Study I and II and the Health Professional Follow-up Study demonstrate beta-blockers and diuretics are independently associated with a higher risk of type 2 diabetes. By contrast, angiotensin converting enzyme (ACE) inhibitors, angiotensin II type 1 (AT1) receptor blockers (ARBs), and calcium channel blockers are not [4-6]. Mechanisms may relate to the common ability of ACE inhibitors, ARBs, and calcium channel blockers to target the vicious synergy between endothelial dysfunction and insulin resistance.
Endothelial dysfunction associated with diabetes, obesity, metabolic syndrome, and other insulin resistant states is characterized by impaired insulin-stimulated nitric oxide (NO) release from endothelium with decreased blood flow and reduced delivery of substrates and hormones to insulin target tissues [7]. Thus, improvement in endothelial function is predicted to improve insulin sensitivity. This may be why ACE inhibitors, ARBs, and calcium channel blockers are not associated with a higher risk of type 2 diabetes. NO plays a pivotal role in maintaining vascular health and protecting against vascular injury under pathological conditions. Angiotensin II activates the AT1 receptor resulting in superoxide anion generation, oxidative stress, and endothelial dysfunction [8,9]. ACE inhibitors and ARBs diminish production of intracellular superoxide anions by reducing activity of angiotensin II-dependent oxidases in the endothelium and vascular smooth muscle. This protects endothelium-derived NO from oxidant degradation to inert or toxic molecules [10]. Calcium channel blockers also activate both NO synthase in vitro and enhance both NO and adenosine production in vivo [11].
Adiponectin and leptin are adipocytokines secreted specifically by adipose cells [12,13]. In humans, plasma levels of adiponectin are negatively correlated with adiposity and insulin resistance. Indeed, decreased plasma adiponectin levels are observed in patients with diabetes [14]. We recently reported that ramipril, candesartan, or efonidipine increase adiponectin levels and insulin sensitivity in patients without changing body mass index [15-17]. Thus, decreased levels of adiponectin may influence the development of insulin resistance rather than simply serving as a biomarker for insulin sensitivity. Leptin may play an important role in atherosclerotic lesion formation and progression [13,18]. Resistin exerts direct effects to promote endothelial cell activation and upregulates adhesion molecules and chemokines [19]. Plasma resistin levels are correlated with markers of inflammation and are predictive of coronary atherosclerosis in humans in some studies [20]. Thus, adiponectin, leptin, and resistin may represent important links between metabolic signals, inflammation, and atherosclerosis.
In a recent clinical trial in hypertensive patients [e.g. Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA)], fewer individuals on the amlodipine plus perindopril regimen had a primary endpoint of non-fatal myocardial infarction or fatal coronary heart disease, total cardiovascular events and procedures, and all-cause mortality when compared with atenolol plus bendroflumethiazide regimen [21]. The incidence of developing diabetes was also less on the amlodipine-based regimen. ASCOT-BPLA investigators suggested that these effects may not be entirely explained by better control of blood pressure [22]. Therefore, we investigated effects of several different classes of anti-hypertensive drugs on endothelial function, adipocytokine profiles, and other metabolic parameters in patients with hypertension.
2. Methods
2.1. Study population and design
We used a randomized, single-blind, placebo-controlled, parallel study design. We recruited patients from a primary care setting in the Vascular Medicine and Atherosclerosis Unit, Cardiology, Gil Medical Center, Gachon University. We recruited patients from April 23, 2004 to July 7, 2005. We used WHO/ISH definitions for hypertension (systolic and diastolic blood pressure ≥140 or ≥90 mm Hg, respectively). We included patients with mild to moderate hypertension (systolic blood pressure <180 and diastolic blood pressure <110 mm Hg) ranging in age from 30 to 70 years. We excluded patients with severe hypertension, type 2 diabetes, unstable angina, acute myocardial infarction, or renal insufficiency. Blood pressure measured in the right arm in the sitting position using a standard sphygmomanometer with appropriate sized cuff was recorded as the mean of 2 successive readings (subjects were seated for at least 10 min prior to measurements) [15-17,23]. Before and during the study period a dietitian educated patients to maintain a low salt diet. A research nurse counted pills at the end of treatment to monitor compliance. In order to minimize acute side effects, study medications were titrated from 50 to 100 mg of atenolol, from 5 to 10 mg of amlodipine, 25 to 50 mg of hydrochlorothiazide, 5 to 10 mg of ramipril, and 8 to 16 mg of candesartan upwards over a 2 week period if no hypotension (systolic blood pressure <100 mm Hg) was noted (run in period). This was followed by a 3-week washout period. At the end of the washout period, participants were randomly assigned to either placebo or one of several anti-hypertensive drugs (atenolol 100 mg, amlodipine 10 mg, hydrochlorothiazide 50 mg, ramipril 10 mg, or candesartan 16 mg) daily during 8 weeks. Allocation concealement was achieved by using envelopes with the collaboration of a statistician. The patients were seen at 14-day intervals or more frequently during the study. One hundred eighty-six mild to moderate hypertensive patients were considered eligible for this study. One patient on amlodipine and one patient on ramipril experienced severe facial flushing and dry coughing, respectively and they were withdrawn from the study. Thus, data from total 184 patients were analyzed (Fig. 1). The number of current smokers in each group were 9 (placebo), 9 (atenolol), 8 (amlodipine), 10 (thiazide), 9 (ramipril), and 9 (candesartan), respectively. No additional medications including anti-hypertensive drugs, aspirin, or nonsteroidal anti-inflammatory drugs were allowed during the study period to avoid confounding effects of other drugs. This study was approved by the Gil Hospital Institute Review Board and all participants gave written, informed consent.
Fig. 1.
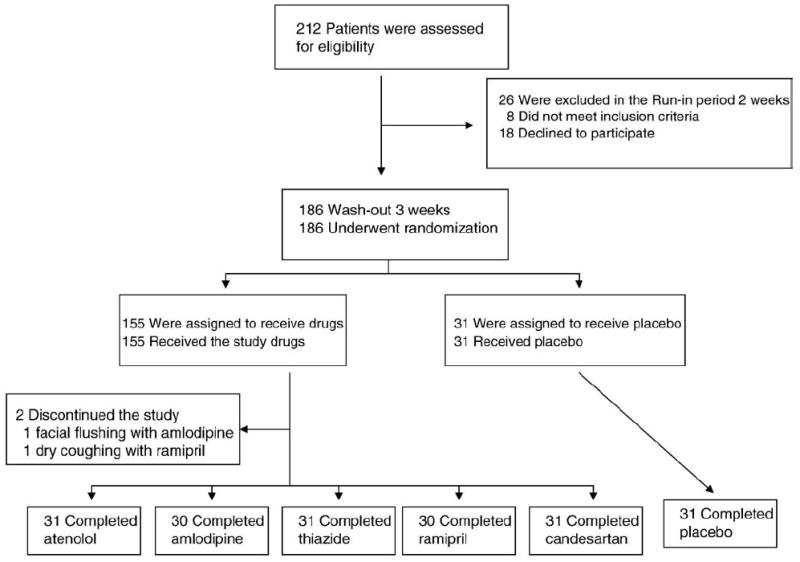
Flow chart.
2.2. Laboratory assays and vascular studies
Blood samples for laboratory assays were obtained at approximately 8:00 a.m. following overnight fasting before and at the end of each 8 week treatment period. These samples were immediately coded so that investigators performing laboratory assays were blinded to subject identity or study sequence. Assays for plasma insulin, malondialdehyde, adiponectin, leptin, and resistin were performed in duplicate by immunoradiometric assay (INSULIN-RIABEAD® II, SRL, Inc, Tokyo, Japan) or by ELISA (BIOXYTECH® LPO-586, OxisResearch, Portland, Oregon; R & D Systems, Inc., Minneapolis, Minnesota, USA). Assays for high sensitivity C-reactive protein levels were performed by latex agglutination (CRP-Latex(II)®, Denka-Seiken, Tokyo, Japan) as previously described [15-17,23,24]. The interassay and intraassay coefficients of variation were <6%. Quantitative Insulin-Sensitivity Check Index (QUICKI), a surrogate index of insulin sensitivity, was calculated as follows (insulin is expressed in μU/ml and glucose in mg/dl): QUICKI=1/[log (insulin)+log(glucose)] [25]. Imaging studies of the right brachial artery were performed using an ATL HDI 3000 ultrasound machine (Bothell, WA, USA) equipped with a 10 MHz linear-array transducer, based on a previously published technique [15-17,23,24].
2.3. Statistical analysis
Data are expressed as mean±SEM or median (range:25%–75%). After testing data for normality, we used Student’s paired t or Wilcoxon Signed Rank test to compare values between baseline and treatment at 2 months, as reported in Table 1. We used one way analysis of variance (ANOVA) or Kruskal–Wallis ANOVA on Ranks to compare baseline or treatment effects among treatment groups. Post-hoc comparisons between different treatment pairs were made using the Student–Newman–Keuls multiple comparison procedures. Pearson or Spearman correlation coefficient analysis was used to assess associations between measured parameters. We calculated that 30 subjects would provide 80% power for detecting an absolute increase of 1.5% or greater in flowmediated dilation of the brachial artery between baseline and candesartan, with α = 0.05 based on our previous studies [24]. The comparison of endothelium-dependent dilation was prospectively designated as the primary end-point of the study. All other comparisons were considered secondary. A value of P<0.05 was considered to represent statistical significance. We used SigmaStat version 2.0 (SPSS Inc.) for statistical data analyses.
Table 1.
Effects of placebo or anti-hypertensive drugs in patients with hypertension
| Variables | Placebo (n = 31) |
Atenolol (n =31) |
Amlodipine (n =30) |
Thiazide (n =31) |
Ramipril (n =30) |
Candesartan (n =31) |
ANOVA | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | Treatment | Baseline | Treatment | Baseline | Treatment | Baseline | Treatment | Baseline | Treatment | Baseline | Treatment | ||
| Age (years) | 46±2 | 49±2 | 51±2 | 48±2 | 46±1 | 47±2 | 0.244 | ||||||
| Male:Female | 21:10 | 21:10 | 19:11 | 20:11 | 20:10 | 22:9 | |||||||
| Body mass index | 25.03±0.56 | 24.96±0.52 | 24.90±0.41 | 24.69±0.39 | 25.11±0.36 | 24.91±0.34 | 25.30±0.44 | 25.11±0.40 | 25.14±0.45 | 25.04±0.43 | 25.21±0.43 | 25.12±0.40 | 0.795 |
| Heart rate (bpm) | 79±2 | 76±2 | 81±2 | 61±1 a | 81±2 | 83±2 | 82±2 | 81±2 | 82±2 | 79±2 | 81±2 | 81±2 | <0.001 |
| Systolic blood pressure (mm Hg) | 152±3 | 145±3 a | 156±1 | 135±2 a, b | 155±1 | 132±2 a, b | 153±1 | 137±2 a, b | 155±1 | 143±2 a, b, c, e, f | 156±1 | 136±2 a, b | <0.001 |
| Diastolic blood pressure (mm Hg) | 93±2 | 89±2 a | 96±1 | 84±1 a, b | 96±1 | 84±1 a, b | 94±1 | 86±2 a | 94±1 | 87±1 a | 94±1 | 84±1 a, b | <0.001 |
| Lipids (mg/dl) | |||||||||||||
| Total cholesterol (mg/dl) | 189±6 | 184±6 | 193±5 | 188±5 | 191±5 | 190±5 | 191±5 | 200±6 | 187±5 | 186±4 | 192±4 | 184±5 | 0.173 |
| Triglycerides (mg/dl) | 166±18 | 172±16 | 169±15 | 189±15 | 174±13 | 153±13 d | 168±18 | 191±20 | 161±17 | 142±16 d | 172±21 | 152±16 d | 0.005 |
| HDL cholesterol (mg/dl) | 47±2 | 44±2 | 47±2 | 44±2 | 47±2 | 52±2 a, b, c | 46±2 | 46±2 | 47±2 | 49±2 | 48±2 | 50±2 | 0.011 |
| LDL cholesterol (mg/dl) | 110±5 | 106±5 | 113±5 | 108±5 | 109±5 | 107±5 | 112±4 | 116±6 | 108±5 | 109±4 | 110±5 | 104±5 | 0.867 |
| Vasomotor | |||||||||||||
| Flow-mediated dilation (%) | 3.82±0.26 | 4.44±0.26 a | 3.99±0.34 | 4.79±0.35 a | 3.57±0.27 | 4.79±0.30 a | 3.63±0.22 | 4.34±0.25 a | 3.70±0.28 | 5.36±0.34 a, b, c, d | 3.48±0.24 | 5.10±0.32 a, b, c, d | <0.001 |
| Nitroglycerin dilation (%) | 14.49±0.79 | 14.36±0.80 | 13.75±0.69 | 13.43±0.76 | 13.43±0.62 | 12.86±0.65 | 14.08±0.62 | 14.55±0.67 | 15.50±0.85 | 15.48±0.82 | 15.01±0.78 | 15.55±0.79 | 0.218 |
| Malondialdehyde (μM) | 1.23±0.06 | 1.21±0.05 | 1.15±0.05 | 1.16±0.07 | 1.21±0.06 | 1.14±0.07 | 1.20±0.08 | 1.20±0.08 | 1.18±0.06 | 1.10±0.07 a | 1.10±0.07 | 1.00±0.07 a | 0.279 |
| C-reactive protein (mg/l) | 0.85 (0.57–2.20) | 1.28 a (0.57–2.66) | 0.90 (0.70–1.78) | 1.00 (0.63–2.05) | 0.80 (0.53–1.40) | 0.70 (0.50–2.00) | 0.90 (0.50–1.90) | 1.00 (0.60–1.75) | 1.29 (0.64–1.93) | 0.86 (0.64–2.30) | 1.07 (0.54–1.92) | 0.90 (0.40–1.43) | 0.151 |
| Insulin resistance | |||||||||||||
| Adiponectin (μg/ml) | 4.0±0.4 | 3.9±0.4 | 4.5±0.5 | 3.6±0.4 a | 4.5±0.5 | 4.7±0.5 c | 3.8±0.5 | 3.2±0.3 | 3.7±0.4 | 4.2±0.4 a, c, d | 3.9±0.4 | 4.5±0.5 a, c, d | <0.001 |
| Insulin (μU/ml) | 7.98±0.80 | 8.33±0.86 | 8.19±0.69 | 8.67±0.65 | 9.09±0.72 | 7.65±0.65 | 8.84±1.20 | 8.97±0.72 | 8.42±1.04 | 7.55±1.05 | 9.70±0.88 | 8.42±1.10 | 0.369 |
| Glucose (mg/dl) | 97±2 | 96±2 | 103±3 | 106±3 | 106±3 | 105±3 | 104±3 | 109±4 | 101±2 | 103±3 | 104±2 | 101±3 | 0.243 |
| QUICKI | 0.358±0.006 | 0.357±0.006 | 0.352±0.005 | 0.345±0.004 | 0.345±0.005 | 0.353±0.005 | 0.353±0.007 | 0.344±0.005 | 0.382±0.016 | 0.396±0.018 | 0.348±0.008 | 0.362±0.008 c | 0.015 |
| Leptin (ng/ml) | 6.2±1.1 | 6.2±1.0 | 5.5±0.8 | 6.2±1.0 | 5.9±0.8 | 5.0±0.7 a, b, c, d | 6.3±0.9 | 6.3±0.9 | 5.9±0.8 | 5.3±0.9 a, b, c | 6.8±1.1 | 5.9±1.0 a, b, c | <0.001 |
| Resistin (ng/ml) | 9.5±0.8 | 9.6±0.8 | 10.5±0.8 | 10.1±0.7 | 9.6±0.8 | 8.7±0.6 a | 9.1±0.8 | 9.2±1.0 | 8.5±0.7 | 9.4±0.8 a, e | 8.7±0.7 | 9.7±0.8 a, c, e | 0.001 |
*P<0.05, +P<0.01, ‡P<0.001 for comparison with each baseline value.
Data are expressed as means±SEM or median (range 25% to 75%).
There were no significant differences among each baseline values.
Student’s paired t test or Wilcoxon Signed Rank test for comparison with baseline vs. treatment value.
Student’s unpaired t test or Mann–Whitney Rank Sum test for comparison with each baseline value or each treatment value.
Quantitative Insulin-Sensitivity Check Index (QUICKI)=1/[log (insulin)+log (glucose)]25.
P<0.05 for comparison with the baseline value.
P<0.05 for comparison with the value after therapy with placebo.
P<0.05 for comparison with the value after therapy with atenolol.
P<0.05 for comparison with the value after therapy with thiazide.
P<0.05 for comparison with the value after therapy with amlodipine.
P<0.05 for comparison with the value after therapy with candesartan.
3. Results
The baseline patient characteristics are reported in Table 1. The distribution of the variables was not statistically different among study groups after randomization.
3.1. Blood pressure and lipids
All therapies significantly reduced systolic and diastolic blood pressure from baseline. Atenolol, amlodipine, and candesartan therapies significantly reduced systolic and diastolic blood pressure when compared with placebo (both P<0.001 by ANOVA; Fig. 1). Atenolol, amlodipine, and candesartan therapies significantly reduced systolic blood pressure when compared with ramipril (P<0.05 by ANOVA; Fig. 2). Atenolol and thiazide therapies significantly increased triglycerides levels from baseline and the magnitude of increasing triglycerides levels following atenolol and thiazide therapies was significantly greater than that following ramipril or candesartan (P=0.005 by ANOVA; Fig. 3). Amlodipine therapy significantly increased high-density lipoprotein (HDL) cholesterol levels from baseline and the magnitude of increasing HDL cholesterol levels was significantly greater than that following atenolol or placebo (P=0.011 by ANOVA; Fig. 3). No significant changes in other lipid profiles were noted with any of the therapies.
Fig. 2.
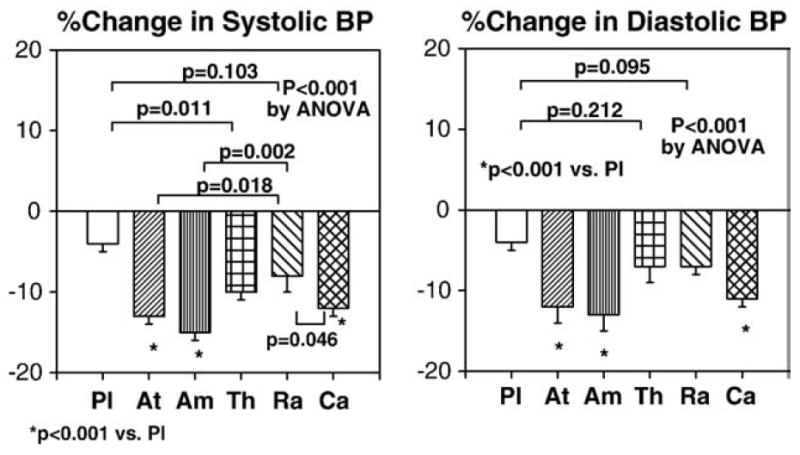
Atenolol, amlodipine, and candesartan therapies significantly reduced systolic and diastolic blood pressure compared with placebo. Atenolol, amlodipine, and candesartan therapies significantly reduced systolic blood pressure compared with ramipril. Pl=placebo, At=atenolol, Am=amlodipine, Th=thiazide, Ra=ramipril, Ca=candesartan. Standard error of the mean is identified by the bars.
Fig. 3.
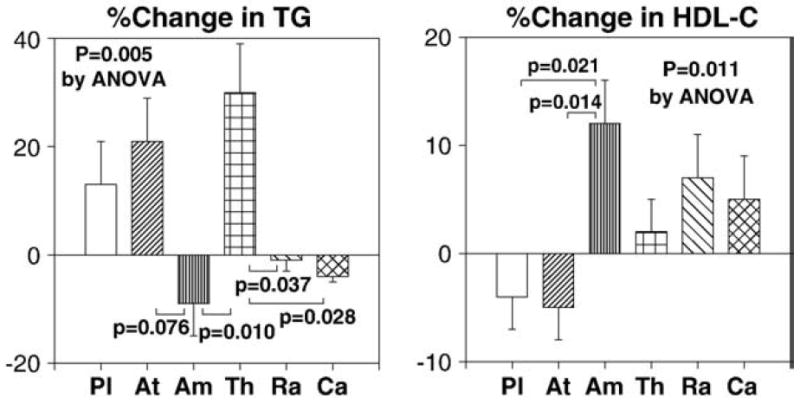
The magnitude of increasing triglyceride levels following atenolol and thiazide therapies was significantly greater than that following ramipril or candesartan. The magnitude of increasing HDL cholesterol levels following amlodipine therapy was significantly greater than that following atenolol or placebo. Pl=placebo, At=atenolol, Am=amlodipine, Th=thiazide, Ra=ramipril, Ca=candesartan. Standard error of the mean is identified by the bars.
3.2. Vasomotor function and malondialdehyde
All therapies significantly improved flow-mediated dilator response to hyperemia (FMD) from baseline. Ramipril and candesartan therapies significantly improved FMD greater than placebo, atenolol or thiazide therapies (P<0.001 by ANOVA; Fig. 4). By contrast, atenolol and thiazide therapies did not significantly improve FMD greater than placebo. Ramipril and candesartan therapies significantly reduced plasma malondialdehyde levels from baseline. However, the magnitude of decreasing malondialdehyde levels following ramipril and candesartan therapies was not significantly different when compared with other therapies (P=0.279 by ANOVA; Fig. 4). Brachial artery dilator responses to nitroglycerin were not significantly different between any of the therapies. In addition, no significant changes in high sensitivity C-reactive protein were noted with any of the therapies (P=0.151 by ANOVA).
Fig. 4.
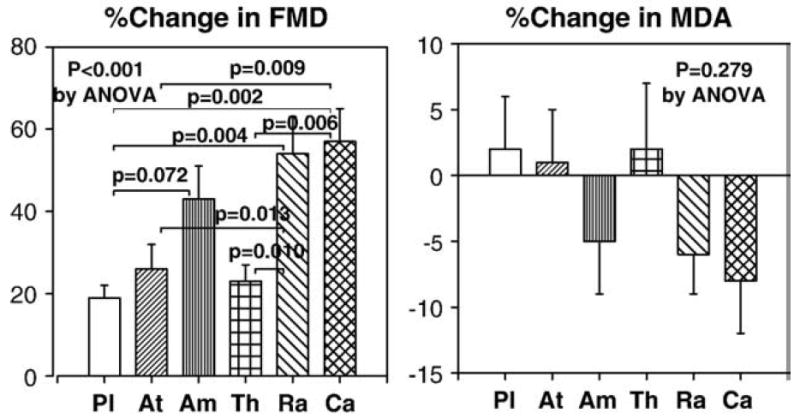
Ramipril and candesartan therapies significantly improved flow-mediated dilator response to hyperemia (FMD) greater than placebo, atenolol or thiazide therapies. By contrast, atenolol and thiazide therapies did not significantly improve FMD greater than placebo. Ramipril and candesartan therapies significantly reduced plasma malondialdehyde levels from baseline. However, the magnitude of decreasing malondialdehyde levels was not significantly greater compared with other therapies. Pl=placebo, At=atenolol, Am=amlodipine, Th=thiazide, Ra=ramipril, Ca=candesartan. Standard error of the mean is identified by the bars.
3.3. Adipocytokines and insulin resistance
None of the therapies changed insulin or glucose levels from baseline. Ramipril and candesartan therapies significantly increased adiponectin levels from baseline. Atenolol therapy significantly decreased adiponectin levels from baseline. Ramipril and candesartan therapies significantly increased adiponectin levels to a greater extent than atenolol or thiazide therapies (P<0.001 by ANOVA; Fig. 5). Amlodipine therapy significantly increased adiponectin levels to a greater extent than atenolol therapy (P<0.05; Fig. 5). None of the therapies significantly increased insulin sensitivity (as assessed by QUICKI) from baseline. However, ramipril and candesartan therapies significantly increased insulin sensitivity to a greater extent than atenolol or thiazide therapies (P= 0.015 by ANOVA; Fig. 5). We observed significant inverse correlations between baseline body mass index and baseline adiponectin levels (r=−0.344 before placebo; r=−0.393 before atenolol; r=−0.429 before amlodipine; r=−0.468 before thiazide; r=−0.401 before ramipril; r=−0.505 before candesartan).
Fig. 5.
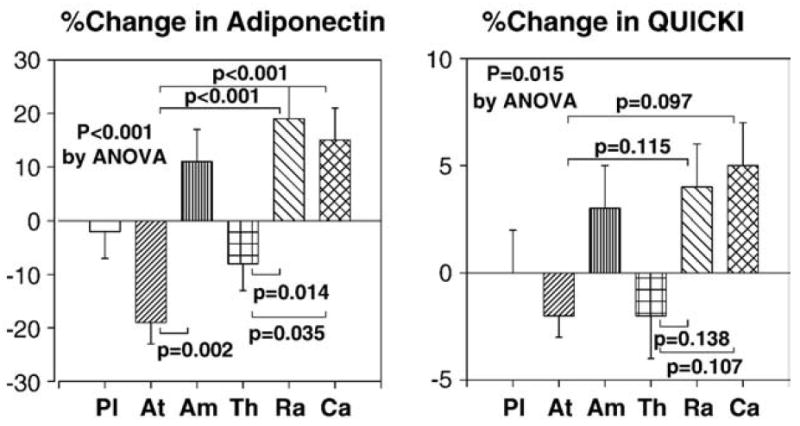
Ramipril and candesartan therapies significantly increased adiponectin levels to greater extent than atenolol or thiazide therapies. Amlodipine therapy significantly increased adiponectin levels to a greater extent than atenolol therapy. Ramipril and candesartan therapies significantly increased insulin sensitivity (as assessed by QUICKI) to a greater extent than atenolol or thiazide therapies. Pl=placebo, At=atenolol, Am=amlodipine, Th=thiazide, Ra=ramipril, Ca=candesartan. Standard error of the mean is identified by the bars.
Ramipril, candesartan, and amlodipine therapies significantly decreased leptin levels from baseline. Atenolol therapy increased leptin levels from baseline and thiazide therapy did not change leptin levels. Ramipril, candesartan, and amlodipine therapies significantly decreased leptin levels to a greater extent when compared with atenolol or thiazide therapies (P<0.001 by ANOVA; Fig. 6). Ramipril and candesartan therapies significantly increased resistin levels from baseline. Amlodipine therapies significantly decreased resistin levels to a greater extent than ramipril or candesartan therapies (P=0.001 by ANOVA; Fig. 6). We observed significant correlations between baseline body mass index and baseline leptin levels (r=0.358 before placebo; r=0.338 before atenolol; r=0.428 before ramipril; r=0.439 before candesartan). However, we did not observe significant correlations between baseline resistin levels and baseline body mass index or insulin sensitivity.
Fig. 6.
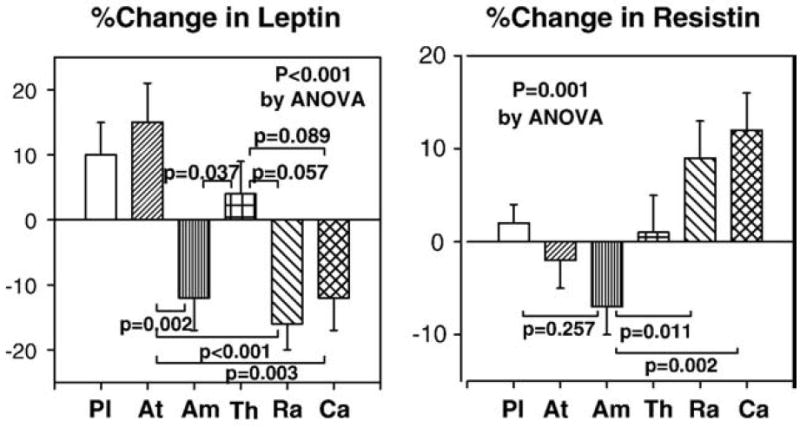
Ramipril, candesartan, and amlodipine therapies significantly decreased leptin levels to a greater extent when compared with atenolol or thiazide therapies. Amlodipine therapies significantly decreased resistin levels to a greater extent than ramipril or candesartan therapies. Pl=placebo, At=atenolol, Am=amlodipine, Th=thiazide, Ra=ramipril, Ca=candesartan. Standard error of the mean is identified by the bars.
We observed significant correlations between percent changes in adiponectin levels and percent changes in insulin or QUICKI following therapy (r=−0.426 and 0.342 after ramipril; r=−0.547 and 0.602 after candesartan). We also observed significant correlations between percent changes in adiponectin levels and percent changes in HDL cholesterol or QUICKI following amlodipine (r=0.450 and 0.380). We investigated whether changes in percent flow-mediated dilator response to hyperemia, plasma levels of malondialdehyde, adiponectin, leptin, and resistin, or insulin resistance were related to reduction of systolic or diastolic blood pressure. There were no significant correlations between changes in these parameters and reduction of systolic blood pressure (−0.226≤r≤0.075) or reduction of diastolic blood pressure (−0.206≤r≤0.145).
4. Discussion
As expected, we observed that all anti-hypertensive drugs improved blood pressure. However, ramipril and candesartan improved blood pressure, endothelial dysfunction, and plasma adiponectin and leptin levels to a greater extent than atenolol or thiazide therapy. Despite a reduction of blood pressure, atenolol and thiazide did not improve endothelial dysfunction and reduced plasma adiponectin and leptin levels.
The ASCOT-BPLA study demonstrated that when compared with the atenolol plus bendroflumethiazide regimen, fewer individuals on the amlodipine plus perindopril regimen had primary endpoint composites in hypertensive patients. The incidence of developing diabetes was less on the amlodipine-based regimen. The ASCOT-BPLA investigators suggested that these effects might not be entirely explained by better control of blood pressure [22]. In further analyses, investigators assessed to what extent these differences were due to differences in blood pressure and other variables noted after randomization. No temporal link between differences in blood pressure and different event rates were observed. Multivariate adjustment accounted for about half of the differences in coronary events and for about 40% of the differences in stroke events between the treatment regimens tested in ASCOT-BPLA (but residual differences were no longer significant). It remains possible that differential effects of the two treatment regimens on other variables also contributed, particularly for stroke [22].
One important mechanism underlying endothelial dysfunction in cardiovascular and metabolic diseases is increased oxidative stress which decreases endothelial NO production and bioavailability leading to impaired endothelium-dependent vasodilation [26]. ACE inhibitors, ARBs, and calcium channel blockers reduce oxidative stress in the vasculature and improve endothelium-dependent vasodilation [15,24,27]. However, the effects of amlodipine on endothelium-dependent vasodilation are controversial. Some studies demonstrate that amlodipine improves endothelium-dependent vasodilation [28,29] while others do not [30,31]. In the current study, ramipril and candesartan therapies significantly improved FMD to a greater extent than placebo. Amlodipine therapy tended to improve FMD greater than placebo. By contrast, atenolol and thiazide therapies did not significantly improve FMD to a greater extent than placebo. We reasoned these differences would be caused by different effects of anti-hypertensive drugs on endothelium-dependent vasodilation, biomarkers of oxidant stress or inflammation. In our study, ramipril and candesartan therapies significantly reduced plasma malondialdehyde levels from baseline. However, the magnitude of decreasing malondialdehyde levels was not significantly greater compared with other therapies. Also, no significant changes in high sensitivity C-reactive protein were noted with any of the therapies.
Due to reciprocal relationships between endothelial dysfunction and insulin resistance [1,2], we hypothesized that improvements in endothelial dysfunction would be accompanied by simultaneous improvement in metabolic parameters. Hypertension is an insulin-resistant condition associated with an increased incidence of diabetes. Abnormal glucose metabolism is diagnosed in two-thirds of patients with essential hypertension [3]. In our study, atenolol and thiazide therapies significantly increased triglyceride levels from baseline and the magnitude of increasing triglyceride levels following atenolol and thiazide therapies was significantly greater than that following ramipril or candesartan therapies. Amlodipine therapy significantly increased HDL cholesterol levels from baseline and the magnitude of increasing HDL cholesterol levels was significantly greater than that following atenolol or placebo therapies. Verapamil increases ATP-binding cassette transporter A1 (a major source of plasma HDL) expression and thereby increases apoA-I-mediated cellular lipid release and production of HDL, another potential anti-oxidant mechanism for calcium channel blockers [32].
Adiponectin is an adipose-derived factor that augments and mimics metabolic and vascular actions of insulin [12]. In our study, ramipril and candesartan therapies significantly increased adiponectin levels without a corresponding change in body weight or body mass index when compared with atenolol or thiazide therapies. Increasing adiponectin levels would be predicted to improve both insulin sensitivity and endothelial function by multiple mechanisms [12,33]. Regulation of metabolic homeostasis and hemodynamic homeostasis may be coupled by vascular actions of insulin to stimulate production of NO [34]. In the current study, there were significant correlations between percent changes in adiponectin levels and percent changes in insulin or QUICKI following ramipril or candesartan. There may be additional mechanisms to improve insulin sensitivity and adiponectin levels in addition to improving endothelial function by blockade of the renin angiotensin system. For example, cross-talk between angiotensin II receptor signaling and insulin signaling pathways may contribute to insulin resistance [35]. In addition, ramipril and candesartan may have direct effects to augment insulin-stimulated glucose uptake, promote adipogenesis [36], and induce peroxisome proliferator-activated receptor-γ activity that promotes differentiation of adipocytes [37]. Recently, it was reported that quinapril increases insulin-stimulated endothelial function and vascular expression of adiponectin in patients with type 2 diabetes [38]. In our study, amlodipine significantly increased adiponectin levels when compared with atenolol. There were significant correlations between percent changes in adiponectin levels and percent changes in HDL-cholesterol and QUICKI following amlodipine therapy. Most clinical studies have demonstrated that long-acting, dihydropiridine calcium channel blockers improve insulin-resistance in hypertensive patients and rats without decreasing accumulation of abdominal fat or serum levels of glucose or insulin [39-41]. However, in some studies amlodipine does not improve insulin sensitivity in obese hypertensive patients [42]. We assessed the surrogate index of insulin sensitivity, QUICKI. It is important to note that the primary outcomes of our study did not include insulin sensitivity. Thus, we used a surrogate index to measure insulin sensitivity rather than the reference standard glucose clamp. Using QUICKI, we observed a trend towards small improvement of insulin sensitivity after ramipril, candesartan, and amlodipine therapies that did not reach statistical significance. Our current observations are similar to other studies [43].
Angiotensin II increases leptin secretion from cultured human fat cells. Candesartan abolishes the effect of angiotensin II to promote leptin production [44]. Leptin may potentiate pressor effects of hyperinsulinemia in insulin resistant states. Therefore, interactions between angiotensin II and insulin with leptin may have deleterious cardiovascular effects in obesity. Additionally, leptin appears to stimulate vascular inflammation, oxidative stress, and vascular smooth muscle hypertrophy. These actions may contribute to the pathogenesis of hypertension, atherosclerosis, and left ventricular hypertrophy [13,18,45]. In the current study, ramipril, candesartan, and amlodipine therapies significantly reduced plasma leptin levels when compared with atenolol therapy.
Serum resistin is increased in type 2 diabetic or obese subjects [46,47]. Resistin reduces glucose uptake in differentiated preadipocytes [46] and promotes endothelial cell activation and upregulates adhesion molecules and chemokines [19]. In this fashion, resistin may be mechanistically linked to cardiovascular diseases in the metabolic syndrome. Serum resistin levels are associated with the presence and severity of coronary artery disease and significant correlations between resistin levels and both fasting insulin levels and insulin resistance [48]. In the current study, ramipril and candesartan significantly increased plasma resistin levels relative to baseline measurements, however, amlodipine significantly decreased plasma resistin levels. But, resistin levels were not correlated with insulin sensitivity or body mass index. This is consistent with other reports that circulating resistin levels are not correlated with the insulin sensitivity, body mass index, or blood pressure [46,48-50].
It is possible that some of our results may be explained by differences in the magnitude of change in blood pressure. For example, using a higher dose of ramipril to achieve comparable blood pressure reduction when compared with amlodipine may slightly alter some of our study outcomes. However, other studies have demonstrated that endothelial and metabolic benefits of anti-hypertensive therapy are due to factors beyond blood pressure-lowering effects [16,17,22,24,29-31]. In the current study, we used the maximum allowable dose of each drug in patients to investigate the clinically practical effects of each drug. We used a single blind design, which may introduce a potential bias. Since withdrawn patients are not included in the analyses, our results may underestimate observed differences. However, this seems unlikely because only 2 patients were withdrawn in our study.
The important clinical implication of our study is that, consistent with recent meta-analyses [51] and discussion of evaluation of clinical trials [52-54], we observed significant differential effects of anti-hypertensive drugs on endothelial dysfunction and plasma adipocytokine levels in hypertensive patients. Thus, our study suggests that the particular therapeutic strategy used to target blood pressure may be an additional important factor to consider in addition to simply monitoring improvement in a biomarker per se. Design of future clinical trials should take into account comparison of therapeutic strategies in addition to simple comparisons of biomarkers.
Acknowledgments
This study was partly supported by grants from established investigator award (2005-1, 2006-1), Gil Medical Center, Gachon University and Korean Society of Hypertension (KSH-2004, 2005).
Footnotes
We presented in part in the American Heart Association 2006, Chicago, IL, November 12-15, 2006 and in the European Society of Cardiology 2007, Vienna, Austria, September 1–5, 2007.
References
- 1.Kim J, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 2.Han SH, Quon MJ, Koh KK. Reciprocal relationships between abnormal metabolic parameters and endothelial dysfunction. Curr Opin Lipidol. 2007;18:58–65. doi: 10.1097/MOL.0b013e328012b627. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Puig J, Ruilope LM, Luque M, et al. AVANT Study Group Investigators. Glucose metabolism in patients with essential hypertension. Am J Med. 2006;119:318–26. doi: 10.1016/j.amjmed.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Yusuf S, Sleight P, Pogue J, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–53. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- 5.Yusuf S, Ostergren JB, Gerstein HC, et al. Candesartan in heart failure assessment of reduction in mortality and morbidity program investigators. Effects of candesartan on the development of a new diagnosis of diabetes mellitus in patients with heart failure. Circulation. 2005;112:48–53. doi: 10.1161/CIRCULATIONAHA.104.528166. [DOI] [PubMed] [Google Scholar]
- 6.Taylor EN, Hu FB, Curhan GC. Antihypertensive medications and the risk of incident type 2 diabetes. Diabetes Care. 2006;29:1065–70. doi: 10.2337/diacare.2951065. [DOI] [PubMed] [Google Scholar]
- 7.Vincent MA, Montagnani M, Quon MJ. Molecular and physiologic actions of insulin related to production of nitric oxide in vascular endothelium. Curr Diab Rep. 2003;3:279–88. doi: 10.1007/s11892-003-0018-9. [DOI] [PubMed] [Google Scholar]
- 8.Fukai T, Siegfried MR, Ushio-Fukai M, Griendling KK, Harrison DG. Modulation of extracellular superoxide dismutase expression by angiotensin II and hypertension. Circ Res. 1999;85:23–8. doi: 10.1161/01.res.85.1.23. [DOI] [PubMed] [Google Scholar]
- 9.Pueyo ME, Gonzalez W, Nicoletti A, et al. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol. 2000;20:645–51. doi: 10.1161/01.atv.20.3.645. [DOI] [PubMed] [Google Scholar]
- 10.Nickenig G, Harrison DG. The AT1-type angiotensin receptor in oxidative stress and atherogenesis: part I: oxidative stress and atherogenesis. Circulation. 2002;105:393–6. doi: 10.1161/hc0302.102618. [DOI] [PubMed] [Google Scholar]
- 11.Asano Y, Kim J, Ogai A, et al. A calcium channel blocker activates both ecto-5(’)-nucleotidase and NO synthase in HUVEC. Biochem Biophys Res Commun. 2003;311:625–8. doi: 10.1016/j.bbrc.2003.10.036. [DOI] [PubMed] [Google Scholar]
- 12.Han SH, Quon MJ, Kim JA, Koh KK. Adiponectin and cardiovascular disease: response to therapeutic interventions. J Am Coll Cardiol. 2007;49:531–8. doi: 10.1016/j.jacc.2006.08.061. [DOI] [PubMed] [Google Scholar]
- 13.Koh KK, Park SM, Quon MJ. Leptin and cardiovascular disease: response to therapeutic interventions. Circulation. 2008;117:3238–49. doi: 10.1161/CIRCULATIONAHA.107.741645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu JG, Javorschi S, Hevener AL, et al. The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes. 2002;51:2968–74. doi: 10.2337/diabetes.51.10.2968. [DOI] [PubMed] [Google Scholar]
- 15.Koh KK, Quon MJ, Han SH, et al. Vascular and metabolic effects of combined therapy with ramipril and simvastatin in patients with type 2 diabetes. Hypertension. 2005;45:1088–93. doi: 10.1161/01.HYP.0000166722.91714.ba. [DOI] [PubMed] [Google Scholar]
- 16.Koh KK, Quon MJ, Han SH, et al. Anti-inflammatory and metabolic effects of candesartan in hypertensive patients. Int J Cardiol. 2006;108:96–100. doi: 10.1016/j.ijcard.2005.07.040. [DOI] [PubMed] [Google Scholar]
- 17.Koh KK, Quon MJ, Lee SJ, et al. Efonidipine simultaneously improves blood pressure, endothelial function, and metabolic parameters in non-diabetic patients with hypertension. Diabetes Care. 2007;30:1605–7. doi: 10.2337/dc06-2267. [DOI] [PubMed] [Google Scholar]
- 18.Correia ML, Haynes WG. Leptin, obesity and cardiovascular disease. Curr Opin Nephrol Hypertens. 2004;13:215–23. doi: 10.1097/00041552-200403000-00010. [DOI] [PubMed] [Google Scholar]
- 19.Verma S, Li SH, Wang CH, et al. Resistin promotes endothelial cell activation: further evidence of adipokine-endothelial interaction. Circulation. 2003;108:736–40. doi: 10.1161/01.CIR.0000084503.91330.49. [DOI] [PubMed] [Google Scholar]
- 20.Reilly MP, Lehrke M, Wolfe ML, et al. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. 2005;111:932–9. doi: 10.1161/01.CIR.0000155620.10387.43. [DOI] [PubMed] [Google Scholar]
- 21.Dahlof B, Sever PS, Poulter NR, et al. ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet. 2005;366:895–906. doi: 10.1016/S0140-6736(05)67185-1. [DOI] [PubMed] [Google Scholar]
- 22.Poulter NR, Wedel H, Dahlof B, et al. ASCOT Investigators. Role of blood pressure and other variables in the differential cardiovascular event rates noted in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA) Lancet. 2005;366:907–13. doi: 10.1016/S0140-6736(05)67186-3. [DOI] [PubMed] [Google Scholar]
- 23.Koh KK, Quon MJ, Han SH, et al. Additive beneficial effects of losartan combined with simvastatin in the treatment of hypercholesterolemic, hypertensive patients. Circulation. 2004;110:3687–92. doi: 10.1161/01.CIR.0000143085.86697.13. [DOI] [PubMed] [Google Scholar]
- 24.Koh KK, Ahn JY, Han SH, et al. Pleiotropic effects of angiotensin II receptor blocker in hypertensive patients. J Am Coll Cardiol. 2003;42:905–10. doi: 10.1016/s0735-1097(03)00846-5. [DOI] [PubMed] [Google Scholar]
- 25.Katz A, Nambi SS, Mather K, et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab. 2000;85:2402–10. doi: 10.1210/jcem.85.7.6661. [DOI] [PubMed] [Google Scholar]
- 26.Koh KK. Effects of statins on vascular wall: vasomotor function, inflammation, and plaque stability. Cardiovasc Res. 2000;47:648–57. doi: 10.1016/s0008-6363(00)00146-2. [DOI] [PubMed] [Google Scholar]
- 27.Zhou MS, Jaimes EA, Raij L. Inhibition of oxidative stress and improvement of endothelial function by amlodipine in angiotensin II-infused rats. Am J Hypertens. 2004;17:167–71. doi: 10.1016/j.amjhyper.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 28.Berkels R, Taubert D, Bartels H, et al. Amlodipine increases endothelial nitric oxide by dual mechanisms. Pharmacology. 2004;70:39–45. doi: 10.1159/000074241. [DOI] [PubMed] [Google Scholar]
- 29.Schiffrin EL, Pu Q, Park JB. Effect of amlodipine compared to atenolol on small arteries of previously untreated essential hypertensive patients. Am J Hypertens. 2002;15(2 Pt 1):105–10. doi: 10.1016/s0895-7061(01)02290-7. [DOI] [PubMed] [Google Scholar]
- 30.Anderson TJ, Elstein E, Haber H, Charbonneau F. Comparative study of ACE-inhibition, angiotensin II antagonism, and calcium channel blockade on flow-mediated vasodilation in patients with coronary disease (BANFF study) J Am Coll Cardiol. 2000;35:60–6. doi: 10.1016/s0735-1097(99)00537-9. [DOI] [PubMed] [Google Scholar]
- 31.Ghiadoni L, Magagna A, Versari D, et al. Different effect of antihypertensive drugs on conduit artery endothelial function. Hypertension. 2003;41:1281–6. doi: 10.1161/01.HYP.0000070956.57418.22. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki S, Nishimaki-Mogami T, Tamehiro N, et al. Verapamil increases the apolipoprotein-mediated release of cellular cholesterol by induction of ABCA1 expression via Liver X receptor-independent mechanism. Arterioscler Thromb Vasc Biol. 2004;24:519–25. doi: 10.1161/01.ATV.0000117178.94087.ba. [DOI] [PubMed] [Google Scholar]
- 33.Koh KK, Han SH, Quon MJ. Inflammatory markers and the metabolic syndrome: insights from therapeutic interventions. J Am Coll Cardiol. 2005;46:1978–85. doi: 10.1016/j.jacc.2005.06.082. [DOI] [PubMed] [Google Scholar]
- 34.Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem. 2003;278:45021–6. doi: 10.1074/jbc.M307878200. [DOI] [PubMed] [Google Scholar]
- 35.Folli F, Kahn CR, Hansen H, Bouchie JL, Feener EP. Angiotensin II inhibits insulin signaling in aortic smooth muscle cells at multiple levels. A potential role for serine phosphorylation in insulin/angiotensin II crosstalk. J Clin Invest. 1997;100:2158–69. doi: 10.1172/JCI119752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma AM, Janke J, Gorzelniak K, Engeli S, Luft FC. Angiotensin blockade prevents type 2 diabetes by formation of fat cells. Hypertension. 2002;40:609–11. doi: 10.1161/01.hyp.0000036448.44066.53. [DOI] [PubMed] [Google Scholar]
- 37.Schupp M, Janke J, Clasen R, Unger T, Kintscher U. Angiotensin type 1 receptor blockers induce peroxisome proliferator-activated receptor-γ activity. Circulation. 2004;109:2054–7. doi: 10.1161/01.CIR.0000127955.36250.65. [DOI] [PubMed] [Google Scholar]
- 38.Hermann TS, Li W, Dominguez H, et al. Quinapril treatment increases insulin-stimulated endothelial function and adiponectin gene expression in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91:1001–8. doi: 10.1210/jc.2005-1231. [DOI] [PubMed] [Google Scholar]
- 39.Harada N, Ohnaka M, Sakamoto S, Niwa Y, Nakaya Y. Cilnidipine improves insulin sensitivity in the Otsuka Long–Evans Tokushima fatty rat, a model of spontaneous NIDDM. Cardiovasc Drugs Ther. 1999;13:519–23. doi: 10.1023/a:1007827720807. [DOI] [PubMed] [Google Scholar]
- 40.Koyama Y, Kodama K, Suzuki M, Harano Y. Improvement of insulin sensitivity by a long-acting nifedipine preparation (nifedipine-CR) in patients with essential hypertension. Am J Hypertens. 2002;15:927–31. doi: 10.1016/s0895-7061(02)03019-4. [DOI] [PubMed] [Google Scholar]
- 41.Lender D, Arauz-Pacheco C, Breen L, et al. A double blind comparison of the effects of amlodipine and enalapril on insulin sensitivity in hypertensive patients. Am J Hypertens. 1999;12:298–303. doi: 10.1016/s0895-7061(98)00259-3. [DOI] [PubMed] [Google Scholar]
- 42.de Courten M, Ferrari P, Schneider M, et al. Lack of effect of long-term amlodipine on insulin sensitivity and plasma insulin in obese patients with essential hypertension. Eur J Clin Pharmacol. 1993;44:457–62. doi: 10.1007/BF00315543. [DOI] [PubMed] [Google Scholar]
- 43.Yilmaz MI, Sonmez A, Caglar K, et al. Effect of antihypertensive agents on plasma adiponectin levels in hypertensive patients with metabolic syndrome. Nephrology (Carlton) 2007;12:147–53. doi: 10.1111/j.1440-1797.2007.00764.x. [DOI] [PubMed] [Google Scholar]
- 44.Skurk T, van Harmelen V, Blum WF, Hauner H. Angiotensin II promotes leptin production in cultured human fat cells by an ERK1/2-dependent pathway. Obes Res. 2005;13:969–73. doi: 10.1038/oby.2005.113. [DOI] [PubMed] [Google Scholar]
- 45.Werner N, Nickenig G. From fat fighter to risk factor: the zigzag trek of leptin. Arterioscler Thromb Vasc Biol. 2004;24:7–9. doi: 10.1161/01.ATV.0000110908.43721.ad. [DOI] [PubMed] [Google Scholar]
- 46.McTernan PG, Fisher FM, Valsamakis G, et al. Resistin and type 2 diabetes: regulation of resistin expression by insulin and rosiglitazone and the effects of recombinant resistin on lipid and glucose metabolism in human differentiated adipocytes. J Clin Endocrinol Metab. 2003;88:6098–106. doi: 10.1210/jc.2003-030898. [DOI] [PubMed] [Google Scholar]
- 47.Degawa-Yamauchi M, Bovenkerk JE, Juliar BE, et al. Serum resistin (FIZZ3) protein is increased in obese humans. J Clin Endocrinol Metab. 2003;88:5452–5. doi: 10.1210/jc.2002-021808. [DOI] [PubMed] [Google Scholar]
- 48.Ohmori R, Momiyama Y, Kato R, et al. Associations between serum resistin levels and insulin resistance, inflammation, and coronary artery disease. J Am Coll Cardiol. 2005;46:379–80. doi: 10.1016/j.jacc.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 49.Furuhashi M, Ura N, Higashiura K, Murakami H, Shimamoto K. Circulating resistin levels in essential hypertension. Clin Endocrinol (Oxf) 2003;59:507–10. doi: 10.1046/j.1365-2265.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 50.Youn BS, Yu KY, Park HJ, et al. Plasma resistin concentrations measured by enzyme-linked immunosorbent assay using a newly developed monoclonal antibody are elevated in individuals with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2004;89:150–6. doi: 10.1210/jc.2003-031121. [DOI] [PubMed] [Google Scholar]
- 51.Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet. 2007;369:201–7. doi: 10.1016/S0140-6736(07)60108-1. [DOI] [PubMed] [Google Scholar]
- 52.Cutler JA, Davis BR. Thiazide-type diuretics and beta-adrenergic blockers as first-line drug treatments for hypertension. Circulation. 2008;117:2691–704. doi: 10.1161/CIRCULATIONAHA.107.709931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter BL, Einhorn PT, Brands M, et al. Thiazide-induced dysglycemia: call for research from a working group from the national heart, lung, and blood institute. Hypertension. 2008;52:30–6. doi: 10.1161/HYPERTENSIONAHA.108.114389. [DOI] [PubMed] [Google Scholar]
- 54.Krumholz HM, Lee TH. Redefining quality—implications of recent clinical trials. N Engl J Med. 2008;358:2537–9. doi: 10.1056/NEJMp0803740. [DOI] [PubMed] [Google Scholar]