

Abstract
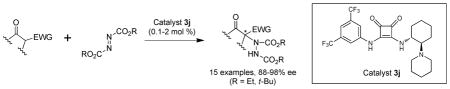
Catalytic enantioselective α-hydrazination of 1,3-dicarbonyl compounds with azodicarboxylates was investigated in the presence of our newly developed hydrogen bonding catalyst, squaramide 3j. High yields and high enantioselectivities were achieved with low catalyst loading under mild conditions.
Hydrogen bond donor catalysis for asymmetric synthesis has attracted intense research efforts in recent years.1 With the focus on substrate generality, catalyst practicality, and mildness of reaction conditions, our laboratory has been involved in the investigation of enantioselective transformations catalyzed by simple chiral hydrogen bond donors.2 Recently we have developed a new family of catalysts based on the squaramide scaffold and demonstrated their utility in C-C bond3a and C-P bond3b formation.4 In both cases nitroalkenes were successfully activated towards 1,4-addition reactions. In order to assess the range of electrophiles that would be compatible with squaramide-based catalysts, we have begun to examine other classes of reactants. In particular, we were drawn to electrophilic amination reactions, an important class of transformations that offers alternative and often attractive routes for the synthesis of nitrogen-containing compounds.5 Among them, the asymmetric α-hydrazination of carbonyl compounds with azodicarboxylates is a powerful tool to install protected amino groups and set chiral centers during a synthetic sequence. When α-monosubstituted-1,3-dicarbonyl compounds are used as nucleophiles, quaternary α-amino acid derivatives are obtained as products.6 These derivatives are of special interest for the synthesis of designer peptides with specific conformational and biological properties7 and as building blocks of potential therapeutic agents such as metabotropic glutamate receptor ligands.8
Although several examples of catalytic enantioselective α-hydrazination of 1,3-dicarbonyl compounds have already been reported,9,10,11 there are still challenging issues that need to be addressed, particularly in the area of organocatalysis. These challenges include enantioselectivity, substrate generality, efficiency and ease of catalyst preparation, catalyst loading, and reaction time. Herein we describe our investigation of catalytic enantioselective α-hydrazination of 1,3-dicarbonyl compounds using a newly developed chiral squaramide catalyst. We report a wide substrate scope with several examples of reactions that proceed at ambient temperature and afford products in 93–96% ee.
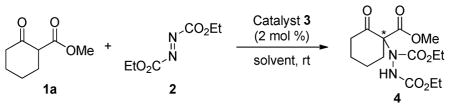
To confirm the ability of squaramides to catalyze the α-hydrazination reaction, a series of catalysts with various substituents on the two vinylogous amide nitrogen atoms was prepared and tested in the addition of β-ketoester 1a to diethyl azodicarboxylate (2, Table 1). In this regard, the ease of synthesis and the modular nature of chiral squaramides facilitates the fine-tuning of their activity. It should be noted that the catalysts shown in Figure 1 are all readily prepared, in two to three steps from commercially available dimethyl squarate.12 Catalyst 3a, which was highly effective in the reported conjugate addition to nitroalkenes,3a did not function well for the present transformation (entry 1). Catalyst 3b bearing an amine derived from the pseudo-enantiomeric cinchona alkaloid also gave an unsatisfactory result but, interestingly, a product enriched in the same enantiomer as 3a (entry 2). Other chiral moieties were examined, and we were delighted to find that catalyst 3e possessing the (R,R)-1,2-diaminocyclohexane unit successfully promoted the reaction in high yield and high enantioselectivity (entry 5). The poor reactivity seen with catalyst 3d (entry 4) is consistent with the expectation that a bifunctional catalyst, possessing a basic amino group, is required to promote the reaction. The amino group in catalyst 3e serves to generate the active nucleophilic anion, and the resulting ammonium ion is expected to organize the transition state geometry through additional hydrogen bonding.13 Further investigation of the effect of the tertiary amino group led to the discovery of another suitable catalyst, 3j, which contains a piperidinyl group (entries 5–10). Evaluation of the effect of the left part of the squaramide catalyst showed bis(trifluoromethyl)phenyl group to be the superior substituent (entries 10–12).
Table 1.
Catalyst Screeninga
 | ||||
|---|---|---|---|---|
| entry | catalystb | time (h) | yieldc (%) | eed (%) |
| 1 | 3a | 15 | 58 | −10 |
| 2 | 3b | 15 | 65 | −29 |
| 3 | 3c | 60 | 71 | −5 |
| 4 | 3d | 48 | 22 | 0 |
| 5 | 3e | 0.5 | 98 | 88 |
| 6 | 3f | 1 | 83 | 69 |
| 7 | 3g | 1 | 78 | 69 |
| 8 | 3h | 30 | 81 | 32 |
| 9 | 3i | 24 | 70 | 25 |
| 10 | 3j | 0.5 | 90 | 89 |
| 11 | 3k | 3 | 91 | 88 |
| 12 | 3l | 2 | 84 | 66 |
| 13 | 3e | 0.5 | 98 | 88 |
| 14 | 3j | 0.5 | 90 | 89 |
| 15e | 3e | 2 | 96 | 90 |
| 16e | 3j | 1 | 97 | 92 |
| 17f | 3j | 2 | 97 | 95 |
| 18g | 3j | 0.5 | 91 | 87 |
| 19h | 3j | 2.5 | 88 | 83 |
Reactions were performed with 0.75 mmol of 1a, 0.5 mmol of 2, and 2 mol % of catalyst 3 in 0.5 mL of toluene at rt, unless otherwise stated.
See Figure 1 for structures of catalysts.
Isolated yield.
Determined by chiral HPLC.
Reaction conducted at 0 °C.
Reaction conducted at −20 °C.
Reaction conducted in CH2Cl2.
Reaction conducted in THF.
Figure 1.

Structures of Catalysts
Since 3e and 3j provided products with similar enantioselectivities, they were investigated further to identify the optimal catalyst. Comparison of the catalytic ability of both catalysts by conducting the reactions at low temperature indicated that 3j was superior to 3e (entries 13–16). Enantioselectivity was improved to 95% ee by simply lowering reaction temperature to −20 °C (entry 17). A brief survey of solvents identified toluene to be somewhat better for this reaction than methylene chloride or THF (entries 10, 18, 19).
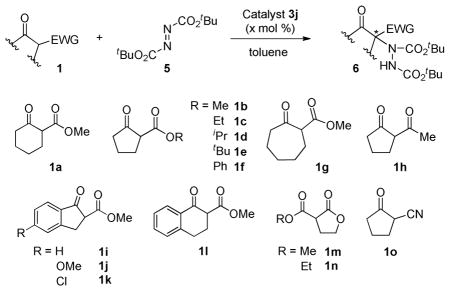
The scope of catalytic enantioselective α-hydrazination of various active methylene compound, using 3j as the catalyst, was evaluated next (Table 2). These reactions were carried out using di-tert-butyl azodicarboxylate (5) as the hydrazination reagent, rather than diethyl azodicarboxylate (2). Preliminary studies had shown that both gave products with similar enantioselectivities, but the former had the advantage that the Boc group can be easily removed, if needed. However, in a small number of cases diethyl azodicarboxylate (2) was used because higher enantioselectivity was observed (1a and 1h, entries 1, 2, 12). A wide variety of cyclic β-ketoesters, 1,3-diketone, malonates, and α-cyanoketone reacted successfully with azodicarboxylates under the optimized conditions, affording the corresponding products with high to excellent enantioselectivities. The range of reactants described is one of the most extensive among the reported organocatalyzed α-hydrazination of 1,3-dicarbonyl compounds. In particular, cyclic malonates 1m and 1n represent a new type of substrates for the reaction.
Table 2.
Substrate Scopea
 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate | x(mol %) | temp(°C) | time(h) | yieldb(%) | eec (%) |
| 1d | 1a | 2 | 0 | 1 | 97 | 92 |
| 2d | 1a | 2 | −20 | 2 | 97 | 95 |
| 3 | 1b | 1 | rt | 0.5 | 97 | 96 |
| 4 | 1b | 0.1 | rt | 20 | 98 | 95 |
| 5 | 1b | 1 | −20 | 1 | 99 | 98 |
| 6 | 1c | 1 | rt | 1 | 96 | 96 |
| 7 | 1d | 1 | rt | 2.5 | 95 | 96 |
| 8 | 1e | 1 | rt | 17 | quant. | 94 |
| 9 | 1e | 1 | rt | 4 | 95 | 93 |
| 10 | 1f | 2 | rt | 23 | 92 | 95 |
| 11 | 1g | 2 | rt | 6 | 96 | 94 |
| 12d | 1h | 2 | −20 | 0.5 | 98 | 91 |
| 13e | 1i | 1 | −40 | 0.5 | quant. | 88 |
| 14e | 1j | 1 | −40 | 1 | 95 | 90 |
| 15e | 1k | 1 | −40 | 2 | 97 | 88 |
| 16 | 1l | 1 | −20 | 28 | quant. | 90 |
| 17e | 1m | 1 | 0 | 0.5 | 90 | 98 |
| 18 | 1n | 1 | rt | 3 | 93 | 95 |
| 19e | 1o | 1 | 0 | 14 | 83 | 90 |
Reactions were performed with 0.55 mmol or 0.75 mmol of 1, 0.5 mmol of 5, and 0.1–2 mol % of catalyst 3j in 0.5 mL of toluene at the indicated temperature, unless otherwise stated.
Isolated yield.
Enantiomeric excess was determined by chiral HPLC. Absolute configuration was determined to be (S) for 6b, 6c, and 6e, see text and Supporting Information for details.
Diethyl azodicarboxylate (2) was used.
1.0 mL of toluene was used.
It is noteworthy that for a number of substrates excellent enantioselectivities were obtained (93–96% ee, entries 3–4, 6–11, 18) at room temperature, without the need to resort to low temperature. Moreover, the catalyst loading could be reduced to 0.1 mol % without significant deterioration of enantioselectivity (entry 4). The reaction was not affected significantly by electronic and steric variations of the ester group (entries 3–10). Likewise, for the indanone containing substrates, electronic perturbations of the aromatic ring had little effect on enantioselectivity (entries 13–15). Interestingly, β-ketoesters 1a and 1g, bearing six and seven membered rings (entries 1, 2, 11), reacted at a satisfactory rate at temperatures between −20 °C and room temperature under the present catalysis method. By comparison, previous reports indicated that such substrates required longer reaction times (> 24 h).10a,b,d,e
The assignment of absolute stereochemistry of the enantiomerically enriched product can provide important information for mechanistic consideration and further improvement of catalyst design. A survey of the literature showed that (R) configuration was assigned to the quaternary center of (−)-6c10e and (+)-6e.9g Our catalyst afforded (+)-6c and (+)-6e, which would be assigned (S) and (R) stereochemistry, respectively, based on correlation with the aforementioned data. It appeared highly improbable that steric difference in the ester groups—ethyl vs tert-butyl—should result in opposite enantioselection. Further, closer inspection showed the literature reports to be less than definitive with regard to the stereochemistry of similar hydrazination products.12 Thus, we sought to provide unequivocal assignment of the absolute stereochemistry of our hydrazination products. Compound 6b was converted to a derivative that contains a (1S)-camphanic amide,12 and the X-ray crystal-structure analysis provided clear proof that our product has the (S) configuration. The absolute stereochemistries of (+)-6c and (+)-6e were then assigned as (S) by correlation.12
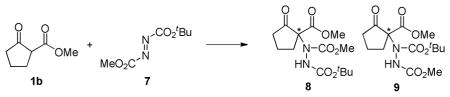
In order to get some insight on the nature of electrophile activation by the squaramide catalyst, and thereby the observed asymmetric induction, we also examined the hydrazination reaction of unsymmetrical azodicarboxylates. Prior work had established that unsymmetrically substituted azodicarboxylates react with nucleophiles in a regioselective manner, resulting in addition to the more electron deficient nitrogen.14 We reasoned that steric differences in the azodicarboxylates might result in a selective dual hydrogen bond, presumably to the less hindered carbonyl and the adjacent nitrogen, which in turn would induce electronic differentiation of the two azo-nitrogens. Interestingly, the reaction of sterically differentiated asymmetric azodicarboxylate 7 with β-ketoester 1b in the presence of squaramide 3j provided the same nitrogen regioselectivity as the uncatalyzed reaction, at room temperature and at 0 °C (Table 3). Moreover, the yield and enantioselectivity with the unsymmterical azodicarboxylate were similar to that observed with di-tert-butyl azodicarboxylate (5). Thus, it appears that the steric difference between a methoxy and tert-butoxy group is not sufficient to induce selectivity in its interaction with the hydrogen bonding catalyst.15
Table 3.
Reaction of β-Ketoester 1b with Asymmetric Azodicarboxylate 7
 | ||||||
|---|---|---|---|---|---|---|
| entry | temp(°C) | methoda | time(h) | yieldb(%) | ratioc | eed(%) |
| 1 | rt | racemic | 20 | 79 | 62:38 | - |
| 2 | rt | asymmetric | 0.5 | 90 | 66:34 | 95/94 |
| 3 | 0 | racemic | 24 | 78 | 66:34 | - |
| 4 | 0 | asymmetric | 1 | 94 | 66:34 | 97/97 |
Method “racemic”: Reaction of 0.5 mmol of 1b, 0.53 mmol of 7, and 50 mol % of KOAc in 5 mL of CH2Cl2. Method “asymmetric”: Reaction of 0.55 mmol of 1b, 0.5 mmol of 7, and 1 mol % of 3j in 0.5 mL of toluene.
Isolated yield.
Unassigned ratio of regioisomers 8 and 9 determined by HPLC.
Determined by chiral HPLC and presented as major regioisomer/minor regioisomer.
In conclusion, the results above demonstrate squaramide 3j to be a highly effective catalyst for the enantioselective α-hydrazination of 1,3-dicarbonyl compounds. All reactions can be conducted under mild conditions, with low catalyst loading, and afford the products in generally excellent yields and enantioselectivities. These results not only provide further demonstration of squaramides as highly effective hydrogen bonding catalysts but also calibration of their usefulness versus other hydrogen bond donor catalysts for the preparation of optically active nitrogen compounds. Further applications of chiral squaramide catalysts, particularly for the development of new enantioselective reactions, are under investigation.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health (R01GM069990). T.Y.L. thanks the Hong Kong Croucher Foundation for a postdoctoral fellowship. We also thank Professor Leo Paquette (Ohio State University) for a generous supply of squaric acid and dimethyl squarate, Ye Zhu (University of Chicago) for assisting with the synthesis of catalysts, and Dr. Ian Steele (University of Chicago) for obtaining the crystal structure of the derivative of 6b.
Footnotes
Supporting Information Available: Procedures for catalyst preparation and hydrazination reaction, discussion and detail of the assignment of absolute stereochemistry, and analytical data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews on hydrogen-bond donor catalysis, see: Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r.McGilvra JD, Gondi VB, Rawal VH. In: Enantioselective Organocatalysis. Dalko PI, editor. Wiley-VCH; Weinheim: 2007. pp. 189–254.Yu X, Wang W. Chem Asian J. 2008;3:516–532. doi: 10.1002/asia.200700415.
- 2.(a) Huang Y, Unni AK, Thadani AN, Rawal VH. Nature. 2003;424:146. doi: 10.1038/424146a. [DOI] [PubMed] [Google Scholar]; (b) Thadani AN, Stankovic AR, Rawal VH. Proc Natl Acad Sci USA. 2004;101:5846–5850. doi: 10.1073/pnas.0308545101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McGilvra JD, Unni AK, Modi K, Rawal VH. Angew Chem Int Ed. 2006;45:6130–6133. doi: 10.1002/anie.200601638. [DOI] [PubMed] [Google Scholar]; (d) Gondi VB, Hagihara K, Rawal VH. Angew Chem Int Ed. 2009;48:776–779. doi: 10.1002/anie.200804244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Malerich JP, Hagihara K, Rawal VH. J Am Chem Soc. 2008;130:14416–14417. doi: 10.1021/ja805693p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu Y, Malerich JP, Rawal VH. Angew Chem Int Ed. 2010;49:153–156. doi: 10.1002/anie.200904779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Since the first publication from our laboratory in 2008, other reports of squaramide-based organocatalysts have appeared: Cheon CH, Yamamoto H. Tetrahedron Lett. 2009;50:3555–3558.Lee JW, Ryu TH, Oh JS, Bae HY, Jang HB, Song CE. Chem Commun. 2009:7224–7226. doi: 10.1039/b917882a.
- 5.(a) Greck C, Genêt JP. Synlett. 1997:741–748. [Google Scholar]; (b) Genet JP, Greck C, Lavergne D. In: Modern Amination Methods. Ricci A, editor. Wiley-VCH; Weinheim: 2000. pp. 65–102. [Google Scholar]; (c) Dembech P, Seconi G, Ricci A. Chem Eur J. 2000;6:1281–1286. doi: 10.1002/(sici)1521-3765(20000417)6:8<1281::aid-chem1281>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]; (d) Duthaler RO. Angew Chem Int Ed. 2003;42:975–978. doi: 10.1002/anie.200390283. [DOI] [PubMed] [Google Scholar]; (e) Greck C, Drouillat B, Thomassigny C. Eur J Org Chem. 2004:1377–1385. [Google Scholar]; (f) Erdik E. Tetrahedron. 2004;60:8747–8782. [Google Scholar]; (g) Janey JM. Angew Chem Int Ed. 2005;44:4292–4300. doi: 10.1002/anie.200462314. [DOI] [PubMed] [Google Scholar]; (h) Nair V, Biju AT, Mathew SC, Babu BP. Chem Asian J. 2008;3:810–820. doi: 10.1002/asia.200700341. [DOI] [PubMed] [Google Scholar]
- 6.For recent reviews on the stereoselective synthesis of quaternary α-amino acids, see: Vogt H, Bräse S. Org Biol Chem. 2007;5:406–430. doi: 10.1039/b611091f.Cativiela C, Díaz-de-Villegas MD. Tetrahedron: Asymmetry. 2007;18:569–623.Cativiela C, Ordóñez M. Tetrahedron: Asymmetry. 2009;20:1–63. doi: 10.1016/j.tetasy.2009.01.002.
- 7.(a) Gante J. Angew Chem Int Ed. 1994;33:1699–1720. [Google Scholar]; (b) Venkatraman J, Shankaramma SC, Balaram P. Chem Rev. 2001;101:3131–3152. doi: 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]
- 8.For a review, see: Schoepp DD, Jane DE, Monn JA. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1.
- 9.For examples of Lewis acid catalysis, see: Juhl K, Jørgensen KA. J Am Chem Soc. 2002;124:2420–2021. doi: 10.1021/ja0175486.Marigo M, Juhl K, Jørgensen KA. Angew Chem Int Ed. 2003;42:1367–1369. doi: 10.1002/anie.200390350.Ma S, Jiao N, Zheng Z, Ma Z, Lu Z, Ye L, Deng Y, Chen G. Org Lett. 2004;6:2193–2196. doi: 10.1021/ol0493498.Foltz C, Stecker B, Marconi G, Bellemin-Laponnaz S, Wadepohl H, Gade LH. Chem Commun. 2005:5115–5117. doi: 10.1039/b509571a.Huber DP, Stanek K, Togni A. Tetrahedron: Asymmetry. 2006;17:658–664.Kang YK, Kim DY. Tetrahedron Lett. 2006;47:4565–4568.Comelles J, Pericas À, Moreno-Mañas M, Vallribera A, Drudis-Solé G, Lledos A, Parella T, Roglans A, García-Granda S, Roces-Fernández L. J Org Chem. 2007;72:2077–2087. doi: 10.1021/jo0622678.Mashiko T, Hara K, Tanaka D, Fujiwara Y, Kumagai N, Shibasaki M. J Am Chem Soc. 2007;129:11342–11343. doi: 10.1021/ja0752585.Mashiko T, Kumagai N, Shibasaki M. Org Lett. 2008;10:2725–2728. doi: 10.1021/ol8008446.Hasegawa Y, Watanabe M, Gridnev ID, Ikariya T. J Am Chem Soc. 2008;130:2158–2159. doi: 10.1021/ja710273s.Mashiko T, Kumagai N, Shibasaki M. J Am Chem Soc. 2009;131:14990–14999. doi: 10.1021/ja9052653.
- 10.For examples of H-bonding catalysis, see: Saaby S, Bella M, Jørgensen KA. J Am Chem Soc. 2004;126:8120–8121. doi: 10.1021/ja047704j.Pihko PM, Pohjakallio A. Synlett. 2004:2115–2118.Liu X, Li H, Deng L. Org Lett. 2005;7:167–169. doi: 10.1021/ol048190w.Xu X, Yabuta T, Yuan P, Takemoto Y. Synlett. 2006:137–140.Terada M, Nakano M, Ube H. J Am Chem Soc. 2006;126:16044–16045. doi: 10.1021/ja066808m.Kim SM, Lee JH, Kim DY. Synlett. 2008:2659–2662.Jung SH, Kim DY. Tetrahedron Lett. 2008;49:5527–5530.Mang JY, Kim DY. Bull Korean Chem Soc. 2008;29:2091–2092.Liu X, Sun B, Deng L. Synlett. 2009:1685–1689. doi: 10.1055/s-0029-1217334.
- 11.For examples of ammonium and phosphonium salt catalysis, see: He R, Wang X, Hashimoto T, Maruoka K. Angew Chem Int Ed. 2008;47:9466–9468. doi: 10.1002/anie.200804140.Lan Q, Wang X, He R, Ding C, Maruoka K. Tetrahedron Lett. 2009;50:3280–3282.
- 12.See Supporting Information for details.
- 13.(a) For the discussion of a proposed mechanism of reactions using related bifunctional thiourea catalysts, see: Okino T, Hoashi Y, Furukawa T, Xu X, Takemoto Y. J Am Chem Soc. 2005;127:119–125. doi: 10.1021/ja044370p.For a theoretical study, see: Hamza A, Schubert G, Soós T, Pápai I. J Am Chem Soc. 2006;128:13151–13160. doi: 10.1021/ja063201x.
- 14.(a) Yamamoto Y, Yumoto M, Yamada J. Tetraheron Lett. 1991;32:3079–3082. [Google Scholar]; (b) Mitchell H, Leblanc Y. J Org Chem. 1994;59:682–687. [Google Scholar]; (c) Evans DA, Johnson DS. Org Lett. 1999;1:595–598. doi: 10.1021/ol990113r. [DOI] [PubMed] [Google Scholar]
- 15.Jørgensen had also reported low regioselectivity (~2:1) in a hydrogen bond donor catalyzed reaction of an asymmetric azodicarboxylate: Brandes S, Bella M, Kjœrsgaard A, Jørgensen KA. Angew Chem Int Ed. 2006;45:1147–1151. doi: 10.1002/anie.200503042.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.