

Abstract
Many research projects will lead to understanding tissue and/or cell responses to extracellular influences either from soluble factors or the surrounding extracellular matrix. These types of investigations will require the understanding of signal transduction. This particular cell biological field has literally exploded with information and new technical approaches in the past 10 years. This article is directed toward investigators interested in using these new approaches to study their systems. An overview of the general principles of signal transduction events including the types of receptors and intracellular signaling events is followed by an introduction to methods for visualizing signal transduction. This is followed by an introduction to biochemical analysis and an example of combining several approaches to understanding a tissue response to extracellular matrix stimulus.
Keywords: extracellular matrix, ECM, Rho, green fluorescent protein, GFP, FRET, FRAP, GST assays, methods, signal transduction, morphology, imaging
INTRODUCTION
Cellular signaling is a complex ballet of molecules interacting and stimulating surrounding proteins, lipids, and ions, resulting in cytoskeletal reorganization, modulation of differentiation, and induction of gene expression. The choreography of events in signaling pathways has been a hot topic in the past few years. The intermolecular reactions should be viewed as a fluid dynamic process in which multiple events may be occurring at the same time. Many of the present experimental procedures document signal transduction snapshots of individual events, such as tyrosine phosphorylation, without the benefit of visualizing the whole stage simultaneously. However, newer assays using fluorescently tagged transfected proteins combined with live cell imaging analysis will both confirm and modify current perceptions of signal transduction events. We have been focusing on integrin-mediated pathways, but many of the experimental approaches described in this publication can be used for any signal transduction cascade. Therefore, this article will naturally be directed toward how integrins and growth factors initiate and propagate their signals.
Before discussing specific approaches to visualizing and biochemically analyzing signal transduction events, we will provide suggestions on how to understand the literature, explain the general principles, and describe simple cellular behavior assays. Both historical and current procedures will be explained, including the necessary reagents and laboratory equipment, with the goal of demonstrating that morphologically equipped laboratories have the ability to conduct these experiments with minimal investment in time and funds.
Many cell and developmental biologists who are currently analyzing signal transduction in their systems have a similar story. We really did not start our careers expecting to be talking in three letter words and explaining cascades of reactions that led to our observations. One of us started her scientific career studying how the cytoskeleton controlled cell shape throughout the development of the optic vesicle (Svoboda and O’Shea, 1984, 1987), and then continued examining the role of the actin cytoskeleton in corneal epithelial responses to extracellular matrix (ECM) (Svoboda and Hay, 1987; Svoboda, 1992; Khoory et al., 1993; Yeh and Svoboda, 1994; Hirsch et al., 1996; Svoboda et al., 1999a). After several years of describing the morphologic changes in cells, it was apparent that an understanding was needed regarding how the changes in cytoskeletal proteins were controlled. This investigation led down the path into signal transduction (Svoboda et al., 1999b; Chu et al., 2000). In this publication, our objective is to relate how a laboratory equipped to examine subtle morphologic changes can use new tools to determine which signal transduction molecules may be responsible for the changes.
GETTING STARTED
One of the biggest hurdles to learning signal transduction is understanding the language. This area of cell biology is full of jargon and acronyms that will quickly trip up the newcomer (See Box 1 for a list of common acronyms). Therefore, the first objective is to learn the terminology. The first word of caution is do not expect the names of proteins to be logical, as many were named for their first known function, but now have been shown to have a wider distribution and functional activity. In addition, there may be multiple names for the same protein or the same names (or abbreviations) used for different proteins, adding to the confusion and frustration to a new investigator. Many Web sites have been generated recently that cover the basic concepts (http://www.unige.ch/sciences/biochimie/Lafont/WA_CB.html or http://geo.nihs.go.jp/csndb/) and are good places to start understanding the different signal pathways. Many vendors have produced informative Web sites and general literature that should not be overlooked. A recent historical review gives the perspective and nomenclature in the field of signal transduction (Hunter, 2000) and may be another possible starting point. Several good review articles on the pathways of interest (Mecham, 1991; Meredith et al., 1996; Cary and Guan, 1999; Gonzalez-Amaro and Sanchez-Madrid, 1999; Duong et al., 2000; Ridley, 2000a,b; Roovers and Assoian, 2000) combined with a few colleagues or students are the ingredients needed to start a journal club. The journal club should start by reading the chapters in a modern cell biology text (Lodish et al., 2000), or a monograph on the specific subject, followed by some review articles, then tackle at least one current article a week. I am suggesting this route because the learning curve takes approximately 3–6 months just to understand the language of selective signal transduction cascades. As an example, I am interested in cell–matrix interactions, and the understanding of these cascades have become increasingly complicated in the past 3 years; a recent literature search (1998 –April of 2001), by using integrin and signal transduction as key words produced thousands of citations and over a hundred reviews.
BOX 1. Signal Transduction-Related Abbreviations Used in this Article.
- ACM
actin cortical mat
- bax
bcl-associated × protein that accelerates cell death (apoptosis)
- BL
basal lamina
- BMP
bone morphogenetic protein, a member of the TGFβ family
- CAM
cell adhesion molecules
- cAMP
cyclic AMP
- cAPKs
cyclic AMP-dependent kinases, also called protein kinase A (PKA)
- CD
cytochalasin D
- CGMP
cyclic GMP
- cLSM
confocal laser scanning microscope
- COL
type I collagen
- DAG
diacylglycerol
- Dia
p140 Diaphanous
- ECL
enhanced chemiluminescence
- ECM
extracellular matrix
- EGF
epidermal growth factor
- Erk
extracellular signal regulated protein or externally regulated kinases
- F-actin
filamentous actin
- FAK
focal adhesion kinase
- FGF
fibroblast growth factor
- FN
fibronectin
- FRAP
fluorescence recovery/redistribution after photo-bleaching
- FRET
fluorescence resonance energy transfer
- GAP
GTPase activating protein
- GDI
guanine-nucleotide dissociation inhibitor
- GEF
guanine exchange factors
- GFP
green fluorescent protein; CFP (CyanFP); YFP (yellowFP); and DsRed are variations that emit signal at different wavelengths
- GPCR
G-protein–coupled receptors
- Grb2
growth factor receptor bound protein 2
- GST
glutathione-S-transferase
- HRP
horseradish peroxidase
- ICE
interleukin-1 converting enzyme
- JNK/SAPK
Jun N-terminal kinase also known as SAPK, stress-activated protein kinase
- LPA
lysophosphatidic acid
- MAP kinase
mitogen-activated protein kinase
- MAPKK
mitogen-activated protein kinase kinase, also known as MEK
- MAPKKK
mitogen-activated protein kinase-kinase-kinase or MEKK
- PDGF
platelet-derived growth factor
- P190RhoGAP
190-kDa protein that functions as a GAP for Rho
- PAX
paxillin
- PI3kinase
phosphatidylinositol 3 kinase
- PIP2
phosphatidylinositol (4,5)-bisphosphate
- p-tyr
phosphotyrosine
- PY-20
an antibody that recognizes tyrosine phosphorylated proteins
- ROCK
Rho associated coiled-coil containing protein kinase also known as p160 Rhokinase
- Rho
a small GTPase family that includes Rac and cdc42
- SH2
Src homology 2 domains
- SH3
Src homology 3 domain
- Smad
sma and mad homology signal proteins renamed 1996, act in the TGFβ pathway
- SOS1
son of sevenless
- Src
a kinase that was first described in the Rous sarcoma virus, however, it is also found in normal cells
- TEM
transmission electron microscopy
- TGFβ
transforming growth factor beta
- TUNEL
Tdt-mediated dUTP-X nick-end labeling for apoptosis
GENERAL PRINCIPLES OF SIGNAL TRANSDUCTION EVENTS
Receptors
Intracellular signaling is triggered by a cell surface event such as a receptor–ligand interaction, cell–cell contact, or cell–ECM contact (Figure 1) (Sastry and Burridge, 2000). The contact with other cells or the environment can stimulate many cellular reactions, including proliferation, motility, differentiation, or even programmed cell death, termed apoptosis. The receptor or cell surface binding proteins are classified by the protein structure and ligand characteristics. The basic protein domains for a receptor are an extracellular ligand binding domain, membrane spanning domain, and a cytoplasmic domain (Figure 1). Most modern cell biology textbooks list at least four types of cell surface receptors, including the G-protein-coupled receptors, ion-channel receptors, tyrosine kinase-linked receptors, and receptors with intrinsic enzymatic activity (Lodish et al., 2000). The G-protein–coupled receptors (GPCR) are characterized by multiple transmembrane domains (usually seven) that wind the protein in and out of the membrane in a serpentine conformation (Figure 1, GPCR). The ion-channel receptors are closely related and actually open a membrane channel when the ligand binds. Many cytokine receptors are in the tyrosine kinase-linked class, as they lack intrinsic activity, but when the ligand binds, intracellular tyrosine kinases become activated to generate cellular changes. The classic growth factor receptors do have intrinsic kinase activity and, therefore, make up the fourth class of receptors, the receptor tyrosine kinases, or receptor serine/threonine kinases (Figure 1). These receptors usually have one transmembrane domain and at least two molecules must dimerize to activate the signal.
Figure 1.

Many different types of receptors are on cells that contribute to some of the same signal pathways. In this illustration, G-protein–coupled receptor (GPCR); receptor tyrosine kinase, cell adhesion molecules (CAMs), selectins, integrins, syndecans, and cadherins are represented with some of their intracellular signal molecules identified. This image was used to show that multiple signal pathways control focal adhesion assembly by coordinating regulation of RhoA. Serum factors such as LPA signal by means of GPCRs to activate guanine-nucleotide exchange factors (GEFs) for RhoA. Growth factors (platelet-derived growth factor [PDGF], epidermal growth factor [EGF]) act also, transmitting stimulatory and inhibitory signals to RhoA. Integrins transmit positive and negative signals to RhoA. Initial integrin-substrate interactions decrease RhoGTP but activate p190RhoGAP through cSrc. As the cell spreads, integrins activate RhoA. Downstream of RhoA, p160ROCK stimulates actin-myosin contraction and increased integrin clustering. Adapted with permission from Sastry and Burridge, 2000.
In addition to these classic receptor classes, cells can respond to their extracellular environment through integrin receptors or proteoglycan receptors such as the syndecan family (Figure 1). Single transmembrane domains with large extracellular and much smaller cytoplasmic domains characterize these receptors. The syndecan molecules have long glycosaminoglycan chains that assist in sequestering the fibroblast growth factors close to the cell membrane (Couchman and Woods, 1999; Richardson et al., 1999). The integrin receptors are heterodimers composed of α and β subunits. The family is very large with at least 25 integrin heterodimers, including 19 α subunits and 8 β subunits identified (Humphries, 2000). The integrins do not have kinase activity, but upon binding to ECM molecules, some associated proteins become activated by autophosphorylation and then phosphorylate surrounding proteins to generate signals for specific cellular activities that usually change the actin cytoskeleton. One of the first proteins to become phosphorylated is focal adhesion kinase (FAK).
Another concept in recent literature is that different signaling molecules may be sequestered in membrane microdomains, termed lipid rafts (Chapman et al., 1999; Giancotti and Ruoslahti, 1999) that may facilitate crosstalk between different receptors and signaling pathways (Schwartz and Baron, 1999; O’Neill et al., 2000; Ridley, 2000a,b; Sastry and Burridge, 2000).
Intracellular Signaling Proteins
One basic principle of signal transduction is that the proteins exist in at least two states: activated and inactivated. At this time, we know of several methods to turn the signals on and off, including phosphorylation, dephosphorylation, intracellular location, nucleotides (ADP/ATP or GDP/GTP) cycles, and calcium/ion levels. In this section, an overview of different methods of producing intracellular signaling will be briefly explained.
One of the first signal transduction mechanisms described was the GPCR cascade that generates the classic second messengers cyclic AMP (cAMP), cyclic GMP (cGMP), diacylglycerol (DAG), phosphoinositols, and calcium (Ca+2). When G-protein linked or hormone receptors become activated, they trigger a series of events at the cell surface that cause transient increases in these second messenger molecules (Lodish et al., 2000). As with other signaling events, there is a transient increase in the active form of the molecule followed by a rapid decrease to produce a “signal.” Briefly, when the ligand binds to the receptor, it causes a conformational change that allows the G-protein α subunit to bind to the cytoplasmic domain of the receptor (Figure 1). This interaction causes the exchange of GDP for GTP on the α subunit and the disassociation of the βγ subunits from the α subunit. The activated (GTP bound) α subunit interacts with adenylyl cyclase, the membrane-bound enzyme responsible for producing cAMP. After activating adenylyl cyclase, the α subunit reverts to the GDP state and reassociates with the βγ subunits.
Not only is the number of GPCRs very large, but the number of α subunits are also numerous, providing a wide variety of signals to the cell. Once the G proteins are activated, the signal can be amplified by interactions with other proteins as illustrated in Figure 1. Classically, adenylyl cyclase produces cAMP that activates other kinases, termed the cyclic AMP-dependent kinases (cAPKs) also called protein kinase A (PKA). These kinases can phosphorylate several substrates, depending on the specific stimulus (Lodish et al., 2000) to amplify the signal from the cell surface. One effect of activation is the release of calcium from intracellular stores such as the rough endoplasmic reticulum and mitochondria. Because free calcium levels in cells are maintained at very low levels, the rapid increase in calcium levels from these intracellular stores has been a way to visualize signal events. Calcium can be labeled with either quantitative ratiometric dyes or single-wavelength dyes to monitor these rapid changes in cells after stimulation (Nuccitelli, 1994). Rapid sequestering of the free calcium ions by molecules such as calmodulin that bind several calcium ions turns off the calcium signal.
Phosphorylation and dephosphorylation provide another mechanism for signal on and off switches. The enzymes that phosphorylate other proteins are called kinases. The common amino acids that become phosphorylated are tryosine, threonine, and serine. As stated previously, the phosphorylated proteins may be activated by adding phosphate molecules, and deactivated by removing the phosphates, a function usually performed by another enzyme class, the phosphatases. Some proteins can phosphorylate themselves (“autophosphorylation”); once activated, they can phosphorylate surrounding substrates. An example of this type of protein was discussed previously in describing the integrin molecules; focal adhesion kinase (FAK) becomes phosphorylated when integrins bind to their ligand, ECM. Once FAK becomes phosphorylated, it will activate or phosphorylate paxillin, an actin-associated protein, and another kinase, Src (Cary and Guan, 1999). Src can also start activating surrounding proteins, creating an amplification of the original signal. To complicate things, some proteins are inactive in the phosphorylated state and become active after dephosphorylation. Therefore, it is important to understand the possible changes in the proteins before starting an investigation. Cells may contain a constant amount of a given protein in a pool, but the protein has to be in an active state to produce a signal that will change the proteins around it (Svoboda et al., 1999a,b). It is important to remember that, just because an mRNA for a given protein is expressed, it does not indicate that the protein is produced or that it is activated.
Another way that proteins get activated involves binding to a nucleotide such as ATP or GTP. The protein is generally in an inactive state if the ADP or GDP nucleotide is bound and becomes activated when the ATP or GTP is bound. An example of this type of activation is the small G-protein families, Ras and Rho (Bokoch, 2000; Ridley, 2000a; Sastry and Burridge, 2000; Schwartz and Shattil, 2000; Settleman, 2000; Symons and Settleman, 2000). These proteins alternate between the GTP-bound active form (on) and the GDP-bound inactive form (off) to regulate other downstream kinases. Many other proteins regulate the “on” and “off” state of the small G-proteins. The guanine-nucleotide exchange factors (GEFs) are the “on” signal, as they add GTP to the protein. GTP-activating proteins (GAPs) are the “off” signal, as they remove a phosphate to deactivate the protein. Guanine-nucleotide dissociation inhibitor proteins (GDIs) sequester the inactive protein in the cytoplasmic pool. We have shown that one of these regulatory proteins (p190RhoGAP; 190-kDA protein that functions as a GAP for Rho) becomes phosphorylated very quickly in embryonic epithelia in response to cells binding ECM (Svoboda et al., 1999b). We have also shown that decreasing Rho protein levels or activity decreased other integrin signaling molecules.
Another way to determine whether a protein is activated is to determine its intracellular location. Some proteins move to specific intracellular structures, such as focal adhesions (Sastry and Burridge, 2000; Turner, 2000) when they become activated. Paxillin, α-actinin, and talin accumulate at the focal adhesions in both migratory and stationary cells. In addition, integrin molecules become clustered at the focal adhesion of fibroblast cells in culture.
Many proteins move to the plasma membrane when they become activated. The movement to the plasma membrane may take several steps, including release from a cytoplasmic chaperone protein, acquiring a lipid tail, or both. The small G proteins (such as Rho) require both the release from the GDI protein (Read et al., 2000; Michaelson et al., 2001) and a lipid tail to move to the membrane. So, in addition to having a GTP bound to the protein, the protein itself moves to the site of action. Many of the small GTPase proteins (Ras, Raf, Rac) follow similar intracellular translocation patterns upon activation.
Other activated proteins move to the nucleus and usually act as transcription factors. Examples of this type of intracellular translocation are some of the MAP kinase proteins (Kortenjann and Shaw, 1995; Treisman, 1996; Roovers and Assoian, 2000). MAP kinases can respond to a variety of extracellular signals, including osmotic stress, heat shock, cytokines, and mitogens (Garrington and Johnson, 1999). Two of the MAP kinases, the extracellular signal-regulated kinases (erk-1 and erk-2, also referred to as erk-1/2), translocate to the nucleus after activation (Figure 2) to regulate the expression of various transcription factors (c-myc, c-fos, and c-jun, tcf, srf, elk-1, apl, atfn) (Kortenjann and Shaw, 1995; Garrington and Johnson, 1999). Activation of the MAP kinase pathways has been identified as a mechanism used by integrins to regulate gene expression leading to cell shape changes during cell spreading or migration (Robinson and Cobb, 1997; Schwartz and Baron, 1999; Schwartz and Shattil, 2000), and as a cross-talk pathway between integrins and growth factors (Schwartz and Baron, 1999; Sastry and Burridge, 2000).
Figure 2.

Fibroblast cells cultured on plastic substrate, serum starved for 24 h, and then stimulated with fibronectin (FN). A: Anti-phosphotyrosine Western blot from cells (0) 30 s, 2, 5, 10, 15, and 60 min after FN stimulation in the MAP kinase molecular weight region and double-labeled immunohistochemistry for erk 1 (green) and anti phosphotyrosine (red). B: In unstimulated cells, the erk distribution appeared diffuse throughout the cells. C: Whereas after 15 min, many proteins are phosphorylated (red) and the erk appears to be moving more centrally with some double-labeled proteins (yellow) moving toward the nucleus. D: At 45 min, the nuclei clearly contain both erk and phosphotyrosine (yellow nuclei).
Sometimes the signal protein needs to bind another protein, a chaperone, before translocating to the nucleus. Transforming growth factor beta (TGF-β) proteins activate a class of proteins called Smads. These growth factors bind to cell surface receptors (TβRI and TβRII) and activate specific Smad proteins (Smad 2 or 3) that then bind to a chaperone protein, Smad 4 before translocating to the nucleus to act as transcription factors (Giorgio and Hemmati-Brivanlou, 1999; Massague, 2000; Schiffer et al., 2000; Zimmerman and Padgett, 2000). Similarly, bone morphogenetic proteins (BMPs) bind to BMP receptors and activate Smads 1 or 5, which bind Smad 4 to translocate to the nucleus.
In summary, intracellular responses to cell surface receptors are complicated and poorly understood. Several studies have established reciprocal linkages between ECM-integrins, growth factor signaling, cell–cell adhesion molecules, specialized membrane domains (lipid rafts) and G-protein–linked receptors (Sastry and Burridge, 2000). In addition, cross-talk has been established between the ECM and intracellular mitogen stimulated pathways, the small G-proteins, and the phosphoinositols (Porter and Hogg, 1998; Ossowski and Aguirre-Ghiso, 2000). The cell’s microenvironment and the resulting tissue profoundly influence each of these linkages. Thus, for a cell to achieve a differentiated phenotype or respond to microenvironment changes, the ECM molecules and their receptors must integrate both form and function. In contrast, mutated genes and aberrant interactions with the microenvironment may degrade this integration, possibly resulting in malignant transformation or abnormal development (Boudreau and Bissell, 1998). Recently, it has also become apparent that integrins regulate Rho GTPases and vice versa. Integrins and GTPases, therefore, might be organized into complex signaling cascades that regulate cell behavior (Schwartz and Baron, 1999; Sastry and Burridge, 2000; Schwartz and Shattil, 2000).
CELL BIOLOGIC ANALYSIS OF SIGNAL TRANSDUCTION
In the following sections, some specific methods that have been used to establish the signaling mechanisms will be explored.
Blocking Specific Kinases With Inhibitors
Signal transduction proteins have been the targets of many pharmacologic agents over the past 20 years. Many inhibitors are available for both generalized and specific interactions with either receptors or specific kinases; we have provided a short inhibitor list as an example of the variety available (Table 1). The reader is cautioned to be extremely careful when using the inhibitors to include carrier controls and a titration analysis for each inhibitor on their system. Many inhibitors may be specific for one kinase at low doses, but as the dose increases, they may affect a wider class of molecules. Many act as competitive inhibitors for ATP binding sites and are reversible; therefore, a constant level of inhibitor is required throughout the experimental time both before and after cellular stimulation. The assessment of inhibitor effects should include morphologic, biochemical, and behavioral analysis as described here.
TABLE 1.
KINASE AND EGFR INHIBITORS
| Kinase | Inhibitor | Action |
|---|---|---|
| MAP Kinase | ||
| MEK/ERK | PD98059 | Competitive inhibition of ATP binding site |
| Olomoucine | Competitive inhibition of ATP binding site | |
| MEK1 | PD184352 | Directly affects MEK1 |
| ERK2 | 5-Iodotubercidin | Competitive inhibition of ATP binding site |
| c-Raf | ZM336372 | Competitive inhibition of ATP binding site |
| MEK1/MEK2 | U0126 | Non-competitive with respect to MEK substrate, ERK, |
| p38 MAPK | SB202190 | Potent, cell-permeable; inhibits phosphorylation of myelin basic protein no effect ERK or JNK |
| PI3 Kinase | LY294002 | Competitive inhibition of ATP binding site |
| Wortmannin | Competitive inhibition of ATP binding site; irreversible inhibitor, induces conformational change in catalytic domain | |
| FAK | echistatin | |
| Src | PP2 | Potent and selective inhibitor for SRC family; inhibits activation of FAK and its phosphorylation at Tyr577 |
| Herbimycin A | Potent and cell-permeable inhibitor of SRC family | |
| Rho | C3 Exoenzyme | ADP ribosylation of all Rho protein |
| p160 ROCK | Y27632 | Tyrosine phosphorylation inhibition |
| MLCK | ML-7 | Cell-permeable and highly selective inhibitor |
| ML-9 | Cell-permeable and highly selective inhibitor | |
| A3 Hydrochloride | Alkyl chain derivative | |
| EGF-Receptor | AG1478 | Selective inhibitor abolishes ERK activation |
| Compound 56 | Most potent and specific inhibitor of tyrosine kinase activity | |
| PD174265 | Reversible, potent, and cell-permeable inhibitor of EGF-R | |
| FGF-Receptor | SU5402 | Inhibits FGF-R kinase activity, also inhibits aFGF induced tyrosine phosphorylation of ERK1 & 2-does NOT inhibit EGF-R kinase activity |
Cell Behavior Assays
To study the signal transduction events in cells, cell behavior assays need to be developed that can be quick, easy, and preferably used on living tissues and cells. These assays may determine the differences in cytoskeletal elements, cell shape changes, matrix binding, and migration or differentiation characteristics. Attachment, spreading, and migration assays were developed over 25 years ago to determine whether cells reacted differently to alternate substrates. These assays are now being used to determine whether down- or up-regulating specific signal transduction proteins change the cellular behavior (Berrier et al., 2000).
Signal transduction proteins are modulated by specific inhibitors to intracellular kinases or cell surface receptors. A representative sample of some inhibitors and how they interfere with protein function is shown in Table 1; however, many more inhibitors have been developed, as these proteins may be pharmacologic targets. Another way to change the activity of specific proteins is by transfecting or microinjecting cells with plasmids that express either inactive or constitutively active proteins (Berrier et al., 2000). Alternately, blocking synthesis with antisense oligonucleotides specific for the mRNA of a selected protein can down-regulate specific proteins.
We routinely use two assays in our corneal epithelial research project: actin reorganization and ECM binding (Svoboda et al., 1999b). Corneal epithelia respond to ECM molecules by reorganizing the basal actin cytoskeleton into long actin bundles (Figure 4), termed the actin cortical mat (ACM). Once the cellular behavior was established, then we selectively decreased the activity of individual or groups of proteins (by using specific inhibitors or antisense oligonucleotides) to establish whether those proteins are necessary for the actin to reorganize. Our results show that many signal transduction pathways contribute to this cytoskeletal rearrangement (Figure 3).
Figure 4.

Single optical sections through the basal region of epithelial tissues that were isolated without the basal lamina, then cultured in control media (B) or type I collagen (COL, D). The epithelia were fixed and stained with fluorescein isothiocyanate–phalloidin then viewed on the Leica CLSM. Single optical sections were obtained from the optical plane indicated by the arrows in the drawings of the corneal epithelial tissue (A,C). Individual basal cells were approximately 8 –10 μm in diameter. Phalloidin also labels the F-actin associated with lateral cell membranes but not nuclei. Actin staining through the cytoplasmic blebs appears punctate in the en face optical section (B, arrowheads). D: Epithelium cultured in the presence of COL for 2 h have extensive actin bundles that line up from cell to cell (arrowheads) across the 100-μm field. A,C: Schematic drawings. Adapted with permission from (Svoboda et al., 1999b). Scale bar = 10 μm.
Figure 3.

Schematic drawing of our proposed signaling hypothesis. The stages of epithelial response to extracellular matrix (ECM) may be divided into four parts: (1) first contact, (2) signal amplification, (3) actin polymerization and contraction, (4) integrin clustering leading to increased ECM binding and further signal amplification. RhoGTPases increase actin organization and contraction through several mechanisms as indicated in the diagram. Adapted with permission from (Chu et al., 2000).
The ECM-stimulated changes in actin cytoskeleton organization have been well documented in this tissue by using transmission electron microscopy (TEM) (Sugrue and Hay, 1981, 1986; Svoboda and Hay, 1987) and confocal microscopy (Svoboda, 1992; Svoboda et al., 1999b; Chu et al., 2000). In addition to the time saved by using a fluorescent phalloidin staining method compared with TEM, we found that different ECM molecules stimulated distinct actin bundle configurations (Svoboda et al., 1999b). Also, we demonstrated that this tissue reorganizes the actin cytoskeleton in response to bombesin or lysophosphatidic acid (LPA) (Svoboda et al., 1999b), which bind to the classic 7 membrane spanning G-protein–type receptors. The equipment needed to morphologically assess the actin cytoskeleton includes a microscope, either a TEM or a confocal microscope. As this tissue is between 20 and 30 microns thick, we need to either physically section the material for TEM or optically section the tissue with confocal microscopy. The actin was stained with fluorescently tagged phalloidin for the confocal microscope, whereas traditional TEM fixation and staining were used to determine the organization of the actin by using TEM analysis.
The advantages of confocal microscopy include time and field size. The phalloidin staining takes approximately 30 min, allowing tissues to be viewed within an hour of experimental treatment. The tissues are scanned en face with field sizes as large as 300 square microns. In contrast, the fixation, embedding, sectioning, and staining preparation for TEM takes a minimum of 3 days and often may take up to a week between the end of the experiment and morphologic analysis. In addition, even at low-power observation, only very small fields of cells can be observed. Many times while viewing TEM samples, we had to search for actin bundles, as the specific section may not have included the bundle. Furthermore, fine actin bundles were obscured from view by the embedding medium, plastic.
The second morphologic assay we use, ECM binding to the basal epithelial surface, also depends on confocal microscopy for analysis. In this assay, we fluorescently label ECM molecules (collagen or fibronectin), or more recently, we have purchased labeled type IV collagen or gelatin (Molecular Probes, Inc.) or collagen-coated beads. The labeled ECM molecules are incubated with the tissues for designated times. The tissue is then rinsed, stained with phalloidin, and viewed with the confocal microscope (Figure 5). The binding of the ECM over a temporal sequence is established, then used as an assay to determine whether selective inhibitors block ECM binding in a specified time (Chu et al., 2000).
Figure 5.

Corneal epithelia were isolated without the basal lamina, incubated with media containing fluorescein isothiocyanate–fibronectin (FITC-FN) for a short time intervals (5 to 30 min). The epithelia were rinsed, fixed, and viewed on the confocal microscope in the xz optical plane. The intensity of bound FITC-FN was recorded at the basal cell surface (intensity wedge). All settings (pinhole, laser power, voltage, and offset) were the same for all epithelia. Average relative intensity measurements were obtained for each epithelial group (n = 3). The FITC-FN increased from 5 to 30 min as analyzed with NIH Image (graph). Adapted with permission from (Svoboda et al., 1999b).
Other cell behavior changes that can be followed are cell attachment, shape change, migration, proliferation, or survival (Wayner et al., 1991; Hanks et al., 1992; Chen et al., 1994; Mooney et al., 1995; Meredith and Schwartz, 1997; Frisch, 1999). For example, numerous researchers use the in vitro wound-healing model in which the cells are grown to confluence, and then a scrape or wound is placed in the culture dish (Nobes and Hall, 1999; Song et al., 2000). The cells are observed moving over the wound surface under various conditions to determine what proteins are necessary for the cell migration to close the wound. This method is a very easy assay and has the further advantage of living cell observations.
Visualizing Signal Transduction Events
Traditionally, signal transduction events, such as calcium or pH changes, were tracked with intracellular fluorescent indicators. Translocation of specific proteins was tracked with immunohistochemistry (Figure 6), caged proteins, or fluorescent markers for certain plasma membrane lipids. More recently, movement of specific proteins has been tracked by incorporating a fluorescent protein gene, green fluorescent protein (GFP), into genetic vectors encoding the protein to be studied.
Figure 6.

Fibroblast cells cultured on plastic substrate were serum starved for 24 h and then stimulated with fibronectin (FN). The cells were either harvested for Western blot analysis at specific time points, (0, 30 s, 2, 5, 10, 15, and 60 min) or prepared for double-label immunohistochemistry at three time points: 30 s (A,D), 10 min (B,E), or 45 min (C,F). The cells were immunolabeled for p190RhoGAP or FAK (green) and phosphotyrosine (red) and analyzed with confocal microscopy. The images are single optical sections through the basal region of the cells. The yellow areas show the overlap in signal from p190 RhoGAP or FAK and ptyr, indicating the intracellular location of phosphorylated target proteins. In unstimulated cells, the p190RhoGAP and FAK distribution appeared diffuse throughout the cells. Many proteins were phosphorylated (red or yellow) in both samples after 10 min of FN stimulation (B,E). P190RhoGAP appeared to accumulate in the cytoplasm, whereas FAK was located in cell attachment areas. The phosphorylated proteins decreased in the cells after 45 min (C,F).
In this section, the general principles of how these different approaches are used to visualize signal transduction events will be described. However, many articles, reviews, and books that explain the details of each approach have been published. In addition, many companies are developing products designed to optimize visualizing cell biological events with both wide field and confocal microscopy. All of these techniques require high-resolution light microscopes with light sources that can excite fluorescent probes. Most also require digital recordings with additional computer analysis.
Several short courses are available to either introduce the novice or experienced morphologist to these techniques. A few Web sites for further information include the following: The Marine Biological Laboratory has two courses (http://courses.mbl.edu/Courses/about.htm), the W.M. Keck Center for Cellular Imaging also has workshops (http://www.cci.virginia.edu/workshop_fret.html), and the Image Facility at San Antonio has a yearly course (http://www.uthscsa.edu/csb/image/facility.html). In addition to these courses that take from 3–7 days, many national meetings have short workshop courses. The American Association of Anatomists has sponsored imaging workshops at the national meeting for the last 5 years (http://www.anatomy.org).
Calcium and pH
Changes in calcium or pH levels can be accomplished by loading the cells with ratiometric or single-wavelength dyes. These fluorescent probes that show a spectral response upon binding Ca2+ that has enabled researchers to investigate changes in intracellular free Ca2+ concentrations by using fluorescence microscopy, flow cytometry, and fluorescence spectroscopy. Most of these fluorescent indicators are derivatives of the Ca2+ chelators EGTA, APTRA, and BAPTA that were developed by Tsien and his colleagues (Tsien, 1980) and, more recently, through scientists at Molecular Probes, Inc. (Haugland and Johnson, 1999) (http://www.probes.com/handbook).
The common ratiometric dyes for Ca2+ are fura 2 and indo-1. These dyes change wavelength in the presence of different concentrations of ions. Single wavelength dyes include fluo-3, rhod-2, calcium green, calcium orange, calcium crimson, and fura red (Nuccitelli, 1994; Haugland and Johnson, 1999). These compounds require a fluorescent microscope with the ability to record relatively fast changes in emission wavelengths by using enhanced video, charged coupled device (CCD) cameras or very fast confocal microscopes. Many articles and even books have been published on this subject; readers are directed to these publications for more details (Nuccitelli, 1994).
Immunohistochemistry
Immunohistochemistry can also be used to locate signaling proteins. The location itself may indicate whether the protein is activated, but two additional approaches may confirm the state of the target protein. Some vendors are producing antibodies to the signal proteins in their activated state. These antibodies have recognition epitopes that include the phosphate or other activating conformation. Antibodies against active epitopes (anti-active) are becoming available for many common signal transduction proteins. If the anti-active antibody is not available for the protein, then an alternate approach is to double label the cells with an antibody specific for the target protein and another antibody that recognizes all serine, threonine, or tyrosine phosphorylated amino acids (Figure 6). In this approach, the overlap of the two signals may indicate a site where the active protein resides, whereas the single-labeled protein is not activated. This method is even more informative if combined with biochemical evidence (Western blots) that the proteins are becoming phosphorylated in the same time frame as the translocation of the protein (Figure 6). However, the protein colocalization is not conclusive, as the resolution of the confocal microscope is approximately 0.18 microns.
Constructed fusion proteins
Living cells can also be observed with GFP-tagged proteins. Vectors with GFP fused to the protein of interest are transfected into cells, tissues, and transgenic animals to track the expression patterns of the protein. Currently, GFPs have been modified to produce multiple emission wavelengths, such as cyanFP (CFP), yellowFP (YFP), and DsRed (Ayoob et al., 2000; Lansford et al., 2001) so that they can be used in combination to label multiple proteins in the same cell, tissue, or animal (Hakamata et al., 2001).
Transfection with GFP-tagged fusion proteins is a particularly powerful method if the GFP-tagged protein changes cellular location after activation as discussed previously (Zamir et al., 2000; Lansford et al., 2001; Li et al., 2001). A recent study used this method to determine the domain of protein–protein interactions between Rho and RhoGDI proteins by mutating specific amino acids in the GFP-tagged constructs (Michaelson et al., 2001). This approach requires expertise in expression vector plasmid construction, a cell type that can be transfected with the constructs, and a fluorescent microscope to view the cells. Some GFP-constructs recently have become commercially available, so in the future, a laboratory set up for tissue culture and morphology may be able to simply purchase the molecular biology component.
A new twist is the recent report of producing a kinase substrate constructed with a GFP reporter. A fragment of myelin basic protein that contains a single consensus ERK/MAP kinase phosphorylation motif (PRTP, where the threonine is phosphorylated) was fused to GFP. The fused protein transfected into mammalian cells acted as kinase substrate. The GFP-MBP fused protein became phosphorylated after serum stimulation; furthermore, a specific MEK inhibitor blocked this change in phosphorylation (Mandell and Gocan, 2001).
FRET and FRAP
Two imaging techniques that were developed in the 1970s and 1980s recently have re-emerged as useful methods in combination with the GFP fusion protein constructs. Fluorescence resonance energy transfer (FRET) (Matyus, 1992) and fluorescence recovery/redistribution after photobleaching (FRAP) were first developed for wide field epifluorescent microscopes and detected with CCD cameras (Matyus, 1992; Kenworthy, 2001). FRET is the nonradioactive transfer of energy from an excited state donor to a nearby acceptor (Figure 7). The donor in this case is a fluorescent molecule that is excited by a specific wavelength of light; the donor emits a higher light wavelength that excites the fluorescent acceptor molecule. Many fluorescent molecules can be used as donor/acceptor pairs, and many are explained in the Molecular Probes Web site (http://www.probes.com/handbook). Donor/acceptor pair examples are Cy3 and Cy5 or CFP and YFP. The energy transfer is dependent on the distance between the fluorescent molecules. As the acceptor absorbs the donor fluorescence, the donor will quench and its lifetime will decrease. Therefore, several parameters may be measured to determine that energy is actually being transferred including (1) increase in acceptor fluorescence, (2) decrease in donor fluorescence, (3) increase in donor fluorescence after the acceptor is photobleached.
Figure 7.
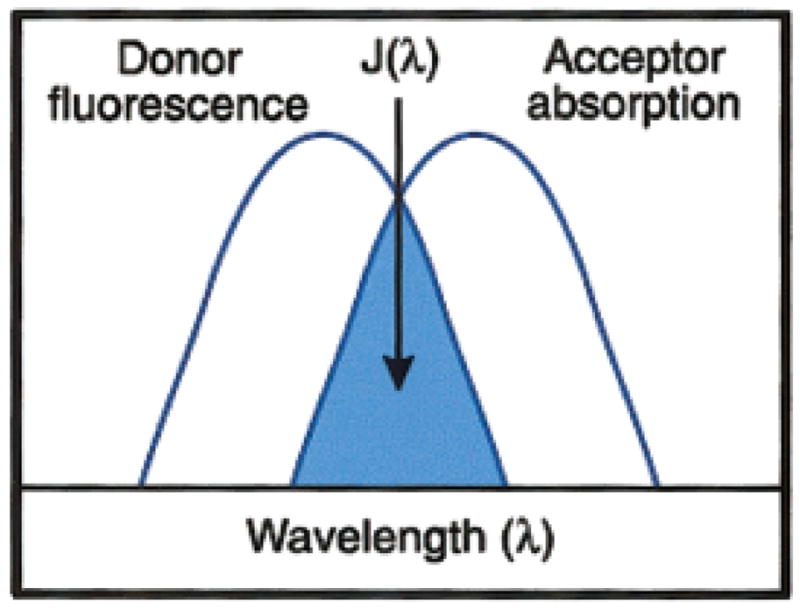
The schematic representation of the fluorescence resonance energy transfer (FRET) spectral overlap integral. Copied with permission from The Handbook of Fluorescent Probes and Research Products by Molecular Probes, Inc. (http://www.probes.com/handbook/). [Color figure can be viewed in the online issue, which is available at www.inter-science.wiley.com.]
FRET is a very powerful light microscopic technique, as it will allow the investigator to determine whether two proteins are within 10 –70 angstroms of each other rather than colocalized with confocal microscopy (200 nm apart). The probes for FRET can be either directly labeled proteins or fusion proteins that contain GFP derivatives (see Box 2 for a specific example) (Sorkin et al., 2000; Mochizuki et al., 2001), directly labeled primary antibodies or secondary antibodies (Fab fragments) (Kam et al., 1995; Dictenberg et al., 1998; Kenworthy, 2001), or caged molecules. Again, several good resources are available to aid the investigator with detailed methods and protocols (Sullivan and Kay, 1999; Kenworthy, 2001).
BOX 2. Examples of FRET Technology in Practice.
Two different groups have recently used this FRET technology to directly study signal transduction in living cells. One group (Sorkin et al., 2000) analyzed the spatial and temporal regulation of epidermal growth factor receptor (EGFR) interactions with the SH2 domain from the adaptor protein Grb2 in living cells. To achieve this goal, they produced three fused proteins. They constructed the growth factor receptor (GFR) fused to CFP in the cytoplasmic domain of the receptor. In addition, they produced two constructs of cytoplasmic proteins fused to YFP. The cells were stably transfected with these fused proteins, then stimulated with the growth factor. Stimulation by the growth factor resulted in the recruitment of a YFP-labeled cytoplasmic adapter protein to cellular compartments that contained GFR-CFP as well as a large increase in the FRET signal. In particular, FRET measurements indicated that activated GFR-CFP interacted with YFP-labeled adapter protein in membrane ruffles and endosomes (Sorkin et al., 2000). This is the first report of the cytoplasmic domain of a receptor directly interacting with adaptor and amplifying proteins.
The second group (Mochizuki et al., 2001) used the combination of CFP and YFP to study the spatiotemporal images of growth factor-induced activation of Ras and Rap1. They made numerous fused protein constructs consisting of the Ras-binding domain of Raf (RafRBS) and a pair of YFP and CFP constructs so that intramolecular binding of GTP-Ras to the fused protein would bring the CFP close enough to the YFP to produce energy transfer. This construct was different as both the donor and acceptor were on the same molecule, so that activation of the signal depended on a conformational change in the protein that occurs when the protein binds to a specific activated G protein. The experiments and controls showed that growth factors activated Ras at the peripheral plasma membrane and Rap1 at the intracellular perinuclear region of COS-1 cells. In PC12 cells, nerve growth factor-induced activation of Ras was initiated at the plasma membrane and transmitted to the whole cell body. By using the FRAP technique, they demonstrated that Ras in neurites turned over rapidly; therefore, the sustained Ras activity in neurites was due to high GTP/GDP exchange rate and/or low GTPase activity, but not to the retention of the active Ras. This is a very powerful and innovative use of this technology to establish the relationship between growth factor stimulation and the intracellular pathways stimulated upon activation of the receptors.
FRAP is based on the common observation that if a region of tissue or cells is exposed to excitation fluorescent wavelength light for an extended period of time, the area will lose its fluorescence or “bleach.” The cells are preloaded with the fluorescent molecule of interest, then a specific region is photobleached and the movement of the fluorescent molecules back into the bleached area (recovery) is measured as described in Box 2 experiment on PC12 cells (Mochizuki et al., 2001). This method allows the investigator to determine the mobility and diffusion of small molecules in the cytoplasm of living cells or record the movement of macromolecules (RNA or large proteins, drugs) into and out of cell organelles such as the nucleus. As these movements may be rapid, it is necessary to capture images in “real time” with CCD cameras or use small areas and special procedures with confocal microscopy. It is important to record the prebleach fluorescent levels, photobleach a selected small area, record the recovery at low light levels to prevent further bleaching, then quantify the time course of recovery for complete analysis (Phair and Misteli, 2000).
Both FRET and FRAP require that the protein/organelle to be studied is fluorescently labeled in either fixed preparations or in living cells. Loading the plasmids, caged molecules, or other markers presents a problem that has been overcome by a variety of methods, including scrape loading, lipid carriers, osmotic shock, or electroporation. All methods require careful examination and analysis of the cells combined with controls for the specific method. For example, it was reported recently that the carriers for lipid transfer might also produce non-specific reflectance or autofluorescence (Guo et al., 2001). In addition, the FRET experiments will require many controls to establish that the change in fluorescence of the acceptor molecule is specific (Sorkin et al., 2000; Mochizuki et al., 2001; Kenworthy, 2001).
In summary, the equipment and reagents needed to viewing signal transduction events are light microscopes with recording devices and software that can calculate and display ratio imaging wavelength emission levels or changes in emission over time. The indicators for some cellular changes, including calcium and pH, can be purchased as esters that easily cross cell membranes. These indicators are sensitive to temperature and oxygen changes and may need special temperature controlled environments for optimum data collection. They may also require special microscope accessories. The GFP-labeled proteins require the transfer of the DNA into the cell and not only successful expression but also that the proteins are in the correct conformation to interact with other proteins and cellular elements. Immunohistochemistry requires that the cells be fixed before labeling, resulting in the disadvantage that the cells are dead. Thus even a temporal sequence may not tell the whole story. New anti-active antibodies have made it possible to locate only the activated forms of some signal proteins. However, double-labeling experiments will allow the visualization of additional proteins that anti-active antibodies are not yet available. It is important to note that not all antibodies work for immunohistochemistry as some have been designed primarily for biochemical analysis by means of Western blots or immunoprecipitation.
BIOCHEMICAL ANALYSIS OF SIGNAL TRANSDUCTION
As we were primarily trained in morphologic techniques, it was relatively easy to add biochemical and molecular biological experiments to our projects. Basically, if you can cook, then you can do biochemistry, as it generally involves following previously identified protocols and recipes. Many of the assays and procedures are being packaged into kits with step-by-step instructions, opening up the procedures to a wider audience. The basic procedures will be briefly described; however, it is recommended that a good laboratory protocol manual be consulted for further details and instructions (e.g., the Current Protocols series; www.currentprotocols.com).
Western Blots
Traditionally, signal transduction events have been described by using biochemical analysis. For example, all tyrosine-phosphorylated proteins can be detected with an anti-phosphotyrosine antibody on a Western blot of cell lysates obtained after stimulation in a temporal sequence (Figure 8A). Alternatively, antibodies specific for other phosphorylated amino acids (serine or threonine) could also be analyzed. The cells were harvested at specific times and placed into a lysis or SDS sample buffer that contains protease and phosphatase inhibitors. The proteins were separated on a mini gel electrophoresis apparatus (Bio-Rad) and then transferred to a membrane (Millipore). Once the proteins are on the membrane, detecting the bound proteins is very similar to immunohistochemistry. After appropriate washing and blocking steps, the membranes were incubated with primary antibodies (in this case, an antibody that recognizes all phosphorylated tyrosine residues). The membrane was washed and incubated with a secondary antibody conjugated to horseradish peroxidase. The labeled antibodies were detected with chemiluminescent exposure to an x-ray film (Figure 8) or an imaging camera (Kodak Imaging Station). The advantage of this type of blot is that it indicates all of the proteins that may be tyrosine phosphorylated and can document how individual proteins may increase or decrease in signal. This blot does not identify the specific proteins.
Figure 8.
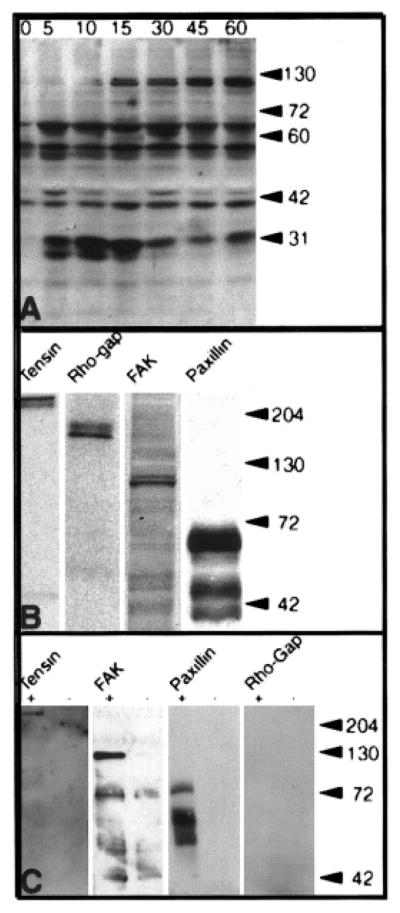
Several proteins were phosphorylated on tyrosine residues after epithelia were incubated in collagen (COL) for 15 min. A: Corneal epithelia (n = 20 –30/group) were isolated without basal lamina and incubated with medium containing COL for various time intervals (0 –60 min). The epithelia were removed from culture and placed directly into Western blot sample buffer. Tyrosine phosphorylated proteins were detected with antibodies to phosphotyrosine (PY-20). Several proteins (molecular mass 125, 60 –70, and 40 –45 and 30 kDa) appeared to increase or decrease phosphorylation with time. The phosphoproteins were identified by several methods: reprobing the same phosphotyrosine blot, separate parallel Western blots B, and immunoprecipitation C. B: Samples of the individual Westerns for each protein demonstrate that a >220-kDa protein was identified as tensin, a 190-kDa protein was p190 Rho-GAP, a 125- to 135-kDa protein was focal adhesion kinase (FAK), and a 60- to 70-kDa protein was paxillin. C: All phosphoproteins were immunoprecipitated from epithelia isolated without basal lamina with PY20 after COL stimulation for 1 h (+) compared with controls (−). The proteins were separated on 7.5% SDS-PAGE gels, and analyzed for tensin, p190RhoGAP, FAK and paxillin (PAX). Tensin, FAK and paxillin co-immunoprecipitated with PY-20, however, p190RhoGAP did not, indicating that the epitope was masked or the protein was not phosphorylated. Adapted with permission from (Svoboda et al., 1999b).
To identify the individual proteins, either this same blot can be stripped and reprobed with another primary antibody (Figure 8B), or a sister blot can be probed with the specific antibody. Unfortunately, those sequential Western blots may not be conclusive; therefore, the current recommended procedure is to either obtain an antibody that only recognizes the activated protein (Figure 9) or immunoprecipitate all tyrosine phosphorylated proteins, and then do a Western blot for the target protein (Figure 8C). Alternatively, the target protein can be immunoprecipitated then all tyrosine-phosphorylated proteins determined with a Western blot (Figure 8D). This approach has the advantage that, if the target protein is bound to other proteins in a complex, multiple proteins and their interactions may be identified.
Figure 9.

Western blot analysis of epithelial extracts cultured with or without collagen (COL) (no treatment, NT, 2 h) to detect MAP kinase (erk-1 and erk-2) activity (A), total erk-2 (B), total MEK-2 (C), with or without pretreatment (24 h) with the MEK inhibitor, PD98059 (10 μM, 50 μM, 100 μM). All inhibitor-treatment groups were COL stimulated for 2 h without inhibitor. A431 fibroblasts served as the positive tissue control. Activated erk-1 or erk-2 (denoted by asterisks) were detected with an antibody that only binds to phosphorylated erk-1 and 2 (A). Phosphorylated erk-1 (44 kDa) and erk-2 (42 kDa) increased over time with COL stimulation in control and COL-stimulated epithelia (A). Pretreating the epithelia with the inhibitor did not appear to effect erk-2, but erk-1 appeared to decrease when compared with collagen-treated controls. Total erk-2 and MEK were determined by reprobing the same membrane (B,C). The 42-kDa protein was erk-2, whereas the 44-kDa protein was MEK-2 (C). The total protein levels of erk-2 and MEK-2 appeared constant in control and COL-stimulated epithelia (C). Adapted with permission from Chu et al., 2000.
The equipment needed for Western blots includes the electrophoresis apparatus and transfer equipment. We recommend getting a mini gel, as it is more economical, uses fewer reagents, and requires less cellular protein. We also purchase the prepoured gels in the appropriate concentration of acrylamide. Although it is more economical to pour the gels, the reagents are toxic and necessary precautions need to be taken. In addition, technical time and consistency from gel to gel are also factors for consideration.
Immunoprecipitation
Immunoprecipitation is relatively easy and requires the same equipment as Western blots. Beads made from a variety of substances, including metals, are purchased with secondary antibodies attached (available from a variety of vendors including Pierce, BioRad, Dynal). In the laboratory, we cross-link primary antibodies to a specific protein or all tyrosine-phosphorylated proteins to the beads. The cells are lysed in buffer containing protease inhibitors and then incubated with the antibody-coated beads. We prefer to use the beads that can be collected with a magnet, as they are more efficient in our experience. The proteins are separated by using SDS electrophoresis, and then the proteins are identified by using the procedures described for Western blots. However, the protein–protein interactions demonstrated by immunoprecipitation and Western blots from cell lysate may not reveal the actual situation in vivo and more experiments are necessary to demonstrate specific interactions.
GST Binding or “Pull-Down” Assays
The glutathione S-transferase (GST) binding or “pull-down” assay is similar to immunoprecipitation, except it determines direct protein–protein interactions. Fragments of proteins or small whole proteins are produced in a bacterial expression system with a GST tag (Cytoskeleton, Upstate Biotechnology). In our experiments, we have purchased GST-labeled proteins such as RhoGDP, RhoGTP, the Rho binding domain of rhotectin (RBD-GST) that selectively bind RhoGTP, and appropriate GST controls. In experiments using RBD-GST, it was demonstrated that the corneal epithelial cells have a biphasic response to collagen (Figure 10). A decrease in RhoGTP after 15 min of collagen stimulation was followed by an increase at 30 min (Figure 10) similar to the response reported by Ren and coworkers in endothelial cells (Ren et al., 1999, 2000).
Figure 10.

Glutathione S-transferase precipitation using Rho binding domain of rhotectin (Upstate Biotechnology). Two concentrations of collagen (COL) were tested (50 and 100 μg/ml) on epithelia isolated without basal lamina and stimulated for 15 or 30 min compared with lysates from tissues incubated without collagen (NT, no treatment). We found that there was a transient decrease in RhoGTP at 15 min, but by 30 min, the tissues treated with 50 μg/ml had a significant increase in RhoGTP.
In another set of experiments, the relationship between Rho and p190Rho-GAP, GST-RhoGDP, and GST-RhoGTP was determined by using affinity precipitations followed by Western blots for p190Rho-GAP (GT-Pase Activating Protein) (Figure 11). Control GST beads precipitated a small amount of p190Rho-GAP (Figure 10). Cell lysates from unstimulated corneal epithelia bound equal amounts of p190Rho-GAP with inactive or active GST-Rho beads. In contrast, in epithelia stimulated with LPA, the GST-RhoGTP beads bound threefold more p190Rho-GAP than the RhoGTP beads mixed with cell lysates from epithelia that were not stimulated with LPA (Figure 11). In addition, an unidentified lower molecular weight protein cross-reacted with the p190Rho-GAP antibody also bound to the GST-RhoGTP beads.
Figure 11.

Western blot analysis of total protein of collagen (COL) -stimulated epithelia over various time points (NT [no treatment], 2, 5, 10, 15, 60, min) to detect FAK (A) and p190RhoGAP (B,C). FAK was present as a single band in control epithelia (NT; A). However, a doublet was detected by 5 min. The molecular weight shift was prominent in FAK by 10 min. After 15 min of COL stimulation, FAK appeared as a doublet of two protein isoforms. By 60 min, most of the detectable protein was present in the phosphorylated form (A). p190RhoGAP had a molecular weight shift in COL (B) and fibronectin (FN) (C) (graph) stimulated epithelia by 5 min and became more prominent by 10 min in the FN-stimulated cells and nearly completely in the higher molecular weight band by 30 min. The COL-stimulated epithelia maintained a doublet through 60 min (B). D: Glutathione S-transferase precipitation of Rho binding proteins (D) by using GST pull-down assays. Cell lysates were added in equal volumes to bovine serum albumin blocked GST beads (10 μl) conjugated to RhoGDP, RhoGTP, or GST controls. Specific proteins were identified with Western blots. Cell lysates from lysophosphatidic acid–stimulated epithelia precipitated more p190 RhoGAP with RhoGTP than either RhoGDP or unstimulated cell lysates with RhoGTP (graph). ECM, extracellular matrix.
The GST fusion proteins can be made with just those protein domains that may interact with other specific proteins. This property of the system makes it a very powerful method to study blots; immunoprecipitation and GST pull-down blots are a common way to compare proteins in different experimental treatment groups. It is difficult to compare blots that were produced on different days, because there are many variables that contribute to the density of the protein bands produced by the procedure; however, it is fair to compare the intensity of different protein bands on the same blot if the gel was loaded with equal amounts of total protein (Figure 11, graphs). To overcome the variability from experiment to experiment, duplicate or triplicate samples from each treatment group could be processed at the same time, or a control group (A4331 cell lysate, Figure 9) can be added as a standard. Many vendors provide a control cell lysate with antibodies as a courtesy or for a nominal cost.
Activity Assays
Another classic way to determine whether a particular kinase is active is to expose the cell lysate to a known substrate for the enzyme in the presence of radioactive phosphate. The products are separated by electrophoresis (with or without immunoprecipitation), then the gel is exposed to x-ray film to determine whether the proteins incorporated the isotope.
Recently, some substrates for enzymes that cleave specific sequences have also been developed to test whether the enzyme is active in living cells. Apoptosis, also known as programmed cell death, causes susceptible cells to undergo a series of enzymatic and morphologic changes. During apoptosis, several signal transduction pathways and specific degradative enzymes become activated. The degradative enzymes are in the caspase family of cysteinyl proteases, also known as ICE-like, because they resemble the first member described, inter-leukin-1 converting enzyme (ICE). These enzymes may cleave essential structural components of the cell, including actin cytoskeletal elements, nuclear lamins, and small nucleoproteins (Whyte, 1996; De Laurenzi and Melino, 2000). These changes eventually lead to nuclear material condensing as the nuclear DNA breaks down and cellular material pinches off, forming apoptotic bodies.
Several companies have developed a class of fluorogenic protease substrates to determine whether the cells contain active caspase-3. One of the caspase-3 substrates (PhiPhiLux, On-coImmunin, Inc.) contains the caspase-3 recognition sequence aspartic acid-glutamic acid-valine-apartic acid (DEVD) in a bifluorophore-derivitized peptide that mimics the structural loop conformation present in the native protease cleavage sites of globular proteins. The compound is not fluorescent unless it is cleaved by endogenous caspase-3. Cells used in an experiment are incubated for the last hour in the presence of the caspase-3 substrate PhiPhiLux then viewed with either confocal or wide-field fluorescent microscopes. Cells that do not contain activated caspase-3 do not have any fluorescence, whereas cells that do have activated caspase-3 will emit light in the wavelength of the substrate. These experiments can be confirmed with a quantitative binding assay for activated caspase-3 (CaspACE Assay, Promega). To test whether the reaction is specific for caspase-3 activity, some tissues can be pretreated with the caspase-3 inhibitor Z-VAD-FMK.
Using a Combination of Western blots, Immunoprecipitation, and GST Pull-Down Assays
Sometimes proteins are particularly difficult to analyze. A protein that we have been investigating, p190RhoGAP cannot be immunoprecipitated with anti-phosphotyrosine (Figure 8C), but we have demonstrated that it is expressed in our model, and a 190-kDa protein becomes activated by tyrosine phosphorylation very quickly after ECM or LPA stimulation (Figure 11). We have used the combination of Western blots demonstrating a molecular weight shift and GST pull-down assays to establish that this protein changes in response to ECM stimulation. The following section describes the data from these experiments in more detail to demonstrate how different experiments provide evidence for protein-protein interactions during signal transduction events.
To understand the timing of this experiment, it is important to know that epithelial sheets require interactions with soluble ECM molecules for a minimum of 15 min for ACM reorganization to occur in 2 h (Svoboda et al., 1999b). In addition, ECM molecules bind to the basal cell surface and immediately initiate tyrosine phosphorylation signaling events as shown by an anti-phosphotyrosine Western blot (Figure 8A). One of the proteins that became phosphorylated very early in response to ECM was p190RhoGAP (Svoboda et al., 1999b). However, attempts to immunoprecipitate the phosphorylated form of this protein failed, because when it became phosphorylated, it bound to other proteins in a complex that masked the phosphorylated tyrosines. To further investigate the role of this protein in the signal cascade, we needed to establish whether p190RhoGAP changed protein levels or molecular weight in response to ECM. Extracts from epithelial sheets cultured in the presence of collagen (COL) or fibronectin (FN) over various times (2–60 min) were analyzed for p190RhoGAP to detect changes in protein levels by using Western blot techniques (Figure 11A–C). The cell extracts were electrophoretically separated on a 7.5% SDS gel overnight (~25 V) to show molecular mass differences in p190RhoGAP. We found that, with increasing exposure to COL, there was a corresponding change in molecular mass in p190Rho-GAP (Figure 11B,C). A high molecular weight shift in p190Rho-GAP from COL-stimulated epithelia was observed as early as 5 min that became more prominent by 30 and decreased by 60 min. The molecular weight shift peaked at 30 min for epithelia treated with FN and COL and could be illustrated by measuring the relative intensity of the higher molecular weight band (Figure 11, graph), which increased in intensity at 30 min in the presence of FN and COL. Comparison of the ratio of the high molecular weight band/lower molecular weight band could also be used to describe the relationship between the two forms of the protein to show that the higher molecular weight form is prominent at 30 min.
To further analyze the relationship between Rho and p190RhoGAP, we performed GST-RhoGDP and GST-RhoGTP affinity precipitations followed by Western blots for p190RhoGAP. These experiments required several treatment groups, including control GST-coated beads, the inactive form of the protein (Rho-GDP), and an active form of the protein (Rho-GTP). In addition, cell lysates from experimental (e.g., lysophosphatidic acid [LPA] treated) and control conditions needed to be compared. The results demonstrated that control GST beads precipitated a small amount of p190Rho-GAP (Figure 11D and densitometry graph). Cell lysates from unstimulated corneal epithelia bound equal amounts of p190Rho-GAP with inactive or active GST-Rho beads. In contrast, in epithelia stimulated with the direct Rho stimulator LPA, the activated GST-Rho beads bound more p190Rho-GAP than the RhoGDP beads. In addition, an unidentified lower molecular weight protein also bound to the GST-RhoGTP beads. This blot was reprobed with an antibody specific for phosphotyrosine, and a small percentage of the p190Rho-GAP protein pulled down by Rho-GTP in the LPA stimulated tissue was tyrosine phosphorylated.
These experiments gave us several pieces of information about the behavior of this protein in our corneal epithelial system. First, p190RhoGAP increased in molecular weight in response to ECM. Second, RhoGTP binds more p190RhoGAP in the presence of LPA than in unstimulated epithelia. Third, some of the phosphorylated p190RhoGAP may bind to RhoGTP. These data provide further evidence that the Rho signal pathway may have a significant role in ECM-and LPA-induced actin reorganization.
Clearly the biochemical assays are very powerful in the analysis of signal transduction events. With these methods, different protein phosphorylation states can be assessed either by using specific anti-active antibodies or combining immunoprecipitation with Western blots. In addition, the GST pull-down assays allow the analysis of specific protein–protein interactions. These approaches provide a means to quantify intracellular changes in response to a specific stimulus. With the addition of specific inhibitors, investigators can determine the sequence of events. However, the weakness of these approaches is that the cells have to be destroyed by homogenization to produce the cell lysates. This requirement necessarily causes the loss of intracellular relationships and the temporal sequence of interactions, as the tissues have to be harvested at finite times. The biochemical analysis requires more tissue and protein than many morphologic approaches. In addition, the laboratory has to be properly equipped with the gel apparatus and transfer equipment. Often, the reagents (antibodies, GST proteins, activity kits, and chemiluminescent materials) are expensive.
CONCLUSION
Future experiments designed to dissect signal transduction pathways and their morphologic effects will require a combination of all of these approaches and surely new ones. The reemergence of FRET and FRAP with custom fusion proteins will revolutionize how signal transduction events are understood, as living cells will be observed. This type of experiment requires the cooperation of cell biologists, molecular biologists, and morphologists to design the experiments, provide the constructs, and visualize the signal transduction events. In the near future, procedures will be perfected to transfect engineered proteins into embryonic tissues, possibly with electroporation, to view signal transduction events in whole tissues.
Acknowledgments
Many students, technicians, and journal club groups have contributed to our understanding of signal transduction events. We thank Dan Orlow and Chia L. Chu for providing Western blots and immunoprecipitation data and Jesus Acevedo, Petra Moessner and Tamara Field for GST pull-down data. We also thank my current signal transduction journal club for critically reading this manuscript. NIH EY08886 has supported this work for 10 years.
Biographies
Dr. Reenstra is the Associate Director of Research for the Department of Emergency Medicine at the Beth Israel Deaconess Medical Center, Harvard Medical School. She is also an instructor while completing her residency in emergency medicine. Dr. Reenstra has focused her research on signal transduction events in dermal fibroblasts and vascular endothelial cells in response to various stimuli, including growth factors and oxygen tension.
Dr. Svoboda is a Professor of Biomedical Sciences and Director of the Cell and Molecular Biology Core Facility at Texas A&M Health Science Center at Baylor College of Dentistry. She also holds an Adjunct Professorship in the Ophthalmology Department of the University of Texas, Southwestern Medical Center. Dr. Svoboda has used several whole tissue developmental models to study cell–matrix interactions, actin reorganization, and signal transduction events.
LITERATURE CITED
- Ayoob J, Shaner N, Sanger J, Sanger J. Expression of green or red fluorescent protein (GFP or DsRed) linked proteins in nonmuscle and muscle cells. Mol Biotechnol. 2000;17:65–71. doi: 10.1385/MB:17:1:65. [DOI] [PubMed] [Google Scholar]
- Berrier AL, Mastrangelo AM, Downward J, Ginsberg M, LaFlamme S. Activated R-Ras, Rac1, PI 3-Kinase and PKC [small element of] can each restore cell spreading inhibited by isolated integrin b1 cytoplasmic domains. J Cell Biol. 2000;151:1549–1560. doi: 10.1083/jcb.151.7.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch GM. Regulation of cell function by Rho family GTPases. Immunol Res. 2000;21:139–148. doi: 10.1385/IR:21:2-3:139. [DOI] [PubMed] [Google Scholar]
- Boudreau N, Bissell MJ. Extracellular matrix signaling: Integration of form and function in normal and malignant cells. Curr Opin Cell Biol. 1998;10:640–646. doi: 10.1016/s0955-0674(98)80040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary LA, Guan JL. Focal adhesion kinase in integrin-mediated signaling. Front Biosci. 1999;4:D102–D113. doi: 10.2741/cary. [DOI] [PubMed] [Google Scholar]
- Chapman HA, Wei Y, Simon DI, Waltz DA. Role of urokinase receptor and caveolin in regulation of integrin signaling. Thromb Haemost. 1999;82:291–297. [PubMed] [Google Scholar]
- Chen Q, Kinch MS, Lin TH, Burridge K, Juliano RL. Integrin-mediated cell adhesion activates mitogen-activated protein kinases. J Biol Chem. 1994;269:26602–26605. [PubMed] [Google Scholar]
- Chu CL, Reenstra RW, Orlow DL, Svoboda KKH. Erk and PI3 kinase are necessary for collagen binding and actin reorganization in avian corneal epithelia. Invest Ophthalmol Vis Sci. 2000;41:3374–3382. [PMC free article] [PubMed] [Google Scholar]
- Couchman JR, Woods A. Syndecan-4 and integrins: Combinatorial signaling in cell adhesion. J Cell Sci. 1999;112:3415–3420. doi: 10.1242/jcs.112.20.3415. [DOI] [PubMed] [Google Scholar]
- De Laurenzi V, Melino G. Apoptosis. The little devil of death. Nature. 2000;406:135–136. doi: 10.1038/35018190. [DOI] [PubMed] [Google Scholar]
- Dictenberg JB, Zimmerman W, Sparks CA, Young A, Vidair C, Zheng Y, Carrington W, Fay FS, Doxsey SJ. Pericentrin and gamma-tubulin form a protein complex and are organized into a novel lattice at the centrosome. J Cell Biol. 1998;141:163–174. doi: 10.1083/jcb.141.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong LT, Lakkakorpi P, Nakamura I, Rodan GA. Integrins and signaling in osteoclast function. Matrix Biol. 2000;19:97–105. doi: 10.1016/s0945-053x(00)00051-2. [DOI] [PubMed] [Google Scholar]
- Frisch SM. Evidence for a function of death-receptor-related, death-domain-containing proteins in anoikis. Curr Biol. 1999;9:1047–1049. doi: 10.1016/s0960-9822(99)80455-2. [DOI] [PubMed] [Google Scholar]
- Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Giorgio L, Hemmati-Brivanlou A. A molecular basis for Smad specificity. Dev Dyn. 1999;214:269–277. doi: 10.1002/(SICI)1097-0177(199903)214:3<269::AID-AJA10>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Amaro R, Sanchez-Madrid F. Cell adhesion molecules: Selectins and integrins. Crit Rev Immunol. 1999;19:389–429. [PubMed] [Google Scholar]
- Guo B, Pearce A, Traulsen K, Rintala A, Lee H. Fluorescence produced by transfection reagents can be confused with green fluorescent proteins in mammalian cells. Biotechniques. 2001;31:314–321. doi: 10.2144/01312st02. [DOI] [PubMed] [Google Scholar]
- Hakamata Y, Tahara K, Uchida H, Sakuma Y, Nakamura M, Kume A, Murakami T, Takahashi M, Takahashi R, Hirabayashi M, Ueda M, Miyoshi I, Kasai N, Kobayashi E. Green fluorescent protein-transgenic rat: A tool for organ transplantation research. Biochem Biophys Res Commun. 2001;286:779–785. doi: 10.1006/bbrc.2001.5452. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Calalb MB, Harper MC, Patel SK. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc Natl Acad Sci U S A. 1992;89:8487–8491. doi: 10.1073/pnas.89.18.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugland RP, Johnson ID. Intracellular ion indicators. In: Mason WT, editor. Fluorescent and luminescent probes for biological activity. Academic Press; Cambridge: 1999. pp. 40–50. [Google Scholar]
- Hirsch M, Chang K, Kao WW-Y, Svoboda KKH. Intracellular distribution of type II collagen mRNA and prolyl 4-hydroxylase in embryonic Avian corneal epithelia. Anat Rec. 1996;244:1–14. doi: 10.1002/(SICI)1097-0185(199601)244:1<1::AID-AR1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Humphries MJ. Integrin structure. Biochem Soc Trans. 2000;28:311–339. [PubMed] [Google Scholar]
- Hunter T. Signaling-2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- Kam Z, Volberg T, Geiger B. Mapping of adherens junction components using microscopic resonance energy transfer imaging. J Cell Sci. 1995;108:1051–1062. doi: 10.1242/jcs.108.3.1051. [DOI] [PubMed] [Google Scholar]
- Kenworthy AK. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods. 2001;24:289–296. doi: 10.1006/meth.2001.1189. [DOI] [PubMed] [Google Scholar]
- Khoory W, Wu E, Svoboda KK. Intracellular relationship between actin and alpha-actinin in a whole corneal epithelial tissue. J Cell Sci. 1993;106:703–717. doi: 10.1242/jcs.106.3.703. [DOI] [PubMed] [Google Scholar]
- Kortenjann M, Shaw PE. The growing family of MAP kinases: Regulation and specificity. Crit Rev Oncog. 1995;6:99–115. [PubMed] [Google Scholar]
- Lansford R, Bearman G, Fraser SE. Resolution of multiple green fluorescent protein color variants and dyes using two-photon microscopy and imaging spectroscopy. J Biomed Optics. 2001;6:311–318. doi: 10.1117/1.1383780. [DOI] [PubMed] [Google Scholar]
- Li HY, Kotaka M, Kostin S, Lee SM, Kok LD, Chan KK, Tsui SK, Schaper J, Zimmermann R, Lee CY, Fung KP, Waye MM. Translocation of a human focal adhesion LIM-only protein, FHL2, during myofibrillogenesis and identification of LIM2 as the principal determinants of FHL2 focal adhesion localization. Cell Motil Cytoskel. 2001;48:11–23. doi: 10.1002/1097-0169(200101)48:1<11::AID-CM2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Molecular cell biology. New York, NY: W.H. Freeman and Co; 2000. [Google Scholar]
- Mandell JW, Gocan NC. A green fluorescent protein kinase substrate allowing detection and localization of intracellular ERK/MAP kinase activity. Anal Biochem. 2001;293:264–268. doi: 10.1006/abio.2001.5138. [DOI] [PubMed] [Google Scholar]
- Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- Matyus L. Fluorescence resonance energy transfer measurements on cell surfaces. A spectroscopic tool for determining protein interactions. J Photochem Photobiol B. 1992;12:323–337. doi: 10.1016/1011-1344(92)85039-w. [DOI] [PubMed] [Google Scholar]
- Mecham RP, editor. Annual Review of Cell Biology. Palo Alto, CA: Annual Reviews Inc; 1991. Laminin receptors. [DOI] [PubMed] [Google Scholar]
- Meredith JE, Schwartz MA. Integrins, adhesion and apoptosis. Trends Cell Biol. 1997;7:146–150. doi: 10.1016/S0962-8924(97)01002-7. [DOI] [PubMed] [Google Scholar]
- Meredith JE, Jr, Winitz S, Lewis JM, Hess S, Ren XD, Renshaw MW, Schwartz MA. The regulation of growth and intracellular signaling by integrins. Endocr Rev. 1996;17:207–220. doi: 10.1210/edrv-17-3-207. [DOI] [PubMed] [Google Scholar]
- Michaelson D, Silletti J, Murphy G, D’Eustachio P, Rush M, Philips MR. Differential localization of Rho GRPases in live cells: Regulation by hypervariable regions and RhoGDI binding. J Cell Biol. 2001;152:111–126. doi: 10.1083/jcb.152.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki N, Yamashita S, Kurokawa K, Ohba Y, Nagai T, Miyawaki A, Matsuda M. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature. 2001;411:1065–1067. doi: 10.1038/35082594. [DOI] [PubMed] [Google Scholar]
- Mooney DJ, Langer R, Ingber DE. Cytoskeletal filament assembly and the control of cell spreading and function by extracellular matrix. J Cell Sci. 1995;108:2311–2320. doi: 10.1242/jcs.108.6.2311. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuccitelli R, editor. A practical guide to the study of calcium in living cells. San Diego: Academic Press; 1994. [Google Scholar]
- O’Neill GM, Fashena SJ, Golemis EA. Integrin signaling: A new Cas(t) of characters enters the stage. Trends Cell Biol. 2000;10:111–119. doi: 10.1016/s0962-8924(99)01714-6. [DOI] [PubMed] [Google Scholar]
- Ossowski L, Aguirre-Ghiso JA. Urokinase receptor and integrin partnership: Coordination of signaling for cell adhesion, migration and growth. Curr Opin Cell Biol. 2000;12:613–620. doi: 10.1016/s0955-0674(00)00140-x. [DOI] [PubMed] [Google Scholar]
- Phair RD, Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature. 2000;404:604–609. doi: 10.1038/35007077. [DOI] [PubMed] [Google Scholar]
- Porter JC, Hogg N. Integrins take partners: Cross-talk between integrins and other membrane receptors. Trends Cell Biol. 1998;8:390–396. doi: 10.1016/s0962-8924(98)01344-0. [DOI] [PubMed] [Google Scholar]
- Read PW, Liu X, Longenecker K, Dipierro CG, Walker LA, Somlyo AV, Somlyo AP, Nakamoto RK. Human RhoA/RhoGDI complex expressed in yeast: GTP exchange is sufficient for translocation of RhoA to liposomes. Protein Sci. 2000;9:376–386. doi: 10.1110/ps.9.2.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X-D, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X-D, Kiosses WB, Sieg DJ, Otey CA, Schlaepfer DD, Schwartz MA. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J Cell Sci. 2000;113:3673–3678. doi: 10.1242/jcs.113.20.3673. [DOI] [PubMed] [Google Scholar]
- Richardson TP, Trinkaus-Randall V, Nugent MA. Regulation of basic fibroblast growth factor binding and activity by cell density and heparan sulfate. J Biol Chem. 1999;274:13534–13540. doi: 10.1074/jbc.274.19.13534. [DOI] [PubMed] [Google Scholar]
- Ridley A. Rho GTPases. Integrating integrin signaling. J Cell Biol. 2000a;150:F107–F109. doi: 10.1083/jcb.150.4.f107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A. What initiates actin polymerization? Genome Biol. 2000b;1 doi: 10.1186/gb-2000-1-1-reviews102. Reviews 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- Roovers K, Assoian RK. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays. 2000;22:818–826. doi: 10.1002/1521-1878(200009)22:9<818::AID-BIES7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Sastry SK, Burridge K. Focal adhesions: A nexus for intracellular signaling and cytoskeletal dynamics. Exp Cell Res. 2000;261:25–36. doi: 10.1006/excr.2000.5043. [DOI] [PubMed] [Google Scholar]
- Schiffer M, von Gersdorff G, Bitzer M, Susztak K, Bottinger EP. Smad proteins and transforming growth factor-beta signaling. Kidney Int Suppl. 2000;77:S45–S52. doi: 10.1046/j.1523-1755.2000.07708.x. [DOI] [PubMed] [Google Scholar]
- Schwartz MA, Baron V. Interactions between mitogenic stimuli, or a thousand and one connections. Curr Opin Cell Biol. 1999;11:197–202. doi: 10.1016/s0955-0674(99)80026-x. [DOI] [PubMed] [Google Scholar]
- Schwartz MA, Shattil SJ. Signaling networks linking integrins and rho family GTPases. Trends Biochem Sci. 2000;25:388–391. doi: 10.1016/s0968-0004(00)01605-4. [DOI] [PubMed] [Google Scholar]
- Settleman J. Getting in shape with Rho. Nat Cell Biol. 2000;2:E7–E9. doi: 10.1038/71390. [DOI] [PubMed] [Google Scholar]
- Song QH, Singh RP, Richardson TP, Nugent MA, Trinkaus-Randall V. Transforming growth factor-beta1 expression in cultured corneal fibroblasts in response to injury. J Cell Biochem. 2000;77:186–199. doi: 10.1002/(sici)1097-4644(20000501)77:2<186::aid-jcb3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Sorkin A, McClure M, Huang F, Carter R. Interaction of EGF receptor and grb2 in living cells visualized by fluorescence resonance energy transfer (FRET) microscopy. Curr Biol. 2000;10:1395–1398. doi: 10.1016/s0960-9822(00)00785-5. [DOI] [PubMed] [Google Scholar]
- Sugrue SP, Hay ED. Response of basal epithelial cell surface and cytoskeleton to solubilized extracellular matrix molecules. J Cell Biol. 1981;91:45–54. doi: 10.1083/jcb.91.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugrue SP, Hay ED. The identification of extracellular matrix (ECM) binding sites on the basal surface of embryonic corneal epithelium and the effect of ECM binding on epithelial collagen production. J Cell Biol. 1986;102:1907–1916. doi: 10.1083/jcb.102.5.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K, Kay S, editors. Methods in cell biology. San Diego, CA: Academic Press; 1999. Methods in cell biology: Green fluorescent proteins. [Google Scholar]
- Svoboda KKH. Embryonic corneal epithelial actin alters distribution in response to laminin. Invest Ophthalmol Vis Sci. 1992;33:324–333. [PMC free article] [PubMed] [Google Scholar]
- Svoboda KKH, Hay ED. Embryonic corneal epithelial interaction with exogenous laminin and basal lamina is F-actin dependent. Dev Biol. 1987;123:455–469. doi: 10.1016/0012-1606(87)90403-9. [DOI] [PubMed] [Google Scholar]
- Svoboda KKH, O’Shea KS. Optic vesicle defects induced by vincristine sulfate: An in vivo and in vitro study in the mouse embryo. Teratology. 1984;29:223–240. doi: 10.1002/tera.1420290209. [DOI] [PubMed] [Google Scholar]
- Svoboda KKH, O’Shea KS. An analysis of cell shape and the neuroepithelial basal lamina during optic vesicle formation in the mouse embryo. Development. 1987;100:185–200. doi: 10.1242/dev.100.2.185. [DOI] [PubMed] [Google Scholar]
- Svoboda KKH, Orlow DL, Ashrafzadeh A, Jirawuthiworavong G. Zyxin and vinculin distribution at the “cell-extracellular matrix attachment complex” (CMAX) in corneal epithelial tissue are actin dependent. Anat Rec. 1999a;254:336–347. doi: 10.1002/(sici)1097-0185(19990301)254:3<336::aid-ar4>3.0.co;2-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda KKH, Orlow DL, Chu CL, Reenstra WR. ECM stimulated actin bundle formation in embryonic corneal epithelia is tyrosine phosphorylation dependent. Anat Rec. 1999b;254:348–359. doi: 10.1002/(sici)1097-0185(19990301)254:3<348::aid-ar5>3.0.co;2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons M, Settleman J. Rho family GTPases: More than simple switches. Trends Cell Biol. 2000;10:415–419. doi: 10.1016/s0962-8924(00)01832-8. [DOI] [PubMed] [Google Scholar]
- Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–215. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- Tsien R. New calcium indicators and buffers with high selectivity against magnesium and protons: Design, synthesis, and properties of prototype structures. Biochemistry. 1980;19:2396–2404. doi: 10.1021/bi00552a018. [DOI] [PubMed] [Google Scholar]
- Turner CE. Paxillin and focal adhesion signalling. Nat Cell Biol. 2000;2:E231–E236. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- Wayner EA, Orlando RA, Cheresh DA. Integrin avb3 and integrin avb5 contribute to cell attachment to vitro-nectin but differentially distribute on the cell surface. J Cell Biol. 1991;113:919–929. doi: 10.1083/jcb.113.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte M. ICE/CED-3 proteases in apoptosis. Trends Cell Biol. 1996;6:245–248. doi: 10.1016/0962-8924(96)20025-x. [DOI] [PubMed] [Google Scholar]
- Yeh B, Svoboda KKH. Intracellular distribution of beta-actin mRNA is polarized in embryonic corneal epithelia. J Cell Sci. 1994;107:105–115. doi: 10.1242/jcs.107.1.105. [DOI] [PubMed] [Google Scholar]
- Zamir E, Katz M, Posen Y, Erez N, Yamada KM, Katz BZ, Lin S, Lin DC, Bershadsky A, Kam Z, Geiger B. Dynamics and segregation of cell–matrix adhesions in cultured fibroblasts. Nat Cell Biol. 2000;2:191–196. doi: 10.1038/35008607. [DOI] [PubMed] [Google Scholar]
- Zimmerman CM, Padgett RW. Transforming growth factor beta signaling mediators and modulators. Gene. 2000;249:17–30. doi: 10.1016/s0378-1119(00)00162-1. [DOI] [PubMed] [Google Scholar]