

Abstract
Background
Metformin, one of most commonly used antidiabetes drugs, is reported to exert its therapeutic effects by activating AMP-activated protein kinase (AMPK); however, the mechanism by which metformin activates AMPK is poorly defined. The objective of the present study was to determine how metformin activates AMPK in endothelial cells.
Methods and Results
Exposure of human umbilical vein endothelial cells or bovine aortic endothelial cells to metformin significantly increased AMPK activity and the phosphorylation of both AMPK at Thr172 and LKB1 at Ser428, an AMPK kinase, which was paralleled by increased activation of protein kinase C (PKC)-ζ, as evidenced by increased activity, phosphorylation (Thr410/40ζ), and nuclear translocation of PKC-ζ. Consistently, either pharmacological or genetic inhibition of PKC-ζ ablated metformin-enhanced phosphorylation of both AMPK-Thr172 and LKB1-Ser428, suggesting that PKC-ζ might act as an upstream kinase for LKB1. Furthermore, adenoviral overexpression of LKB1 kinase-dead mutants abolished but LKB1 wild-type overexpression enhanced the effects of metformin on AMPK in bovine aortic endothelial cells. In addition, metformin increased the phosphorylation and nuclear export of LKB1 into the cytosols as well as the association of AMPK with LKB1 in bovine aortic endothelial cells. Similarly, overexpression of LKB1 wild-type but not LKB1 S428A mutants (serine replaced by alanine) restored the effects of metformin on AMPK in LKB1-deficient HeLa-S3 cells, suggesting that Ser428 phosphorylation of LKB1 is required for metformin-enhanced AMPK activation. Moreover, LKB1 S428A, like kinase-dead LKB1 D194A, abolished metformin-enhanced LKB1 translocation as well as the association of LKB1 with AMPK in HeLa-S3 cells. Finally, inhibition of PKC-ζ abolished metformin-enhanced coimmunoprecipitation of LKB1 with both AMPKα1 and AMPKα2.
Conclusions
We conclude that PKC-ζ phosphorylates LKB1 at Ser428, resulting in LKB1 nuclear export and hence AMPK activation.
Keywords: diabetes mellitus, endothelium, metabolism, molecular biology
Metformin is one of the most widely used antidiabetic agents for type 2 diabetes mellitus. In the UK Prospective Diabetic Study, metformin significantly reduced the incidence of death and cardiovascular events in obese type 2 diabetic patients compared with patients treated with other glucose-lowering reagents but with the same glycemic control,1 suggesting that metformin may have unique protective effects against diabetes-related cardiovascular complications. In addition, metformin is shown to improve vascular endothelial function in diabetic humans.2–7 Although it has been in clinical use for decades, the precise mechanism of action of metformin has yet to be established. Recent studies8,9 demonstrate that metformin activates AMP-activated protein kinase (AMPK) in vitro and in vivo. Furthermore, we10,11 found that clinically relevant concentrations of metformin activated AMPK in cultured endothelial cells and aortas in vivo and ascribed the beneficial effects of metformin on vascular function to AMPK activation. In addition, activation of AMPK by metformin is required for decreased glucose production and increased fatty acid oxidation in hepatocytes9 and for increased glucose uptake in skeletal muscle.8
AMPK is a serine/threonine kinase and an evolutionary conserved regulator of the cellular response to low energy.12,13 It is activated when nutrient supply is limited, ATP generation is impaired, or cellular energy demand is increased.13,14 AMPK has a critical role in many metabolic processes, and, once activated, AMPK phosphorylates a number of proteins that result in increased glucose uptake and metabolism as well as fatty acid oxidation. In addition, AMPK simultaneously inhibits hepatic lipogenesis, cholesterol synthesis, and glucose production15 and is also responsible for increased fatty acid oxidation in response to the adipocyte-derived hormones leptin16,17 and adiponectin.18 Phosphorylation of Thr172 in the activation loop of the catalytic α subunit is a prerequisite of significant kinase activity, and an increase in AMP/ATP ratios further allosterically stimulates the enzyme, resulting in 1000-fold activation.
Recently, at least 2 upstream kinases, LKB1 and calcium/calmodulin-dependent kinase kinase (CaMKK)-β, have been identified as AMPK kinases.19–24 LKB1, a 50-kDa serine/threonine kinase, was originally identified as the product of the gene mutated in the autosomal dominantly inherited Peutz-Jeghers cancer syndrome.25 In resting cells, LKB1 is reported to be predominantly located in the nucleus,26–28 and, like AMPK, LKB1 forms a heterotrimeric complex with regulatory proteins termed STRAD and MO25, which are required for its activation and cytosolic localization.29 LKB1 has been shown to mediate Thr172 phosphorylation of AMPK both in vitro and in intact cells.19,21,23
The mechanism by which metformin activates AMPK remains controversial, although earlier studies30,31 demonstrate that metformin might activate AMPK by decreasing cellular energy by acting as an inhibitor of complex I of the respiratory chain. However, several recent studies32,33 argue against this notion because in these studies metformin activates AMPK without affecting the AMP/ATP ratio. Interestingly, metformin neither activates AMPK in a cell-free assay nor influences the phosphorylation of AMPK by LKB1 in vitro. Furthermore, it was found that in cell lines such as HeLa-S3 cells and A549 cells, which lack LKB1 expression, or in LKB1−/− mouse embryonic fibroblasts, AMPK could not be activated by a variety of agonists and stresses, including metformin.19,23 Mice lacking LKB1 expression have significantly inhibited AMPK activity and blunted increases in glucose transport caused by electrically stimulated muscle contraction and the AMPK-activating compound 5-aminoimidazole-4-carboxamide 1-β-D-ribofuranos-ide (AICAR) and phenformin.34 Importantly, with the use of mice that have LKB1 deleted in liver, it has been reported that metformin required LKB1 in the liver to lower blood glucose.35 Although these studies have established the critical role of LKB1 in AMPK activation, none of these studies have elucidated the upstream signaling that controls LKB1-dependent AMPK activation. In the present study, we have found that metformin activates AMPK by activating PKC-ζ–dependent LKB1 phosphorylation at Ser428, a site that is required for LKB1 nuclear export into the cytosol and its association with AMPK.
Methods
A full description of methods, including methods such as protein kinase assays for PKC-ζ, AMPK, and LKB1, immunocytochemical staining of LKB1, and preparation of subcellular fractions, can be found in the online-only Data Supplement.
Cell Culture and Treatment
Human umbilical vein endothelial cells (HUVEC) were cultured in medium 200 supplemented with low-serum growth supplements. Bovine aortic endothelial cells (BAEC) were maintained in endothelial basal medium with 2% serum and growth factors before use. After they reached confluence, BAEC were serum deprived overnight before the experiments. HeLa-S3 and A549 cells were grown in F-12K medium supplemented with 10% serum. All culture media were supplemented with penicillin (100 U/mL) and streptomycin (100 μg/mL). The cells were treated with 1 mmol/L metformin for 1 hour after adenoviral infection or pretreatment with protein kinase inhibitor.
Plasmid Transfection and Adenovirus Infection
Site-directed mutagenesis of LKB1 was performed as described previously.36 Mutations were confirmed by DNA sequencing, and plasmid DNA was extracted on a large scale with the use of Qiagen EndoFree plasmid maxi kit (catalogue No. 12362) and transfected into HeLa-S3 with the use of the Lipofectamine 2000 kit from Invitrogen (catalogue No. 11668-019), according to the instructions provided by the supplier. Twenty-four hours after transfection, the cells were treated with metformin (1 mmol/L) or vehicle for 1 hour. Both LacZ expression vector and untreated cells were used as control.
BAEC or HUVEC were infected with adenovirus expressing a wild-type PKC-ζ (PKC-ζ-WT) or a dominant negative mutant PKC-ζ (PKC-ζ-DN). A replication-defective adenoviral vector expressing green fluorescence protein (ad-GFP) was used as control. The AMPK-DN adenoviral vector was constructed from AMPK-α2 bearing a mutation altering lysine 45 to arginine (K45R) as described previously.10,11 Cells were infected with the adenovirus at a multiplicity of infection of 100 in serum-free medium overnight. Cells were then washed and incubated in fresh medium without serum for an additional 18 to 24 hours before experimentation. Under these conditions, infection efficiency was typically >75% as determined by GFP expression.
Statistical Analysis
Values are expressed as mean±SE. All of the data were analyzed with a 1-way ANOVA followed by Bonferroni post hoc analyses, except for those obtained from the time course, which were analyzed with repeated-measures ANOVA. A P value of <0.05 is considered statistically significant.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Metformin Induces AMPK and Acetyl Coenzyme A Carboxylase Phosphorylation
Because AMPK activation requires the phosphorylation of Thr172 in the activation loop of α1 and α2 subunits,37,38 AMPK activity was determined in Western blots by monitoring the phosphorylation of both AMPK at Thr172 and its best-characterized downstream kinase, acetyl coenzyme A carboxylase (ACC) at Ser79, by using specific antibodies. AMPK activation was further confirmed by AMPK activity assayed as 32P incorporation into SAMS (HMRSAMSGLHLVKRR) peptides. Confluent HUVEC and BAEC were treated with metformin (1 mmol/L) for 1 hour. As shown in Figure 1A, metformin markedly increased the detection of AMPK-Thr172 and ACC at Ser79 in both HUVEC and BAEC, respectively. Because metformin activated AMPK in both BAEC and HUVEC at a similar potency, we performed most of the experiments in BAEC.
Figure 1.

Inhibition of PKC-ζ attenuates metformin-enhanced AMPK activity. Confluent BAEC or HUVEC, with or without adenovirus infection, were preincubated with PKC-ζ-PS for 30 minutes before being exposed to metformin. After treatment, the cells were lysaed and extracted. Both phosphorylated AMPK-Thr172 and ACC-Ser79 were detected in Western blots by using specific antibodies, as described in Methods. A, Metformin (Met) activates AMPK in BAEC and HUVEC. The blot is a representative of 6 blots obtained from 6 individual experiments. Con indicates control. B and C, Selective inhibition of PKC-ζ attenuated metformin-enhanced AMPK activity (n=6). ♣P<0.05 (control [Con] vs metformin treated), †P<0.05 (metformin vs metformin plus PKC-ζ-PS). D, PKC-ζ-PS dose-dependently inhibits metformin-enhanced AMPK activity in BAEC (n=5). ♣P<0.05 (control vs metformin), †P<0.05 (metformin vs metformin plus PKC-ζ-PS). E and F, Genetic inhibition of PKC-ζ with PKC-ζ-DN, but not the adenovi-rus encoding GFP, attenuated metformin-enhanced phosphorylation of AMPK-Thr172 and ACC-Ser79 in BAEC. The blot is representative of 5 blots from 5 individual experiments (n=5). ♣P<0.05 (compared with control), †P<0.05 (metformin-treated vs metformin plus adenovirus). G, AMPK activity was assayed as described in Methods (n=4). ♣P<0.05 (compared with control), †P<0.05 (metformin-treated vs metformin plus adenovirus).
Inhibition of PKC-ζ Attenuates Metformin-Enhanced AMPK-Thr172 Phosphorylation
We have previously shown that inhibition of PKC-ζ attenuates ONOO−-enhanced AMPK activation in BAEC.36 To establish PKC-ζ as a mediator for metformin-induced activation of AMPK, we first determined whether PKC-ζ–specific pseudosubstrate peptide (PKC-ζ-PS), a synthetic peptide that selectively inhibits PKC-ζ without affecting other PKC isoforms, altered the effects of metformin on AMPK-Thr172 and ACC-Ser79 phosphorylation. As depicted in Figure 1B, PKC-ζ-PS significantly ablated metformin-enhanced phosphorylation of both AMPK-Thr172 (n=5; P<0.01) (Figure 1B and 1C) and ACC-Ser79 (Figure 1B). The inhibitory effect of PKC-ζ-PS was also confirmed by the suppression of AMPK activity. As depicted in Figure 1D, PKC-ζ-PS dose-dependently suppressed metformin-enhanced AMPK activity (Figure 1D), as assayed by the SAMS peptides.
Additional evidence for PKC-ζ–dependent AMPK activation was obtained from genetic inhibition of PKC-ζ. As depicted in Figure 1E and 1F, adenoviral overexpression of either GFP, PKC-ζ-DN, or PKC-ζ-WT did not alter the basal levels of AMPK in BAEC. However, overexpression of PKC-ζ-DN, but not GFP, abolished the effects of metformin on both AMPK and ACC, whereas overexpression of PKC-ζ-WT significantly enhanced metformin-enhanced phosphorylation of both AMPK-Thr172 and ACC-Ser79 (Figure 1E and 1F) and AMPK activity (Figure 1G).
Activation of PKC-ζ by Metformin
We next determined whether metformin activated PKC-ζ in BAEC. The phosphorylation of PKC-ζ at Thr410/403 by upstream kinases such as phosphoinositide 3-kinase (PI-3 kinase)/PDK-1 axis and translocation of PKC-ζ from the cytosol into cytoplasmic membrane are considered critical steps in the activation of PKC-ζ. Thus, PKC-ζ phosphorylation was monitored in total cell lysates in Western blots by using specific antibodies. As shown in Figure 2A, metformin increased PKC-ζ Thr410/403 phosphorylation without altering PKC-ζ expression. Inhibition of PKC-ζ with PKC-ζ-PS abolished metformin-induced PKC-ζ phosphorylation (Figure 2A), indicating a specific inhibition by PKC-ζ-PS.
Figure 2.

Metformin increases PKC-ζ phosphorylation and the translocation of PKC-ζ from cytosol to the membrane. Confluent BAEC were exposed to metformin (1 mmol/L, 1 hour), and the translocation of PKC-ζ and PKC-ζ phosphorylation was assayed as described in Methods. A, Metformin (Met) increased the phosphorylation of PKC-ζ in BAEC. The blot is a representative of 3 blots obtained from 3 independent experiments. Con indicates control. B, PKC-ζ activity was determined as described in Methods (n=4). ♣P<0.05 (compared with control [Con]), †P<0.05 (metformin-treated vs metformin plus adenovirus), #P<0.05 (PKC-ζ-WT vs metformin plus WT adenovirus). C and D, Metformin increases the translocation of PKC-ζ to the membrane (n=5). ♣P<0.05 (control vs metformin-treated). E and F, Metformin increases the translocation of PKC-ζ from cytosol into the nucleus. The blot is a representative of 3 blots from 3 individual experiments (n=5). ♣P<0.05 (control vs metformin-treated). G, Analysis of the purity of subcellular fractions. The subcellular fractions were prepared as described in Methods. Marker enzymes were detected by Western blot with the use of specific antibodies.
We next assayed PKC-ζ activity by using 32P incorporation in PKC-ζ–specific peptides. As expected, overexpression of PKC-ζ-DN, but not GFP, attenuated PKC-ζ activity, whereas overexpression of PKC-ζ-WT increased PKC-ζ activity in BAEC (Figure 2B). In agreement with increased PKC-ζ phosphorylation at Thr410/403 (Figure 2A), metformin significantly increased PKC-ζ activity in BAEC or BAEC infected with GFP. Overexpression of PKC-ζ-DN abolished metformin-enhanced PKC-ζ activation, whereas PKC-ζ-WT increased PKC-ζ activity (Figure 2B). These results implied that metformin activated PKC-ζ.
The translocation of PKC-ζ is considered a critical step in PKC-ζ activation. Exposure of BAEC to metformin significantly increased the presence of PKC-ζ in membrane fractions but lowered the amount of PKC-ζ in the cytosol (Figure 2C and 2D). In parallel, metformin also increased the translocation of PKC-ζ from the cytosol into the nuclei (Figure 2E and 2F). The purity of these subcellular fractions was confirmed by using antibodies against specific protein marker enzymes39,40 of the cytosol (lactate dehydrogenase), plasma membrane (alkaline phosphatase), or nucleus (histone H2AX), respectively. The nuclear histone H2AX was detected only in the nuclear fraction but not in cytosolic or membrane fractions (Figure 2G). Lactate dehydrogenase was detected only in the cytosolic fraction, whereas alkaline phosphatase was found only in the membrane fraction (Figure 2G). Thus, metformin caused cellular redistribution of PKC-ζ from the cytosol to nuclei and membranes.
Protein Kinase A and p90 Ribosomal S6 Kinase Are Not Required in Metformin-Enhanced AMPK Activation
Because both protein kinase A (PKA) and p90 ribosomal S6 kinase (RSK) are reported to phosphorylate LKB1 at Ser428, we next determined whether PKA or RSK was involved in metformin-enhanced phosphorylation of AMPK-Thr172 or LKB1-Ser428. As depicted in Figure 3A and 3B, administration of either H89 (10 μmol/L), a selective PKA inhibitor, or SL0101 (10 μmol/L), a selective RSK inhibitor, did not alter metformin-enhanced phosphorylation of AMPK-Thr172 or LKB1-Ser428 (Figure 3A and 3B). In contrast to increased PKC-ζ phosphorylation, metformin affected neither PKA catalytic subunit (PKAc) phosphorylation at Thr197 nor RSK3 phosphorylation at Thr356/Ser360 (Figure 3C), suggesting that metformin had no effect on PKA or RSK. Further evidence comes from coimmunoprecipitation experiments. As shown in Figure 3D, LKB1 was coimmunoprecipitated with PKCζ but not with either PKA or RSK. Furthermore, metformin markedly increased the coimmunoprecipitation of LKB1 with PKC-ζ (Figure 3D), which was sensitive to PKC-ζ inhibition (Figure 3D), suggesting that PKC-ζ activity was required for the association of PKC-ζ with LKB1. Indeed, alteration of PKC-ζ activity by overexpression of either PKC-ζ-DN or PKC-ζ-WT did not alter the phosphorylation of PKAc or RSK (Figure 3E), suggesting that the effect of PKC-ζ was independent of PKA or RSK in BAEC. Taken together, these results suggest that metformin-enhanced phosphorylation of LKB1 and AMPK was PKC-ζ dependent but independent of PKA or RSK.
Figure 3.

Neither PKA nor RSK is involved in metformin-enhanced AMPK activation in BAEC. A and B, Confluent BAEC were pretreated with protein kinase inhibitors for 30 minutes followed by treatment with metformin (1 mmol/L) for 1 hour, the cells were lysed, and phosphorylation of AMPK and LKB1 was detected by Western blot. The blot is a representative of blots from 3 different experiments. ♣P<0.05 compared with respective control (Con). C, BAEC were treated with metformin (Met) (1 mmol/L) for 1 hour, cell lysate was prepared, and the phosphorylation of PKC-ζ, PKAc, and RSK3 was detected by Western blot with the use of specific antibodies. The blot is a representative of 3 blots obtained from 3 individual experiments. D, LKB1 was immunoprecipitated, and PKC-ζ, PKAc, and RSK3 were detected by Western blotting. The blot is representative of blots from 3 different experiments. E, Confluent BAECs were transfected with PKC-ζ-DN and PKC-ζ-WT adenovirus for 48 hours and treated with metformin for 1 hour. The cells were lysed and analyzed by Western blotting. The blot is a representative of 3 blots obtained from 3 individual experiments.
Metformin-Enhanced AMPK Activation Requires LKB1
At least 2 upstream kinases, including LKB1 and CaMKK-β, have been identified as AMPK kinases.19–24 We next determined whether metformin required LKB1 to activate AMPK in BAEC. As depicted in Figure 4A and 4B, overexpression of the kinase-dead LKB1 mutant LKB1-D194A abolished metformin-enhanced phosphorylation of both AMPK-Thr172 and ACC-Ser79 in BAEC. In contrast, overexpression of LKB1-WT enhanced the effects of metformin on the phosphorylation of both AMPK and ACC. Because overexpression of GFP did not alter the effect of metformin, these results suggested that LKB1 was required for metformin-enhanced AMPK activation in endothelial cells.
Figure 4.

PKC-ζ–dependent LKB1 phosphorylation of LKB1 at serine 428. A and B, Metformin-activated AMPK is LKB1 dependent in BAEC. Adenoviral overexpression of the kinase-dead LKB1 mutant blocks metformin-induced AMPK activation, whereas LKB1 WT overexpression enhanced the effect of LKB1 in BAEC. The blot is a representative of 5 blots from 5 independent experiments. C and D, Metformin-enhanced LKB1 phosphorylation at serine 428 is PKC-ζ dependent. The blot is a representative of 5 blots from 5 independent experiments (n=5). ♣P<0.05 (control [Con] vs metformin), †P<0.05 (metformin vs metformin plus PKC-ζ adenovirus). E, LKB1 activity was measured as described in Methods (n=4). F and G, Time course of PKC-ζ, LKB1, and AMPK phosphorylation. BAEC were treated with metformin (1 mmol/L), and the phosphorylation of PKC-ζ, LKB1, and AMPK was detected at the indicated time by Western blot analysis. The blot is a representative of 3 blots from 3 individual experiments. *♣†P<0.05 compared with respective controls.
Metformin-Enhanced LKB1-Ser428 Phosphorylation Is PKC-ζ Dependent
We have previously demonstrated that ONOO− enhances the phosphorylation of LKB1 at Ser428 via PKC-ζ.36 We next determined whether metformin altered the phosphorylation of LKB1 at Ser428. As depicted in Figure 4C and 4D, metformin significantly increased the phosphorylation of LKB1 at Ser428. Overexpression of PKC-ζ-DN, which ablated metformin-enhanced AMPK activation (Figure 1E through 1G), abolished metformin-enhanced LKB1-Ser428 phosphorylation (Figure 4C and 4D). Conversely, overexpression of GFP or PKC-ζ-WT did not suppress metformin-enhanced LKB1-Ser428 phosphorylation (Figure 4C and 4D). However, alteration of PKC activity by overexpression of PKC-ζ-DN or PKC-ζ-WT did not alter LKB1 activity (Figure 4E). In addition, consistent with our previous results,34,36,41,42 metformin did not alter LKB1 activity in the cells infected with either PKC-ζ-DN or PKC-ζ-WT (Figure 4E). Thus, changes of AMPK-Thr172 phosphorylation cannot be attributed to altered kinase activity of LKB1.
Metformin-Enhanced PKC-ζ Phosphorylation Occurs Before the Phosphorylation of Both LKB1 and AMPK
We next determined the time course of metformin-enhanced phosphorylation of PKC-ζ, LKB1, and AMPK. As shown in Figure 4F and 4G, metformin significantly increased the PKC-ζ phosphorylation within 1 minute. The phosphorylation of LKB1-Ser428 was significantly elevated ≈10 minutes after metformin treatment, whereas a significant increase of AMPK-Thr172 phosphorylation was observed at 30 minutes and reached a peak at 60 minutes (Figure 4F and 4G). These data suggest a chronology of events, with metformin rapidly activating first PKC (Thr410/403), then LKB1 (Ser428), and finally AMPK (Thr172).
Metformin Triggers LKB1 Translocation From Nucleus Into Cytoplasm
Earlier works27,43–45 had indicated that LKB1 is localized mainly in the nucleus, whereas AMPK is localized mainly in the cytoplasm. LKB1 must be exported from the nucleus into cytosols where AMPK is located. We first used immunohistochemical staining to assay whether metformin altered the subcellular localization of LKB1 in HUVEC because all commercially available antibodies against LKB1 could not be used in BAEC. As expected, LKB1 was found mainly in the nucleus of nonstimulated HUVEC (Figure 5A). In metformin-treated HUVEC, LKB1 was detected mainly in the cytosols. Metformin-enhanced LKB1 nuclear export was further confirmed by Western blot analysis of LKB1 in subcellular fractions. As shown in Figure 5B and 5C, metformin significantly increased the protein amount of LKB1 in the cytosol, whereas it was markedly reduced in the nucleus of HUVEC (Figure 5A through 5C). Similarly, LKB1 was predominantly located in the nuclear fractions in BAEC. Exposure of BAEC to metformin reduced the amount of LKB1 in the nuclear fraction, whereas it increased the amount of LKB1 in the cytosols (data not shown). Interestingly, the signals corresponding to LKB1-Ser428 phosphorylation were significantly increased in both cytosol and nuclei of metformin-treated BAEC, whereas the reduced nuclear LKB1 protein content after metformin treatment (Figure 5A through 5C) corresponded to a relatively high LKB1-Ser428 signal (Figure 5D and 5E). These data suggest that LKB1 phosphorylation might occur in the nucleus, resulting in LKB1 translocation from nuclear to cytoplasmic locations.
Figure 5.

Metformin increases the translocation of LKB1 from nucleus into cytosol in HUVEC. A, Immunocytochemical staining of translocation of LKB1 from nucleus to the cytosols caused by metformin in HUVEC. B and C, Metformin (Met) also increases the amount of LKB1 in the cytosol while it decreases LKB1 in the nuclei in HUVEC. The blot is a representative of 5 blots obtained from 5 independent experiments. Con indicates control. D and E, Metformin increases the serine 428 phosphorylation of LKB1 in both nucleus and cytosol. The blot is a representative of 3 blots obtained from 3 independent experiments.
Mutation of LKB1 Serine 428 With Alanine Prevents Metformin-Enhanced LKB1 Translocation
We next determined how metformin increased LKB1 translocation. With the use of site-directed mutagenesis techniques, Ser428 of LKB1 was mutated into alanine (LKB1-S428A, loss of function). In addition, Asp194 of LKB1, which is essential for LKB1 activity, was also replaced with alanine, resulting in a kinase-dead LKB1 mutant (LKB1-D194A). These plasmids were transfected into LKB1-deficient A549 cells. After transfection, the subcellular distributions of LKB1 were detected by using a mouse anti-His Tag antibody. As expected, A549 transfected with Lac-Z did not react with an anti-His Tag antibody (data not shown). Like LKB1 in HUVEC, LKB1 was found mainly in the nucleus of A549 transfected with either LKB1 WT, S428A mutants, or D194A mutants (Figure 6A). Metformin markedly increased LKB1 staining in the cytosol of A549 transfected with LKB1-WT but not in A549 transfected with either LKB1-D194A or LKB1-S428A mutants (Figure 6A). Western blot analysis of LKB1 in the subcellular fractions confirmed that LKB1-WT or LKB1 mutants were all nuclear proteins and that metformin significantly increased the amount of cytosolic LKB1 in A549 cells transfected with LKB1-WT but not LKB1 mutants (Figure 6B). These results suggest that the Ser428 phosphorylation of LKB1 as well as LKB1 activity was required for metformin-enhanced nuclear export.
Figure 6.
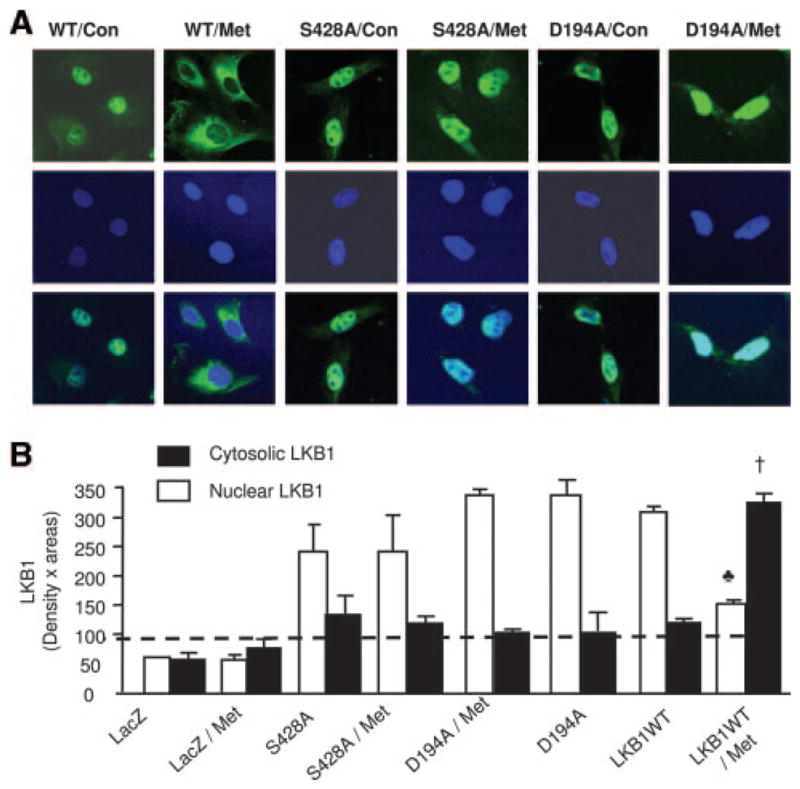
The phosphorylation of LKB1-Ser428 is required for metformin-enhanced translocation of LKB1 and AMPK activation. A, Immunocytochemical staining of metformin (Met)-enhanced LKB1 translocation in A549 cells. The cells were transfected with indicated plasmids, as described in Methods. LKB1 was detected by using a mouse anti-His Tag antibody. The image with Lac-Z was omitted because A549 cells transfected with Lac-Z did not react with the antibody. Con indicates control. B, Western blot detection of LKB1 in A549 cells. The blot is a representative of 5 blots from 5 independent experiments (n=5). ♣P<0.05 (WT vs WT plus metformin), †P<0.05 (WT plus metformin vs WT or S428A plus metformin).
Mutation of Ser428 Into Alanine Abolishes Metformin-Enhanced AMPK Activation
Because LKB1-Ser428 mutation altered its subcellular localization, we next determined whether Ser428 mutation of LKB1 also altered metformin-enhanced AMPK activation. As shown in Figure 7A and 7B, metformin did not activate AMPK in either HeLa-S3 or HeLa-S3 transfected with either LacZ or the D194A mutant of LKB1, confirming the essential role of LKB1 in metformin-enhanced AMPK activation. Conversely, transfection of LKB1-WT significantly increased the phosphorylation of AMPK and ACC in HeLa-S3 cells transfected with WT LKB1 (Figure 7A and 7B). Interestingly, the point mutation of LKB1-Ser428 into alanine, like the kinase-inactive mutant D194A, abolished metformin-enhanced phosphorylation of AMPK. Similarly, overexpression of LKB1-S428A and LKB1-D194A mutants ablated peroxynitrite (ONOO−, 100 μmol/L)-enhanced phosphorylation of AMPK, whereas the mutation of Ser428 into aspartic acid (LKB1-S428D), which mimics phosphorylation of Ser428, increased AMPK phosphorylation as LKB1-WT did (Figure 7C).
Figure 7.

Ser428 phosphorylation of LKB1 is required for metformin-enhanced AMPK activation and coimmunoprecipitation of LKB1 with AMPK. A and B, Metformin activates AMPK in HeLa-S3 overexpressing LKB1-WT but not LKB1-Ser428A mutants. After being transfected with LKB1-WT or kinase-dead LKB1 mutants or the LKB1-Ser428A mutant, HeLa-S3 cells were exposed to metformin (1 mmol/L, 1 hour). AMPK activation was monitored in Western blots with the use of specific antibodies. The blot is a representative of 5 blots from 5 independent experiments. C, Ser428 phosphorylation of LKB1 is required for ONOO−-enhanced AMPK activation. After being transfected with LKB1-WT or LKB1-Ser428A (Ser428 was mutated into alanine, loss of function) or the LKB1-Ser428D (Ser428 replaced by aspartic acid, phosphorylation mimicking), HeLa-S3 cells were exposed to ONOO− (100 μmol/L). AMPK activation was monitored by Western blotting with the use of specific antibodies. The blot is a representative of 5 blots from 5 independent experiments. D, LKB1 activity in LKB1 WT or LKB1-Ser431A mutants. E, Metformin increases the association of LKB1 and AMPK. LKB1 was immunoprecipitated from BAEC, and AMPK was detected in Western blots. Inhibition of PKC-ζ attenuated metformin-enhanced association of LKB1 with AMPK. The blot is a representative of 5 blots from 5 independent experiments. F, Mutation of Ser428 of LKB1 into alanine (S428A) abolishes metformin-enhanced coimmunoprecipitation of LKB1 with AMPK. Metformin increased the coimmunoprecipitation of LKB1 with LKB1 WT but not with LKB1 mutant. The blot is a representative of 3 blots from 3 individual experiments.
We next assayed the activity of mutant LKB1 with Ser428 (Ser431 in mouse) altered to alanine in comparison to WT LKB1, using an artificial peptide substrate. As shown in Figure 7D, LKB1-Ser431A mutants exhibited a 10% reduction of LKB1 activity compared with LKB1 WT.
We next determined whether LKB1-Ser428 phosphorylation increased LKB1 activity. Mouse LKB1-WT or LKB1-S431A (equivalent to human 428) mutants were coexpressed with both MO25α and STRADα in bacteria. Partially purified LKB1 WT or LKB1-S431A complex was coincubated with recombinant PKC-ζ. PKC-ζ–dependent LKB1 phosphorylation and LKB1 activity were assayed by using LKB1tide or p53. PKC-ζ increased 32P incorporation in LKB1 WT paralleled with a 10% increase in LKB1 activity but had less effect on LKB1-S431A mutants. Only LKB1, but not STRADα or MO25α, was phosphorylated by PKC-ζ as determined by 32P autoradiographic gels when PKC-ζ was incubated with LKB1-MO25α-STRADα complex in vitro (data not shown). Because LKB1 S431A mutants expressed a 10% reduction of LKB1 activity, whereas PKC-ζ caused a 10% increase of LKB1 activity, these results suggest that the phosphorylation of LKB1 at Ser428/431 might increase LKB1 activity by 10%. However, such rather small increases of LKB1 activity would only partly explain the strongly elevated AMPK activity after metformin treatment, therefore emphasizing the importance of the nucleocytoplasmic redistribution of LKB1 that is expected to increase AMPK activation in the cytosol.
Mutation of Ser428 Into Alanine Abolishes Metformin-Enhanced Association of LKB1 With AMPK
We have previously shown that ONOO− activates AMPK by increasing the association of LKB1 and AMPK.36 In addition, we found that PKC-ζ coimmunoprecipitated with LKB1 and metformin increased the association of LKB1 with PKC-ζ. We next determined whether phosphorylation of LKB1-Ser428 by PKC-ζ contributed to metformin-enhanced association of AMPK with LKB1 and therefore whether LKB1 was immunoprecipitated from BAEC and stained with AMPK in Western blots. As shown in Figure 7E, metformin showed significantly increased coimmunoprecipitation of LKB1 with AMPKα1 or AMPKα2. Importantly, inhibition of PKC-ζ blunted metformin-enhanced association of LKB1 with AMPK (Figure 7E), suggesting that PKC-ζ enhances the association of LKB1 with AMPK.
We next investigated whether mutations of LKB1-Ser428 into alanine altered the interaction of AMPK and LKB1 in HeLa-S3 cells. Metformin increased the association of AMPK and LKB1 in HeLa-S3 cells transfected with LKB1 WT but not in HeLa-S3 transfected with LacZ or LKB1 mutants (Figure 7F), implying that LKB1-Ser428 phosphorylation is required for metformin-enhanced AMPK activation.
Discussion
In the present study, we have established that LKB1-Ser428 phosphorylation by atypical PKC-ζ plays an essential role in metformin-enhanced AMPK activation. LKB1-Ser428 phosphorylation is required for both its nuclear export of LKB1 into cytosols, in which it forms an active complex with STRAD and MO25 and consequent AMPK phosphorylation. Because various stimuli increased the phosphorylation of LKB1 at Ser428, it is tempting to speculate that LKB1-Ser428 phosphorylation might be a common pathway required for AMPK activation.
LKB1 localization depends on its kinase activity, import into the nucleus, and retention within cytoplasm.27,29 It has been reported that cytoplasmic localization of LKB1 is critical for its normal function.27,29 Several residues including Ser31, Thr336, Thr366, and Ser 428 of LKB1 have been identified as phosphorylation sites, although their physiological functions are largely unknown. In the present study, we have provided evidence that metformin induced AMPK activation by phosphorylating LKB1 at Ser428, which results in translocation of LKB1 from the nucleus to the cytosol and increased association of LKB1 with AMPK. The key findings can be summarized as follows: metformin increases LKB1 phosphorylation at Ser428, induces LKB1 translocation from nucleus to cytoplasm, and enhances coimmunoprecipitation of LKB1 with AMPK. Mutation of Ser428 into alanine prevented translocation of LKB1 from the nucleus to the cytoplasm and abolished metformin-enhanced association of LKB1 with AMPK, thus abolishing AMPK activation. The LKB1-S428A mutation did not affect normal nuclear localization of LKB1 but prevented LKB1 translocation to the cytoplasm after metformin treatment, suggesting that phosphorylation of Ser428 might be essential for LKB1 translocation from the nucleus to the cytoplasm. Thus, Ser428 phosphorylation located in the C-terminus of LKB1 might play a crucial role in regulating AMPK activation. In agreement with this notion, Forcet et al46 demonstrated that C-terminal mutations impair LKB1-mediated activation of AMPK and downstream signaling but do not affect LKB1 autokinase activity or abolish LKB1-induced growth arrest. Our data indicate that mutation of Ser428 of LKB1 to alanine impaired its ability to export from nucleus and subsequent association with AMPK after metformin treatment (Figure 7F), suggesting that phosphorylation of Ser428 might contribute to its binding to AMPK and that mutations of this site into alanine uncouple this process, thereby resulting in reduced enzyme-substrate recognition. These results are in contrast with the previous report that mutation of Ser431 to either alanine to abolish phosphorylation or to glutamate to mimic phosphorylation did not significantly affect the cellular localization or catalytic activity of LKB1 in vitro.47 The reasons for the discrepancy are unknown and might be related to the species differences (human versus mouse) or cell types. In vivo, LKB1 forms a heterotrimeric complex with 2 proteins termed STRAD and MO25.29,48 STRAD is an LKB1-specific adaptor and substrate; when binding to the kinase domain of LKB1, STRAD enhances LKB1 activity >100-fold and anchors LKB1 in the cytosol. MO25 stabilizes the binding of STRAD to LKB1 and cooperates with STRAD to localize LKB1 in the cytoplasm of cells.49 In addition, LKB1 can also be localized in the cytoplasm of cells through an interaction with a cytosolic protein called LIP1 (LKB1 interacting protein-1).50 It is reported that in muscle-specific LKB1 knockout mice, AICAR, phenformin, or muscle contraction did not increase AMPKα2 activity in the skeletal muscle, indicating that LKB1 is the major upstream kinase of AMPK.34 However, these 3 treatments failed to increase LKB1 activity.41 In our study, we found that metformin increased AMPK phosphorylation and activity. Although it did not change LKB1 activity to a great extent, metformin markedly enhanced LKB1 phosphorylation at Ser428; mutation of Ser428 into alanine prevented translocation of LKB1 from nucleus to cytoplasm and abolished metformin-enhanced association of AMPK with LKB1, thus abrogating AMPK activation. Theses results demonstrate that the phosphorylation of LKB1 at Ser428 plays a crucial role in AMPK activation by altering its cellular location or by enabling it to interact with regulatory proteins or protein substrates. In addition, the active form of LKB1 might be as important as Ser428 phosphorylation because the mutation of LKB1 D194 into alanine prevented the translocation of the enzyme. Because LKB1 S431A mutants expressed a 10% reduction of LKB1 activity, whereas PKC-ζ caused a 10% increase of LKB1 activity, metformin-enhanced AMPK activity is likely via the nucleocytoplasmic redistribution rather than an increase of LKB1 activity as a result of LKB1-Ser428 phosphorylation. Moreover, it remains to be determined whether other sites are required for LKB1 translocation.
We have previously reported10,11,51,52 that activation of PI-3 kinase by metformin or ONOO− might enhance AMPK activity by increasing the association of LKB1 with AMPK. Indeed, inhibition of c-Src or PI-3 kinase activity by pharmacological inhibition or dominant negative mutants abolishes metformin- or ONOO−-enhanced AMPK activation in endothelial cells.10,11,53,54 In the present study, we not only confirm but also extend these observations by demonstrating that PKC-ζ, a member of the PI-3 kinase AGC family, plays a critical role in metformin-induced AMPK activation. Indeed, it has been shown that some AMPK agonists, such as AICAR and dinitrophenol, increase PKC-ζ activity in isolated extensor digitorum longus muscle and L6 myotubes.55 In type 2 diabetic patients, metformin increases PKC-ζ activity, accompanied by increased AMPK activity.56 We have also demonstrated that PKC-ζ phosphorylates LKB1 at Ser428 in both in vitro assays and in cultured cells, including endothelial cells, pericytes, adipocytes, and cultured vascular smooth muscle cells.36 Furthermore, inhibition of PKC-ζ attenuates AMPK activation by other stimuli (Z.X., unpublished data, 2008). Because other kinases, including cAMP-dependent PKA and p90 RSK, are reported to phosphorylate LKB1 at Ser428, the Ser428 phosphorylation of LKB1 might be a common pathway for AMPK activation triggered by signaling pathways other than the c-Src/PI-3 kinase axis (Figure 8).
Figure 8.

LKB1 nuclear export is required for metformin-enhanced AMPK activation. In the resting state, LKB1 is mainly located in the nucleus, whereas LKB1 activity required that adaptor proteins MO25 and STRAD as well as AMPK-α1 (the major isoform of AMPK catalytic units in endothelial cells) be located in the cytosol. Metformin (or ONOO−) activates PI-3 kinase, resulting in PDK-1/2 phosphorylation. PDK-1/2 phosphorylates PKC-ζ, resulting in the translocation of the latter into nucleus as well as cytoplasmic membrane. In the nucleus, activated PKC-ζ phosphorylates LKB1 at serine 428 and likely other sites. Phosphorylated LKB1 is exported from the nucleus via an unknown mechanism into the cytosol. LKB1 is subsequently bound to the preexisting adaptors proteins (STRAD and MO25), which recruit and phosphorylate AMPK at Thr172. AMPK is likely also activated by other stimuli, which either activate PKC-ζ or increase LKB1-Ser428 phosphorylation (PKA or RSK90). Overall, our results suggest that the PI-3 kinase-PDK1/2-PKC-ζ pathway plays a critical role in metformin-enhanced AMPK activation.
To summarize, we provide evidence that PKC-ζ plays a critical role in regulating AMPK activity. Our data suggest that metformin-induced AMPK activation requires LKB1 and demonstrate that phosphorylation of Ser428 of LKB1 could affect cellular location of LKB1 and its interaction with AMPK. We conclude that PKC-ζ can regulate AMPK activity by increasing LKB1 phosphorylation, resulting in LKB1 nuclear export and consequent AMPK Thr172 phosphorylation by LKB1. In addition, the Ser428 phosphorylation of LKB1 might be a common pathway for AMPK activation triggered by signal pathways, including PI-3 kinase/PKC-ζ axis, PKA, and RSK.
CLINICAL PERSPECTIVE
Metformin is one of most commonly used drugs for the treatment of type II diabetes mellitus. Unlike traditionally used glucose-lowering reagents, such as sulfonylureas or insulin, metformin improves cardiovascular functions and reduces cardiovascular risks in diabetes. Earlier studies from us and others found that clinically relevant concentrations of metformin activated the AMP-activated protein kinase (AMPK); these studies ascribed the beneficial effects of metformin on vascular function to AMPK activation. The present study was designed to elucidate the mechanism by which metformin activates AMPK. Exposure of cultured endothelial cells to metformin significantly increased the phosphorylation of both AMPK at Thr172 and LKB1 at Ser428, an AMPK kinase, which was paralleled by increased activation of protein kinase C (PKC)-ζ, as evidenced by increased activity, phosphorylation, and nuclear translocation of PKC-ζ. Consistently, either pharmacological or genetic inhibition of PKC-ζ ablated metformin-enhanced phosphorylation of both AMPK at Thr172 and LKB1 at Ser428. Our data suggest that the Ser428 phosphorylation of LKB1 by PKC-ζ is required for metformin-triggered AMPK activation.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grants (HL079584, HL080499, HL074399), a research award from the American Diabetes Association, a research award from the Juvenile Diabetes Research Foundation, a grant from the Oklahoma Center for the Advancement of Science and Technology, and funds from the Paul H. Doris Eaton Travis Chair in Endocrinology of the University of Oklahoma Health Sciences Center (all to Dr Zou). Part of the work was also supported by National Institutes of Health grant 1P20RR024215-01 (Drs Xie and Zou) and a Scientist Development Grant of the American Heart Association (Dr Xie). The work was further supported by the Helmut Horten Foundation (to Dr Neumann) and European Union FP6 contract LSHM-CT-2004-005272 (EXGENESIS).
Footnotes
The online-only Data Supplement, consisting of expanded Methods, is available with this article at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.107.744490/DC1.
Disclosures
None.
References
- 1.UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- 2.Abbasi F, Chu JW, McLaughlin T, Lamendola C, Leary ET, Reaven GM. Effect of metformin treatment on multiple cardiovascular disease risk factors in patients with type 2 diabetes mellitus. Metabolism. 2004;53:159–164. doi: 10.1016/j.metabol.2003.07.020. [DOI] [PubMed] [Google Scholar]
- 3.Verma S, Yao L, Dumont AS, McNeill JH. Metformin treatment corrects vascular insulin resistance in hypertension. J Hypertens. 2000;8:1445–1450. doi: 10.1097/00004872-200018100-00012. [DOI] [PubMed] [Google Scholar]
- 4.Katakam PV, Ujhelyi MR, Hoenig M, Miller AW. Metformin improves vascular function in insulin-resistant rats. Hypertension. 2000;35:108–112. doi: 10.1161/01.hyp.35.1.108. [DOI] [PubMed] [Google Scholar]
- 5.Marfella R, Acampora R, Verrazzo G, Ziccardi P, De RN, Giunta R, Giugliano D. Metformin improves hemodynamic and rheological responses to L-arginine in NIDDM patients. Diabetes Care. 1996;19:934–939. doi: 10.2337/diacare.19.9.934. [DOI] [PubMed] [Google Scholar]
- 6.Petersen JS, DiBona GF. Acute sympathoinhibitory actions of metformin in spontaneously hypertensive rats. Hypertension. 1996;27:619–625. doi: 10.1161/01.hyp.27.3.619. [DOI] [PubMed] [Google Scholar]
- 7.Bhalla RC, Toth KF, Tan E, Bhatty RA, Mathias E, Sharma RV. Vascular effects of metformin: possible mechanisms for its antihypertensive action in the spontaneously hypertensive rat. Am J Hypertens. 1996;9:570–576. doi: 10.1016/0895-7061(95)00356-8. [DOI] [PubMed] [Google Scholar]
- 8.Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, Zhou G, Williamson JM, Ljunqvist O, Efendic S, Moller DE, Thorell A, Goodyear LJ. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51:2074–2081. doi: 10.2337/diabetes.51.7.2074. [DOI] [PubMed] [Google Scholar]
- 9.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis BJ, Xie Z, Viollet B, Zou MH. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55:496–505. doi: 10.2337/diabetes.55.02.06.db05-1064. [DOI] [PubMed] [Google Scholar]
- 11.Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles WG, Schlattner U, Neumann D, Brownlee M, Freeman MB, Goldman MH. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo: role of mitochondrial reactive nitrogen species. J Biol Chem. 2004;279:43940–43951. doi: 10.1074/jbc.M404421200. [DOI] [PubMed] [Google Scholar]
- 12.Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 13.Kemp BE, Stapleton D, Campbell DJ, Chen ZP, Murthy S, Walter M, Gupta A, Adams JJ, Katsis F, van DB, Jennings IG, Iseli T, Michell BJ, Witters LA. AMP-activated protein kinase, super metabolic regulator. Biochem Soc Trans. 2003;31:162–168. doi: 10.1042/bst0310162. [DOI] [PubMed] [Google Scholar]
- 14.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- 15.Hardie DG, Carling D. The AMP-activated protein kinase: fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–273. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 16.Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 17.Atkinson LL, Fischer MA, Lopaschuk GD. Leptin activates cardiac fatty acid oxidation independent of changes in the AMP-activated protein kinase-acetyl-CoA carboxylase-malonyl-CoA axis. J Biol Chem. 2002;277:29424–29430. doi: 10.1074/jbc.M203813200. [DOI] [PubMed] [Google Scholar]
- 18.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 19.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 21.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 24.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la CA, Aaltonen LA. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 26.Tiainen M, Ylikorkala A, Makela TP. Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci U S A. 1999;96:9248–9251. doi: 10.1073/pnas.96.16.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiainen M, Vaahtomeri K, Ylikorkala A, Makela TP. Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1) Hum Mol Genet. 2002;11:1497–1504. doi: 10.1093/hmg/11.13.1497. [DOI] [PubMed] [Google Scholar]
- 28.Nezu J, Oku A, Shimane M. Loss of cytoplasmic retention ability of mutant LKB1 found in Peutz-Jeghers syndrome patients. Biochem Biophys Res Commun. 1999;261:750–755. doi: 10.1006/bbrc.1999.1047. [DOI] [PubMed] [Google Scholar]
- 29.Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, Prescott AR, Clevers HC, Alessi DR. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003;22:5102–5114. doi: 10.1093/emboj/cdg490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- 31.El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 32.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 33.Fryer LG, Parbu-Patel A, Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- 34.Sakamoto K, McCarthy A, Smith D, Green KA, Grahame HD, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, DePinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;10:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie Z, Dong Y, Zhang M, Cui MZ, Cohen RA, Riek U, Neumann D, Schlattner U, Zou MH. Activation of protein kinase C zeta by per-oxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J Biol Chem. 2006;281:6366–6375. doi: 10.1074/jbc.M511178200. [DOI] [PubMed] [Google Scholar]
- 37.Stein SC, Woods A, Jones NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J. 2000;345:437–443. [PMC free article] [PubMed] [Google Scholar]
- 38.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 39.Hoffmann K, Blaudszun J, Brunken C, Hopker WW, Tauber R, Steinhart H. New application of a subcellular fractionation method to kidney and testis for the determination of conjugated linoleic acid in selected cell organelles of healthy and cancerous human tissues. Anal Bioanal Chem. 2005;381:1138–1144. doi: 10.1007/s00216-004-3009-z. [DOI] [PubMed] [Google Scholar]
- 40.Srinivas KS, Chandrasekar G, Srivastava R, Puvanakrishnan R. A novel protocol for the subcellular fractionation of C3A hepatoma cells using sucrose density gradient centrifugation. J Biochem Biophys Methods. 2004;60:23–27. doi: 10.1016/j.jbbm.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 41.Sakamoto K, Goransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. Am J Physiol. 2004;287:E310–E317. doi: 10.1152/ajpendo.00074.2004. [DOI] [PubMed] [Google Scholar]
- 42.Sriwijitkamol A, Coletta DK, Wajcberg E, Balbontin GB, Reyna SM, Barrientes J, Eagan PA, Jenkinson CP, Cersosimo E, DeFronzo RA, Sakamoto K, Musi N. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes. 2007;56:836–848. doi: 10.2337/db06-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, House CM, Fernandez CS, Cox T, Witters LA, Kemp BE. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996;271:611–614. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- 44.Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334:177–187. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- 46.Forcet C, Etienne-Manneville S, Gaude H, Fournier L, Debilly S, Salmi M, Baas A, Olschwang S, Clevers H, Billaud M. Functional analysis of Peutz-Jeghers mutations reveals that the LKB1 C-terminal region exerts a crucial role in regulating both the AMPK pathway and the cell polarity. Hum Mol Genet. 2005;14:1283–1292. doi: 10.1093/hmg/ddi139. [DOI] [PubMed] [Google Scholar]
- 47.Sapkota GP, Kieloch A, Lizcano JM, Lain S, Arthur JS, Williams MR, Morrice N, Deak M, Alessi DR. Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essential for LKB1 to suppress cell growth. J Biol Chem. 2001;276:19469–19482. doi: 10.1074/jbc.M009953200. [DOI] [PubMed] [Google Scholar]
- 48.Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, Alessi DR, Clevers HC. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003;22:3062–3072. doi: 10.1093/emboj/cdg292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, Prescott AR, Clevers HC, Alessi DR. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003;22:5102–5114. doi: 10.1093/emboj/cdg490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith DP, Rayter SI, Niederlander C, Spicer J, Jones CM, Ashworth A. LIP1, a cytoplasmic protein functionally linked to the Peutz-Jeghers syndrome kinase LKB1. Hum Mol Genet. 2001;10:2869–2877. doi: 10.1093/hmg/10.25.2869. [DOI] [PubMed] [Google Scholar]
- 51.Zou MH, Hou XY, Shi CM, Nagata D, Walsh K, Cohen RA. Modulation by peroxynitrite of Akt- and AMP-activated kinase-dependent Ser1179 phosphorylation of endothelial nitric oxide synthase. J Biol Chem. 2002;277:32552–32557. doi: 10.1074/jbc.M204512200. [DOI] [PubMed] [Google Scholar]
- 52.Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, Cohen RA. Activation of 5′-AMP-activated kinase is mediated through c-Src and phosphoinositide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells: role of peroxynitrite. J Biol Chem. 2003;278:34003–34010. doi: 10.1074/jbc.M300215200. [DOI] [PubMed] [Google Scholar]
- 53.Zou MH, Hou XY, Shi CM, Nagata D, Walsh K, Cohen RA. Modulation by peroxynitrite of Akt- and AMP-activated kinase-dependent Ser1179 phosphorylation of endothelial nitric oxide synthase. J Biol Chem. 2002;277:32552–32557. doi: 10.1074/jbc.M204512200. [DOI] [PubMed] [Google Scholar]
- 54.Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, Cohen RA. Activation of 5′-AMP-activated kinase is mediated through c-Src and phosphoinositide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells: role of peroxynitrite. J Biol Chem. 2003;278:34003–34010. doi: 10.1074/jbc.M300215200. [DOI] [PubMed] [Google Scholar]
- 55.Chen HC, Bandyopadhyay G, Sajan MP, Kanoh Y, Standaert M, Farese RV, Jr, Farese RV. Activation of the ERK pathway and atypical protein kinase C isoforms in exercise- and aminoimidazole-4-carboxamide-1-beta-D-riboside (AICAR)-stimulated glucose transport. J Biol Chem. 2002;277:23554–23562. doi: 10.1074/jbc.M201152200. [DOI] [PubMed] [Google Scholar]
- 56.Luna V, Casauban L, Sajan MP, Gomez-Daspet J, Powe JL, Miura A, Rivas J, Standaert ML, Farese RV. Metformin improves atypical protein kinase C activation by insulin and phosphatidylinositol-3,4,5-(PO4)3 in muscle of diabetic subjects. Diabetologia. 2006;49:375–382. doi: 10.1007/s00125-005-0112-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.