

Abstract
Ubiquitin-like proteins (UBLs), such as NEDD8, are transferred to their targets by distinct, parallel, multi-enzyme cascades that involve the sequential action of E1, E2, and E3 enzymes. How do enzymes within a particular UBL conjugation cascade interact with each other? We report here that the unique N-terminal sequence of NEDD8’s E2, Ubc12, selectively recruits NEDD8’s E1 to promote Ubc12~NEDD8 thioester formation. A peptide corresponding to Ubc12’s N-terminus (Ubc12N26) specifically binds and inhibits NEDD8’s E1, the heterodimeric APPBP1-UBA3 complex. The structure of APPBP1-UBA3-Ubc12N26 reveals conserved Ubc12 residues docking in a groove generated by loops conserved in UBA3s but not other E1s, explaining why this interaction is unique to the NEDD8 pathway. These studies define a novel mechanism for E1–E2 interaction and show how enzymes within a particular UBL conjugation cascade can be tethered together by unique protein-protein interactions emanating from their common structural scaffolds.
INTRODUCTION
There are over a dozen ubiquitin-like proteins (UBLs) in higher eukaryotes that function by covalently modifying myriad substrates1. The best understood functional consequence of UBL modification is ubiquitin-mediated proteolysis. However, different UBLs alter the functions of their targets in different ways, for example by changing the target’s subcellular localization, enzymatic activity, or protein-protein interactions. Ubiquitin’s closest relative, NEDD8, does not direct its targets to the proteasome. Rather, NEDD8 is conjugated to the cullin subunits of SCF (Skp1-cullin-F-box) and related ubiquitin ligases to alter their activity2–4. NEDD8 enhances the ability of SCF to multiubiquitinate substrates, and displaces the CAND1 inhibitor of SCF assembly5–8. The NEDD8 pathway is essential in organisms ranging from fission yeast to mammals9,10, and regulates many important biological processes, including cell division2,3,11, signal transduction5, and development12.
It is important to understand how the different UBLs are ligated to their correct targets. Different UBLs are ligated to their targets by parallel but distinct cascades of enzymes that sequentially involve an E1 activating enzyme, an E2 conjugating enzyme, and an E3 ligase13,14. First, the E1 binds the UBL, Mg2+, and ATP and catalyzes adenylation of the UBL’s C-terminus. The E1 then forms a thioester intermediate between its catalytic cysteine and the UBL’s activated C-terminus, and subsequently transfers the thioester-linked UBL to the catalytic cysteine of the E2 enzyme. The E3 recruits the target and facilitates UBL transfer from the E2 to a primary amino group, often from a lysine side-chain, on the target.
In recent years, several general principles have emerged for protein-protein interactions in UBL transfer cascades15–26. Nonetheless, the selective coordination of enzymes within a particular UBL’s cascade remains incompletely understood. Particularly lacking is knowledge of unique protein-protein interactions distinct for the cascades for different UBLs. The NEDD8 pathway has served as a good model for studying UBL-specific protein-protein interaction29–31. Although the enzymes in the NEDD8 pathway are related to those in other UBL transfer cascades, the NEDD8 pathway involves one E1, one E2, and few E3s3,4,27. Moreover, it appears that NEDD8 is ultimately directed to a small number of targets2–4. By contrast, the ubiquitin pathway involves tens of E2s, hundreds of E3s, and thousands of targets. Thus, the relatively minimal nature of the NEDD8 pathway simplifies the identification of protein-protein interactions specific for a particular UBL’s conjugation cascade.
NEDD8’s E2, Ubc12, has a 26-residue N-terminal extension upstream of its ~150 residue conserved E2 core domain. Ubc12’s N-terminal extension is unique to the NEDD8 pathway: Ubc12’s N-terminal sequence is conserved across species, but is not found in other E2s. The function of Ubc12’s N-terminal extension has remained unknown. Here we find a role for Ubc12’s N-terminal extension in cell proliferation: wild-type Ubc12, but not a mutant lacking the N-terminal extension, rescues CSF-1-dependent proliferation of NIH 3T3 cells expressing a mitogenically defective but survival competent human CSF-1 receptor. Biochemical and X-ray crystallographic studies collectively show that Ubc12’s N-terminal extension is a selective peptide motif that optimally recruits NEDD8’s E1 for the NEDD8 transfer cascade.
RESULTS
Ubc12’s N-terminal extension is important for function
The sequence of Ubc12 has two regions: (1) a unique 26-residue N-terminal extension found only in Ubc12 family members and (2) an E2 core domain, a ~150 residue domain conserved among all E2s, which contains the E2 catalytic cysteine (Fig. 1a). To assess whether Ubc12’s N-terminal extension has any functional significance, we tested the function of a deletion mutant lacking residues 2–26, termed Ubc12ΔN, using a biochemical assay for NEDD8 transfer. The ultimate targets of NEDD8 modification include cullin family members2,3, such as human Cul1. Cul1 is modified by NEDD8 at a single lysine, Lys72027. The NEDD8 transfer cascade can be reconstituted in vitro with the APPBP1-UBA3 heterodimeric E1 complex, the E2 Ubc12, the E3 Rbx1, the target Cul1, NEDD8, Mg2+, and ATP27 (Fig. 1b). We monitored NEDD8 transfer using 32P-labelled NEDD8 phosphorylated at an N-terminal Protein Kinase A site with [γ-32P]ATP28. We find that deleting Ubc12’s N-terminal extension significantly hinders Ubc12’s ability to transfer NEDD8 to Cul-1’s Lys720 (Fig. 1c).
Figure 1.
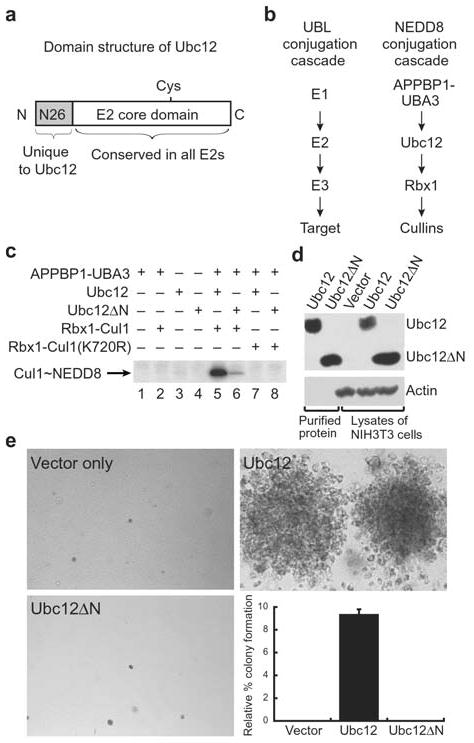
Ubc12’s N-terminal extension is important for function. a, Domain structure of Ubc12. Ubc12 has a unique 26-residue N-terminal extension conserved in Ubc12’s across species but not found in other E2s. Following this sequence is the ~150-residue E2 core conserved in all E2s. The position of Ubc12’s catalytic Cys is shown. b, NEDD8 conjugation involves the sequential action of NEDD8’s E1, the heterodimeric APPBP1-UBA3 complex, NEDD8’s E2, Ubc12, a RING E3 Rbx1, and cullin targets2–4,27. c, NEDD8 conjugation to Cul1 was assayed by incubation with purified APPBP1-UBA3 (E1), Ubc12 or Ubc12ΔN lacking residues 2–26 (E2), Rbx1-Cul1 (E3-target complex, lanes 2, 5, 6) or Rbx1-Cul1 Lys720Arg (target complex mutated at NEDD8 modification site, lanes 7–8) as indicated. Lanes 1–4 are controls, lane 5 shows the complete reaction with wild-type Ubc12 and lane 6 shows the reaction with Ubc12ΔN. Lanes 7–8 show that neither Ubc12 nor Ubc12ΔN modify Cul1 with the Lys720Arg mutation. d, Immunoblot control for expression of Ubc12 and Ubc12ΔN in cell proliferation assay. e, Soft agar colony formation assay of NIH 3T3 cells expressing the mutant human CSF-1R[Tyr809Phe] infected with retroviruses empty (top left) or expressing wild type Ubc12 (top right) or the Ubc12ΔN mutant (bottom left) in the presence of human CSF-1. Relative % colony formation is based on controls expressing wild-type human CSF-1R as a reference for 100% (graph, bottom right).
We next assayed the importance of Ubc12’s N-terminal extension in cell proliferation. The NEDD8 pathway enzymes play an essential role in cell proliferation in organisms ranging from fission yeast to mammals9,10. Consistent with this function, we identified Ubc12 in a genetic screen for proteins involved in cell proliferation (R.M and M.F.R., unpublished results; details of screen to be published elsewhere). This same screen previously identified cell cycle regulatory proteins such as Myc and D-type cyclins29,30. Therefore, we can assay Ubc12’s effects on cell proliferation as follows: ectopic expression of the human macrophage colony-stimulating factor 1 (CSF-1) receptor (CSF-1R) in NIH 3T3 mouse fibroblasts allows the cells to survive, migrate and proliferate in serum-free medium and form colonies in soft agar the presence of human CSF-1 as the sole growth factor31. By contrast, cells expressing CSF1-R with the Tyr809Phe mutation in the T-loop survive and remain arrested in G1, but fail to proliferate and form colonies in soft agar when stimulated with the CSF-1 growth factor29. As with Myc and D-type cyclins29,30, which are other regulators of mitogenesis, retroviral expression of Ubc12 in these cells allows proliferation in soft agar in the presence of human CSF-1 (Fig. 1d, e). Unlike wild-type Ubc12, Ubc12ΔN does not support proliferation, even though the wild-type and mutant Ubc12 proteins are expressed at similar levels (Fig. 1d, e). Therefore, the N-terminal extension of Ubc12 plays an important role in mitogenesis, as well as in vitro.
Ubc12’s N-Extension Interacts Selectively with NEDD8’s E1
We next sought to identify the biochemical basis for Ubc12ΔN’s defect in NEDD8 transfer. Ubc12 is involved in multiple steps in NEDD8 transfer: (1) interaction with NEDD8’s E1, the APPBP1-UBA3 complex, (2) receiving NEDD8 from APPBP1-UBA3’s catalytic cysteine in a transthiolation reaction, (3) interaction with the RING E3 Rbx1, and finally, (4) conjugation of NEDD8 to Cul-1’s Lys720. To identify whether Ubc12ΔN is defective at the first two steps in the reaction, we performed kinetic analyses, monitoring the initial rate of APPBP1-UBA3-dependent transfer of 32P-labeled NEDD8 onto Ubc12’s catalytic cysteine, as a function of Ubc12 concentration (Fig. 2a). The kcat values for forming the Ubc12~NEDD8 and Ubc12ΔN~NEDD8 thioester complexes were similar, suggesting that Ubc12ΔN is chemically competent for the transthiolation reaction (Supplementary Table 1 online). However, the Km for Ubc12ΔN (209±11 nM) is ~25-fold higher than wildtype (5150±153 nM), indicating a defect in binding to APPBP1-UBA3 (Supplementary Table 1 online).
Figure 2.
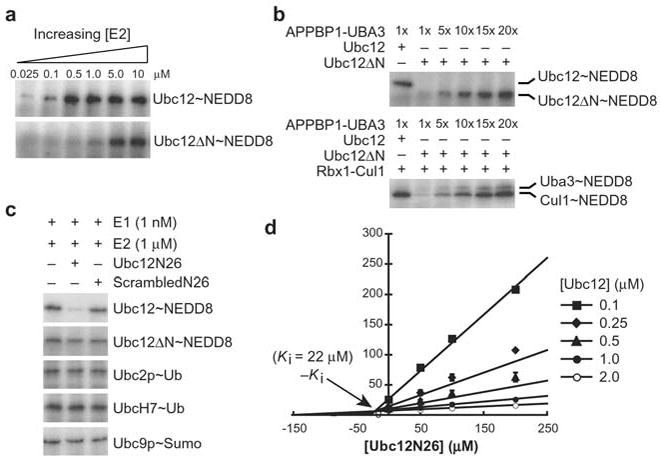
Ubc12’s N-terminus is involved in E1 binding. a, Ubc12ΔN is impaired at forming a thioester complex with NEDD8. Ubc12~NEDD8 and Ubc12ΔN~NEDD8 thioester formation was examined as a function of E2 concentration. 30s time-points are shown for reactions involving 1 nM APPBP1-UBA3 and 25 nM, 100 nM, 500 nM, 1 μM, 5 μM and 10 μM Ubc12 or Ubc12ΔN, from left to right. b, Ubc12ΔN is competent for NEDD8 conjugation to Cul1. Top panel – Ubc12ΔN~NEDD8 thioester formation with increasing concentrations of the APPBP1-UBA3 E1 complex. Bottom panel – Ubc12ΔN-mediated NEDD8 conjugation to Cul-1, with increasing concentrations of APPBP1-UBA3, assayed as in the top panel but with the addition of Rbx1-Cul1. c, 1 mM Ubc12N26 peptide corresponding to the N-terminal 26 residues of Ubc12 inhibits APPBP1-UBA3-catalyzed Ubc12~NEDD8 thioester formation, but not thioester formation between Ubc12ΔN and NEDD8, or E1(ubiquitin)-catalyzed Ubc2p~ubiquitin or UbcH7~ubiquitin thioester formation, or E1(Sumo)-catalyzed Ubc9p~Sumo thioester formation. There is no effect of a peptide with identical composition but scrambled sequence (ScrambledN26). d, Dixon plot analyzing inhibition of APPBP1-UBA3-catalyzed Ubc12~NEDD8 thioester formation by the Ubc12N26 peptide. The Ubc12N26 peptide is a competitive inhibitor with a Ki of 22±5 μM.
To test whether Ubc12ΔN is competent to transfer NEDD8 to Cul1, we examined Ubc12ΔN-mediated Cul1~NEDD8 complex formation in the presence of increased APPBP1-UBA3. These experiments are performed at identical concentrations of NEDD8, Ubc12, Ubc12ΔN, and Rbx1-Cul1, with the only difference being the concentration of the APPBP1-UBA3 E1 complex. 20-fold higher levels of the APPBP1-UBA3 E1 complex are sufficient to allow Ubc12ΔN to form a thioester intermediate with NEDD8, and to ligate NEDD8 to Cul1 to the same degree as wild-type Ubc12 (Fig. 2b). These results suggest that Ubc12ΔN’s primary defect in the NEDD8 pathway arises from its decreased binding to E1.
We tested direct binding to APPBP1-UBA3 with a 26-residue synthetic peptide corresponding to the sequence of Ubc12’s N-terminal extension, referred to as Ubc12N26. A peptide with identical composition but scrambled sequence was used as a control (ScrambledN26). Ubc12N26 competitively inhibits formation of the Ubc12~NEDD8 thioester with a Ki of 22 μM, whereas the scrambled control peptide has no effect (Fig. 2c,d). The Ubc12N26 peptide has no effect on Ubc12ΔN~NEDD8 thioester formation, nor on the ability of the E1s for ubiquitin and Sumo to catalyze E2~ubiquitin and E2~Sumo thioester formation (Fig. 2c). In addition, deletion of Ubc12’s N-terminal extension eliminates Ubc12’s ability to co-elute with APPBP1-UBA3 during gel filtration chromatography (data not shown). These findings suggest that Ubc12’s N-terminal extension binds directly and selectively to APPBP1-UBA3.
Overall Structure of the Ubc12N26 complex with NEDD8’s E1
In order to understand how Ubc12’s N-terminal extension interacts with APPBP1-UBA3, we determined the crystal structure of the APPBP1-UBA3-Ubc12N26 complex at 2.6 Å (Fig. 3 and Table 1). Structures of NEDD8’s heterodimeric E1, the APPBP1-UBA3 complex, alone and in complex with NEDD8 and ATP, have been determined previously26,28. The previous structures reveal that the E1 contains three domains. An adenylation domain containing the ATP-binding site is linked through flexible loops to a domain organized around the catalytic cysteine, and to the C-terminal domain also involved in E2 binding. The domains are organized around two clefts in the middle (Fig. 4). From the direction facing the catalytic cysteine, with the catalytic cysteine domain located above the adenylation domain, “Cleft 1” is on the left and “Cleft 2” is on the right (Fig. 4, middle panels). The large size of the two clefts suggested that they accommodate the E1’s substrates28, and the structure of the APPBP1-UBA3-NEDD8-ATP complex revealed ATP binding in Cleft 1 and the globular domain of NEDD8 binding in Cleft 226. There are no significant conformational changes in structure of APPBP1-UBA3 upon Ubc12N26 peptide binding (0.517 Å rmsd over 917 Cα atoms between apo APPBP1-UBA328 and APPBP1-UBA3-Ubc12N26).
Figure 3.
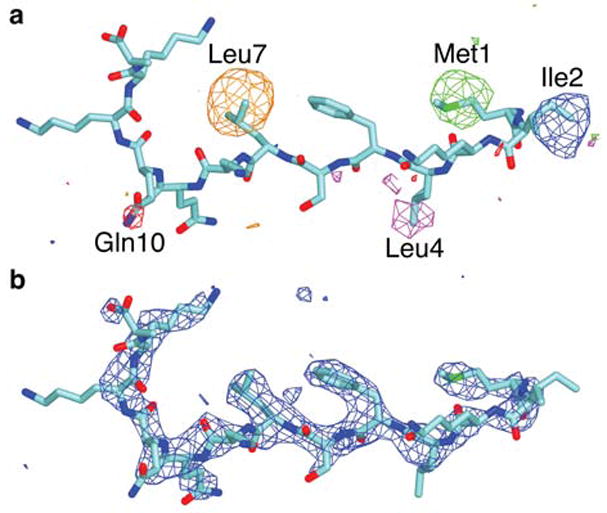
Electron density maps displayed over the Ubc12N26 peptide structure. a, Selenomethionine-scanning of Ubc12N26. Overlay of five different selenium anomalous difference Fourier maps contoured at 3.2σ, obtained from crystals containing Ubc12N26 peptides with each of the following residues substituted one-at-a-time with selenomethionine: Met1 (green map), Ile2 (blue map), Leu4 (magenta map), Leu7 (orange map), and Gln10 (red map). b, Fo-Fc map contoured at 3σ calculated after performing simulated annealing at 4000K on the model lacking the Ubc12N26 peptide.
Table 1.
Data collection and refinement statistics
| APPBP1-UBA3-Ubc12N26 | |
|---|---|
| Data Collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 92.4, 122.8, 195.9 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution limit (A) | 2.6 |
| Rsym | 7.0 (35.6) |
| I/σI | 30.4 (3.6) |
| Completeness (%) | 99.7 (99.7) |
| Mean redundancy | 31.8 |
| Refinement | |
| Resolution (A) | 18.0–2.6 |
| No. reflections | 2,157,076 |
| No. unique reflections | 67,734 |
| Rwork/Rfree | 23.7/27.9 |
| No. atoms | |
| Protein | 14,344 |
| Zinc | 2 |
| Water | 298 |
| B-factors | |
| Protein | 68.3 |
| Zinc | 49.1 |
| Water | 56.0 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.42 |
Figure 4.

Overall architecture of the APPBP1-UBA3-Ubc12N26 complex. Three views of the complex are shown in cartoon (top) and surface representations (bottom), each view with a 40–80° rotation around the y-axis as indicated. APPBP1 is shown in blue, UBA3 in red, and Ubc12N26 in cyan, and the position of the catalytic cysteine (C216A here) in green. The locations of the adenylation domain with its ATP binding site, the catalytic cysteine domain, the C-terminal domain (CTD), and the binding site for NEDD8’s globular domain are indicated. In the middle view, Cleft 1, which binds ATP is on the left, and Cleft 2, which binds the globular domain of NEDD8, is on the right. Residues 1 and 13 at the N- and C-termini of the visible portion of the Ubc12N26 peptide are labeled N and C, respectively. The “top” and “bottom” sides of UBA3’s Ubc12N26 binding groove are labeled.
In the APPBP1-UBA3-Ubc12N26 peptide complex, residues 1–13 of the Ubc12N26 peptide are ordered and adopt an extended structure. The Ubc12N26 peptide docks in a groove in the adenylation domain portion of UBA3 (Fig. 4). This portion of UBA3 has a central 8-stranded mixed β-sheet (strands a–d, g–j). In the previous APPBP1-UBA3-NEDD8-ATP structure, ATP and NEDD8 bind across one surface of the sheet, facing the catalytic cysteine domain. The Ubc12N26 peptide binds across the opposite face of the β-sheet. The N-terminus of Ubc12 approaches the edge of the sheet nearest the binding site for NEDD8’s globular domain. The observed C-terminus of the peptide (Ubc12’s residue 13) approaches the edge of the sheet near the ATP-binding site, and is ~ 55 Å from UBA3’s catalytic cysteine. The peptide binding groove comes from splaying apart at the ends of β-strands a, d, g, h, i, and j (Fig. 4, top left panel). The “top” side of the peptide binding groove comes from loops at the N-termini of β-strands d and g and at the C-termini of β-strands h and j. The “bottom” side of the groove is formed by UBA3’s helix 2 and the N-terminus of the subsequent β-strand a, and the loop preceding β-strand i.
Structural basis for selective interactions with Ubc12N26
In order to understand the selectivity of Ubc12’s N-terminal extension toward NEDD8’s E1, we examined the conservation of the E1 docking groove in the sequences of other UBL activating enzymes. We aligned the sequence of the Ubc12N26-binding domain of UBA3 (residues 9–205 and 288–346) with the corresponding sequences either in UBA3s from different species, or in activating enzymes for the following UBLs: human ubiquitin, Sumo and ISG15, E. coli MoaD and ThiF, and S. cerevisiae Apg8p and Urm1p. Degree of conservation is displayed on the structure in Figure 5. There is a high degree of conservation in the binding groove for Ubc12’s N-terminal extension among UBA3 family members from different species. However, the groove is poorly conserved among activating enzymes for different UBLs, explaining the lack of interaction we observe between the Ubc12N26 peptide and ubiquitin’s and Sumo’s E1s. Thus, the structure of the APPBP1-UBA3-Ubc12N26 peptide complex reveals how within the conserved structural scaffold of an E1, unique surfaces can be generated that mediate distinct, specific protein-protein interactions.
Figure 5.

The Ubc12N26 binding surface is conserved in UBA3s, but not in activating enzymes for other UBLs. The structure of Ubc12’s N-terminal peptide (cyan) is displayed in a surface representation of the Ubc12N26-binding domain of UBA3 (residues 9–205, 288–367), rotated ~90° in x relative to the middle orientation in Figure 4, for a direct view of the peptide contacts. a, The surface colored according to conservation among UBA3s from 8 species: H. sapiens, R. norvegicus, M. musculus, A. thaliana, C. elegans, D. melanogaster, D. rerio and S. pombe. b, The surface colored according to conservation among the corresponding region of activating enzymes for 8 different UBLs: human NEDD8, ubiquitin, Sumo and ISG15, S. cerevisiae Urm1p and Apg8p/Apg12p, and E. coli MoaD and ThiS. White equals 0% identity, yellow 25% identity, orange 50% identity, and brick 75–100% identity.
The Ubc12N26-UBA3 interface
The extended conformation of the Ubc12N26 peptide is stabilized by numerous hydrogen bonds between UBA3 and the peptide backbone, resulting in burial of 950 Å2 of the Ubc12N26 peptide’s surface area. The complex is anchored by burial of two hydrophobic residues from Ubc12, Phe5 and Leu7, in a broad cavity on the top side of the groove (Fig. 6a). In contrast to the Ubc12 portion of this interaction, which is dominated by only the two hydrophobic side-chains, the UBA3 portion of this interaction involves numerous residues. UBA3’s Phe44, Cys49, His139, Ile140, Pro171, Ile174, Pro176, Leu193, Met196, Ile310, Ala311 and Pro317 line the broad cavity that interacts with Ubc12N26’s Phe5 and Leu7. Facing the opposite, bottom side of the groove, Ubc12’s Leu4 makes hydrophobic contacts with UBA3’s His32, Pro33 and Ile316, and Ubc12’s Ser6 forms a hydrogen bond with UBA3’s Ser313. The observed portion of the peptide sequence is anchored at both ends, with Ubc12’s N-terminal Met1 tucking into one end of UBA3’s broad hydrophobic cavity, and Lys12’s side-chain forming hydrogen bonds with the carbonyls from UBA3’s Arg136 and Phe138.
Figure 6.

Contributions of individual residues from Ubc12’s N-terminal peptide to E1 binding. a, Stereoview of interactions between APPBP1-UBA3 and Ubc12’s N-terminal peptide, with the structure rotated ~90° in x relative to the middle orientation in Figure 4 for a direct view of the peptide contacts. APPBP1 is shown in blue, UBA3 in red with side-chains in yellow, and Ubc12N26 in cyan. Nitrogen atoms are highlighted in blue, oxygen atoms in red, sulfur atoms in green, and hydrogen bonds are dashed. b, Effects of alanine substitutions in Ubc12 on the relative binding affinity as a substrate for NEDD8’s E1, APPBP1-UBA3 plotted as (Km of wild-type Ubc12/Km of indicated variant of Ubc12). c, Effects of mutations in UBA3 on the relative binding affinity for Ubc12 as a substrate plotted as Km of Ubc12 (wild-type APPBP1-UBA3/APPBP1-indicated variant of UBA3).
The roles of individual residues in Ubc12’s N-terminal extension were assessed by testing the effects of single alanine substitutions on Ubc12~NEDD8 thioester formation. Mutations of Phe5 and Leu7 have the greatest effect (Fig. 6b and Supplementary Table 1 online), indicating that the burial of these hydrophobic side-chains contributes the bulk of the binding energy. There are smaller effects of mutating Leu4 and Lys12. The Phe5Ala/Leu7Ala double mutation results in a ~20-fold increase in Km, and the triple mutant also containing a Leu4Ala substitution has the same effect on Km as deleting the entire 26-residue N-terminal extension (Fig. 6b and Supplementary Table 1 online). Leu4, Phe5 and Leu7 are conserved as hydrophobic residues among Ubc12 family members from different species, suggesting that their N-terminal extensions will bind their UBA3s in a similar manner.
The broad cavity from UBA3 that recognizes Ubc12’s Phe5 and Leu7 involves numerous hydrophobic side-chains. The roles of different parts of UBA3’s cavity were tested by assaying the effects of mutating side-chains from different regions of the groove (Fig. 6c and Supplementary Table 1 online). All of the mutations affect the Km for Ubc12~NEDD8 thioester formation without affecting kcat. The binding affinity for Ubc12 in the transthiolation reaction further decreases as the number of mutations increases, reflecting the importance of UBA3’s entire broad cavity in recognizing Ubc12’s N-terminal extension.
Optimal spacing between Ubc12’s N-extension and core domain
If Ubc12’s N-terminal peptide and E2 core domain bind the E1 simultaneously, then there should be a minimal length requirement for the linker between the two domains of Ubc12. To address this possibility, we made deletions and insertions between Ubc12’s residues 16 and 23 based on (1) the location of these residues in the sequence, between Ubc12’s N-terminal residues ordered in the crystal structure (residues 1–13) and E2 core domain (Ubc12 residues 27–183), (2) the low sequence complexity of these residues (sequence SAGGTKG), and (3) the finding that mutating the Leu4, Phe5, and Leu7 side-chains has the same effect as deleting the entire N-terminal peptide binding, which suggests that no other side-chains contribute significantly to the binding of Ubc12’s N-terminal 26 residues. Insertions of up to seven Gly-Ser residues and deletions of up to five residues have little effect on the kinetics of Ubc12~NEDD8 thioester formation. There is a small effect of removing six residues (Fig. 7 and Supplementary Table 1 online). In contrast, deleting seven residues is highly detrimental (Fig. 7 and Supplementary Table 1 online). To control for sequence effects, we tested the activity of a 4-residue deletion with three flanking residues mutated to alanines (Δ4A3, Fig. 7b,c). The results indicate that there is not a requirement for the sequence removed in the 7-residue deletion (Fig. 7 and Supplementary Table 1 online). Therefore, the deleterious effect of the 7-residue deletion reflects a minimum length requirement between Ubc12’s E1-docking peptide and E2 core domain.
Figure 7.
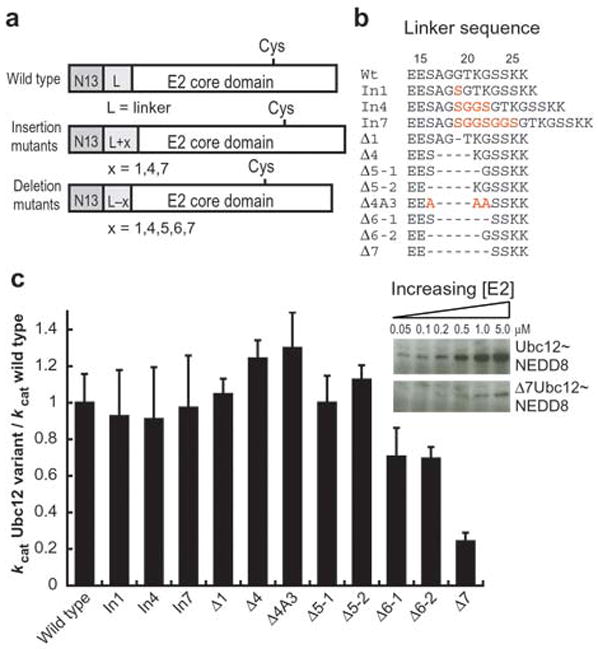
Minimal length requirement for the linker between the E1 docking motif and the E2 core domain in Ubc12. a, Schematic diagram of insertion and deletion mutants used for these experiments. b, Sequences of Ubc12 linkers (residues 14–26 in wild-type Ubc12) for insertion and deletion mutants used for these experiments. Insertions and amino acid changes are highlighted in red, and deletions are denoted by dashes. c, kcat Ubc12 variant/kcat wild-type Ubc12 for the 1, 4, and 7 residue insertion mutants (In1, In4, In7), the 1, 4, 5, 6 and 7-residue deletion mutants (Δ1, Δ4, Δ5–1, Δ5–2, Δ6–1, Δ6–2, Δ7) and an additional alanine-containing mutant to control for sequence requirements (Δ4A3), as indicated. The inset shows Ubc12~NEDD8 thioester formation for wild-type Ubc12 and the 7-residue deletion mutant, Δ7, at concentrations of 0.05, 0.1, 0.2, 0.5, 1 and 5 μM.
In contrast to removing the entire peptide extension, which affects only Km, the 7-residue linker deletion affects both Km and kcat. This is likely a combination of two effects. First, formation of the productive complex would involve Ubc12’s catalytic cysteine approaching APPBP1-UBA3’s catalytic cysteine (Fig. 8a). In the 7-residue deletion mutant, the linker between Ubc12’s catalytic cysteine-containing E2 core domain and N-terminal docking sequence is too short to allow docking of the peptide in the productive complex (Fig. 8b, left panel). Second, docking of Ubc12’s N-terminal peptide in the 7-residue deletion mutant would prevent proper docking of the E2 core domain (Fig. 8b, right panel). Thus, the interaction of Ubc12 with APPBP1-UBA3 is bipartite: both Ubc12’s N-terminal peptide and conserved E2 core domain bind the E1 simultaneously for optimal Ubc12~NEDD8 thioester formation.
Figure 8.

Model for optimal positioning of Ubc12 in the E1 structure for formation of the Ubc12-NEDD8 thioester. APPBP1 is represented in blue, UBA3 in red, NEDD8 in yellow, Ubc12 in cyan, and catalytic cysteines in green. a, Ubc12’s interaction with APPBP1-UBA3 is bipartite: both Ubc12’s N-terminal peptide and conserved E2 core domain must bind the E1 simultaneously for optimal Ubc12~NEDD8 thioester formation. b, Deletions of 6 or more residues from the linker between Ubc12’s N-terminal docking peptide and E2 core domain are deleterious, either preventing docking of the N-terminal docking sequence (left panel) or the E2 core domain (right panel). The minimum length of 8 residues between Ubc12’s N-terminal 13-residue docking peptide and E2 core domain suggests that the E2 core domain binds Cleft 1 in the APPBP1-UBA3 structure.
The distance between Ubc12’s docking peptide and E2 core domain can be estimated based on the minimum number of linker residues required for optimal activity. With an approximation of 3.5 Å per residue for an extended conformation and a minimum requirement for 8 linker residues (equivalent to the 5-residue deletion), the distance is ~28 Å between the C-terminus of the docking peptide and N-terminus of the E2 core domain. With the additional constraint that the E1 and E2 catalytic cysteines will be in proximity in the productive complex, the most likely location for the conserved catalytic E2 core domain to bind the E1 structure is in Cleft 1 (Fig. 8).
DISCUSSION
We find that Ubc12’s unique N-terminal extension tethers Ubc12 selectively to APPBP1-UBA3 via a novel type of E1–E2 interaction. First, deletion of Ubc12’s N-terminal extension reduces the Km, but not kcat, for APPBP1-UBA3-catalyzed Ubc12~NEDD8 thioester formation. Second, a peptide corresponding to Ubc12’s N-terminal 26 residues inhibits NEDD8~Ubc12 thioester formation. This effect is specific to the NEDD8 pathway, because the peptide does not affect thioester formation between ubiquitin or Sumo and their E2s (Fig. 2c). Third, deletion mutations reveal a minimum length requirement for the linker between Ubc12’s docking peptide and E2 core domain. These results suggest that for the NEDD8 pathway, the E1–E2 interaction is bipartite: both Ubc12’s docking peptide and catalytic core domain must bind the E1 simultaneously for optimal transfer of NEDD8 from E1 to E2.
The crystal structure of the APPBP1-UBA3-Ubc12N26 peptide complex reveals that Ubc12’s peptide-like extension interacts with a docking groove generated from unique loop sequences in UBA3. Even though the docking peptide and docking groove sequences are unique to Ubc12 and UBA3 family members, respectively, the adenylation domain portion of UBA3 that contains the docking groove corresponds to the most conserved domain in UBL activating enzymes13,32. Thus, the structure reveals how unique protein-protein interactions, specific for a particular UBL’s pathway, can be generated from a common structural scaffold such as an E1 adenylation domain.
Many post-translational modifications are directed to their targets via multi-enzyme cascades. In addition to UBL modification cascades, some of the best-studied examples of multi-enzyme post-translational modification cascades are serine/threonine phosphorylation pathways, such as MAPK and cyclin-dependent kinase (cdk) cascades. Several characteristics of the interaction between NEDD8’s E1 and the peptide from NEDD8’s E2 are reminiscent of interactions in these pathways. MAP kinases and cdks bind their targets through docking peptide-docking groove interactions33,34. Similar to APPBP1-UBA3’s interactions with Ubc12, the interactions between MAPKs and cdks with the downstream enzymes in their cascades are often bipartite, with both a docking peptide binding distal from the active site and the target’s phosphoacceptor sequence binding at the kinase active site. A primary function of the docking peptide interaction with these kinases is to reduce the Km of the target35. Thus, the APPBP1-UBA3-Ubc12N26 interaction reveals that common design principles underlie these very divergent multi-enzyme post-translational modification cascades.
The E1–E2[N-terminal peptide] interaction found between APPBP1-UBA3 and Ubc12 is likely to be unique to the NEDD8 pathway, because many E2s for other UBLs lack N-terminal extensions. We wonder why the NEDD8 cascade requires additional E1–E2 interactions. Clues to a possible function for this interaction come from the paradigm of docking groove – docking peptide interactions established by serine/threonine phosphorylation cascades. In MAP kinase or cdk cascades, the kinase docking groove is multifunctional, recruiting not only targets, but also regulatory proteins such as other kinases, phosphatases, and inhibitors, which all contain similar docking peptide sequences33,34. For example, part of the function of some cdk inhibitors comes from their ability to displace peptide-like docking sequences in substrates33. Interestingly, a UBA3 binding protein, But1, has recently been identified in fission yeast36. But1’s biological function is consistent with an inhibitory role in the NEDD8 pathway36. The But1 sequence contains many hydrophobic-hydrophobic-X-hydrophobic sequences similar to the Leu4-Phe5-Ser6-Leu7 sequence that we find anchors Ubc12 in UBA3’s docking groove. Future studies will reveal whether But1 or other proteins bind to UBA3 via the Ubc12 docking groove to inhibit NEDD8 conjugation.
Another possible function for the additional APPBP1-UBA3-Ubc12N26 interaction comes from the remarkable specificity of the NEDD8 pathway2–4. NEDD8 modification of cullins is one of many regulatory mechanisms controlling ubiquitin conjugation by SCF and related E3s. Consistent with its important regulatory function, NEDD8 modification of cullins is controlled not only by the conjugation pathway, but also by deconjugation by the COP9 signalosome37, underscoring the importance of precision in this pathway. The additional E1–E2 interaction may serve to ensure the selectivity of the NEDD8 conjugation cascade.
Like Ubc12, many other E2s also have their own distinct extensions at their N- and C-termini, beyond the conserved E2 core domain. The molecular functions of only a handful of these extensions are known38–41, and to date, the structural basis for protein-protein interactions mediated by these extensions remain elusive. One of the best understood extensions is in the E2 Ubc2p, which interacts with the E3, Ubr1p. While the Ubc2p-Ubr1p interaction is likely to involve interactions common to E2-E3 complexes because it requires Ubr1p’s RING domain, Ubc2p’s C-terminal extension is also known to stabilize the interaction with Ubr1p42. Therefore, it is likely that the extensions on other E2s function in a manner analogous to Ubc12’s, strengthening interactions between enzymes in other UBL modification cascades.
The importance of Ubc12’s N-terminal extension is further underscored by the detrimental effect of the Ubc12ΔN deletion in our cell proliferation assay. The best-characterized targets of Ubc12 are cullins2–4,12,9,5,6,10,11,7,8,43, although we do not yet know which targets are important in our assay. Cul1, Cul2, Cul3 and Cul4a-containing ubiquitin ligases are known to promote the degradation of key regulators of cell proliferation, including cyclins, cyclin-dependent kinase inhibitors, proteins involved in DNA replication, proteins involved in mitotic spindle assembly, transcription factors, and proteins involved in signal transduction (reviewed in 43). It will be of interest to determine which targets of Ubc12-mediated NEDD8 conjugation are involved in the CSF-1-dependent proliferation. Our finding that Ubc12ΔN is defective in this cell proliferation assay raises the possibility that APPBP1-UBA3 and Ubc12 may serve as good targets for antimitogenic agents.
METHODS
Protein and peptide preparation
All constructs were generated by standard PCR/ligation molecular biology methods. The entire coding sequence for each construct was verified by automated sequencing. All proteins were expressed as GST-fusions in E. coli strains BL21(DE3), BL21Gold(DE3), or BL21(DE3) RIL codon-enhanced strains (Novagen and Stratagene), from either pGEX4T1 or pGEX2TK (Pharmacia). The proteins were initially purified as GST-fusions by glutathione affinity chromatography, and were subsequently purified to homogeneity following thrombin cleavage using a combination of ion exchange, gel filtration and glutathione affinity chromatography44,45,26,28. A mutant form of APPBP1-UBA3 lacking APPBP1’s 254–258 loop and UBA3’s N-terminal 11 residues, with UBA3’s catalytic Cys216 mutated to Ala was used for crystallization26. Biochemical studies used full-length, wild-type E1s44,45,28 or APPBP1-UBA3 mutants expressed and purified as for the wild-type E1; wild-type or mutant versions of Ubc1228 as indicated; and the truncated active versions of ubiquitin and the UBLs human NEDD8 and S. cerevisiae Sumo (Smt3p) terminating with the sequence Gly-Gly44,26,28. The human Rbx1-Cul1 (split) complex was obtained in soluble form from E. coli by simultaneously coexpressing Rbx1, Cul1’s N-terminal domain (residues 1–411), and Cul1’s C-terminal domain (residues 411–776 at the C-terminus). The complex consists of Cul1’s N- and C-terminal domains associated non-covalently with each other and with Rbx1. The split Cul1-Rbx1 complex was shown previously to possess the same 3-dimensional structure and ubiquitin ligase activity as the Rbx1 complex with Cul1 expressed as a single polypeptide in insect cells45. The 26-residue peptide corresponding to Ubc12’s N-terminus, termed Ubc12N26 (sequence MIKLFSLKQQKKEEESAGGTKGSSKK), the five different selenomethionine-substituted Ubc12N26 peptides, and the ScrambledN26 peptide (sequence MKFQLKEIEAGKSKLKSGTSEKGQKS) were chemically synthesized with C-terminal amide blocking groups. The peptides were purified by reversed-phase HPLC, and their identities confirmed by mass spectrometry. The lyophilized peptides were dissolved in 25 mM HEPES, 150 mM NaCl, 5 mM DTT, pH 7.5 at a concentration of 10 mM.
Biochemical assays
All biochemical assays were performed in 10 μl in 50 mM Tris-HCl (pH 7.6), 50 mM NaCl, 10 mM MgCl2, 5 mM ATP, 1 mM DTT, 0.3 U ml−1 inorganic pyrophosphatase, 0.3 U ml−1 creatine phophatase, 5 mM creatine phosphate, 2 mg ml−1 ovalbumin, with 5 μM NEDD8, ubiquitin or S. cerevisiae Sumo (Smt3p) phosphorylated at the N-terminal PKA site (from pGEX2TK) with [γ-32P]ATP28, at 18°C, which is the ambient room temperature for our dedicated radioactivity laboratory. Reactions were quenched with an equal volume of 2X SDS sample buffer. Reactions were carried out with 1 nM of the appropriate E1, except for the modification of Cul1 by Ubc12ΔN shown in Fig. 2, which used 1, 5, 10, 15, and 20 nM APPBP1-UBA3 as indicated, and 3 μM Rbx1-Cul1. Kinetics of E1–E2 transthiolation were determined from at least 7 different Ubc12 concentrations per curve, ranging from 0 to 50 μM, with reactions stopped after 30 seconds46. The Cul1 modification and peptide inhibition assays were stopped after 60 seconds. Proteins were resolved by SDS-PAGE, dried and visualized by autoradiography. Bands were quantitated using a STORM860 Phorphorimager and ImageQuant software (Molecular Dynamics). Known amounts of 32P-labeled NEDD8 and [γ-32P]ATP were exposed together as standards. kcat and Km for E1–E2 transthiolation were determined from Lineweaver-Burke plots. The Ki of Ubc12N26 for APPBP1-UBA3-catalyzed Ubc12~NEDD8 thioester formation was determined from five different Ubc12 concentrations (0.1–2 μM) with varying concentrations of Ubc12N26 (0–1 mM), and calculated from Dixon plots using SigmaPlot 8.0 software (SPSS).
Cell culture, antibody production and immunoblotting
NIH3T3 mouse fibroblast lines expressing the wild-type or mutant [Y809F] human CSF-1 receptors were previously described31. For identification of Ubc12 in the human CSF-1-dependent mitogenesis assay, cells expressing human CSF-1R [Y809F] were infected with a retroviral MaRX rat cDNA library generously provided by Dr Gregory Hannon (Cold Spring Harbor Labs, NY)47. Infected cells were plated on 100 mm plates and foci of transformed cells were recovered by cylinder cloning. Integrated proviruses were recovered by digestion of the cell’s high molecular weight DNA with the Cre recombinase, as described47, and Ubc12 was recovered multiple times from the soft agar clones. The protein sequences of rat and human Ubc12 are identical, so human Ubc12 and Ubc12ΔN were subcloned into the pBabepuro retroviral vector for preparing high titer retroviruses in 293T cells. NIH3T3 cells expressing the wild type or Y809F mutant human CSF-1R were infected with retroviruses expressing Ubc12 or Ubc12ΔN, and cloned in soft agar two days after infection in the presence or absence of recombinant human CSF-1 (generously provided by Michael Clark). Lysates were also prepared in parallel and 24 μg of each protein lysate or 50 ng of purified bacterially-produced wild-type or mutant Ubc12 were separated by SDS-PAGE. Proteins were analyzed by immunoblotting, with Ubc12 detected by rabbit polyclonal antiserum raised against residues 169–180 of Ubc12 (sequence RGGYIGSTYFER, generated by Rockland Immunochemicals). Anti-actin serum (C-11, Santa Cruz Biotechnology) was used to control for loading.
Crystallization, data collection and structure determination
APPBP1[Δ254–258]-UBA3[ΔN11,C216A] and Ubc12N26 were mixed at 1:5 molar ratio, and crystals were grown at room temperature by hanging and sitting drop vapor diffusion methods by mixing the complex with an equal volume of reservoir solution containing 9.5%-12.5% (v/v) PEG 10k, 0.2 M KCl, 0.1 M MES, 5 mM DTT, pH 6.5. The crystals form in P212121 with a = 92.4 Å, b =122.8 Å, c =195.9 Å, with two complexes in the asymmetric unit. Crystals were flash-frozen in 12.5% (v/v) PEG 10k, 0.2 M KCl, 0.1 M MES, 5 mM DTT, 20% (v/v) MPD, pH 6.5, prior to data collection at the SERCAT beamline at the Advanced Photon Source (APS), at the X25 beamline at the National Synchrotron Light Source, and at the 8.3.1 beamline at the Advanced Light Source. Reflection data were indexed, integrated, and scaled using HKL2000 or DENZO and SCALEPACK48 or Elves49. Initial phases were obtained by molecular replacement using Elves49 with the coordinates of apo APPBP1-UBA328 as a search model, or by refining the apo APPBP1-UBA3 structure against the new data using CNS50. Electron density for the peptide was readily visible after preliminary refinement. Because only 13 of the 26 residues in the Ubc12N26 peptide were visible in the structure, we wished to obtain independent experimental data to verify the structure of the peptide. We grew crystals of the complex with individual selenomethionine substitutions in place of Met1, Ile2, Leu4, Leu7 and Gln10, collected data to better than 3.5 Å resolution at the peak wavelength for the incorporated selenium (~0.9796Å), and identified the locations of the seleniums by anomalous difference fourier analysis, with phases obtained by using the apo APPBP1-UBA3 structure28 as a search model with Elves49. Each selenium is located on the assigned side-chain in the native structure (Fig. 3a), providing experimental validation for the structure of the peptide. The final model was refined at 2.6 Å using CNS50, and it contains two copies of the complex. The electron density is significantly better over one copy, which contains Ubc12N26 residues 1–13, APPBP1 residues 1–253, 259–534, UBA3 residues 12–355, 361–384, and an additional 3 residues due to cloning at the N-terminus and 43 residues in the C-terminal domain built as polyalanine, owing to weak side-chain density and lack of density for loops precluding unambiguous determination of the side-chains in this region. The other copy contains Ubc12N26 residues 4–13, APPBP1 residues 7–199, 211–253, 259–534, UBA3 residues 17–354, 361–384, and an additional 33 residues in the C-terminal domain built as polyalanine. Side-chains lacking electron density were not modeled. The final model has excellent geometry, with no Ramachandran outliers in disallowed regions. Details of refinement are given in Table 1.
Supplementary Material
Acknowledgments
We are grateful to M.S. Podgorski for initial characterization of the Ubc12ΔN mutant, to P.D. Jeffrey, N.P. Pavletich, M. Pagano, T. Izard, H. Walden and other members of the Schulman lab for many helpful discussions, to D.L. Minor for critical reading of the manuscript, to P.J. Murray for assistance with Figure 8, to C. Ross for crystallography support, to G. Hannon for the MaRX library and initial experiments with Ubc12 in cell proliferation assays, to S. Olsen and K. Rakestraw for expert DNA synthesis and sequencing, and to J. Tanamachi and staff at the 8.3.1 beamline at ALS, M. Becker and staff at the X25 beamline at NSLS, and staff at the SERCAT beamline at APS for synchrotron support. This work was supported by American Lebanese Syrian Associated Charities, the NIH (P30CA21765 NCI Cancer Center Core grant to St. Jude, R01GM69530 to BAS, P01CA071907 to MFR), the DOD (DAMD17-03-1-0420), a grant from Phillip and Elizabeth Gross, and a Pew Scholar in Biomedical Sciences Award to BAS, and a St. Jude Special Fellowship to DH.
Footnotes
Coordinates and structure factors for the APPBP1-UBA3-Ubc12N26 structure have been deposited to the RCSB under the PDB accession code 1TT5.
References
- 1.Schwartz DC, Hochstrasser M. A superfamily of protein tags: ubiquitin, SUMO and related modifiers. Trends Biochem Sci. 2003;28:321–328. doi: 10.1016/S0968-0004(03)00113-0. [DOI] [PubMed] [Google Scholar]
- 2.Lammer D, et al. Modification of yeast Cdc53p by the ubiquitin-related protein rub1p affects function of the SCFCdc4 complex. Genes Dev. 1998;12:914–926. doi: 10.1101/gad.12.7.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liakopoulos D, Doenges G, Matuschewski K, Jentsch S. A novel protein modification pathway related to the ubiquitin system. Embo J. 1998;17:2208–2214. doi: 10.1093/emboj/17.8.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osaka F, et al. A new NEDD8-ligating system for cullin-4A. Genes Dev. 1998;12:2263–2268. doi: 10.1101/gad.12.15.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Read MA, et al. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell Biol. 2000;20:2326–2333. doi: 10.1128/mcb.20.7.2326-2333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu K, Chen A, Pan ZQ. Conjugation of Nedd8 to CUL1 enhances the ability of the ROC1-CUL1 complex to promote ubiquitin polymerization. J Biol Chem. 2000;275:32317–32324. doi: 10.1074/jbc.M004847200. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Furukawa M, Matsumoto T, Xiong Y. NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol Cell. 2002;10:1511–1518. doi: 10.1016/s1097-2765(02)00783-9. [DOI] [PubMed] [Google Scholar]
- 8.Zheng J, et al. CAND1 binds to unneddylated CUL1 and regulates the formation of SCF ubiquitin E3 ligase complex. Mol Cell. 2002;10:1519–1526. doi: 10.1016/s1097-2765(02)00784-0. [DOI] [PubMed] [Google Scholar]
- 9.Osaka F, et al. Covalent modifier NEDD8 is essential for SCF ubiquitin-ligase in fission yeast. Embo J. 2000;19:3475–3484. doi: 10.1093/emboj/19.13.3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tateishi K, Omata M, Tanaka K, Chiba T. The NEDD8 system is essential for cell cycle progression and morphogenetic pathway in mice. J Cell Biol. 2001;155:571–579. doi: 10.1083/jcb.200104035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurz T, et al. Cytoskeletal regulation by the Nedd8 ubiquitin-like protein modification pathway. Science. 2002;295:1294–1298. doi: 10.1126/science.1067765. [DOI] [PubMed] [Google Scholar]
- 12.Pozo JC, Timpte C, Tan S, Callis J, Estelle M. The ubiquitin-related protein RUB1 and auxin response in Arabidopsis. Science. 1998;280:1760–1763. doi: 10.1126/science.280.5370.1760. [DOI] [PubMed] [Google Scholar]
- 13.Hochstrasser M. Evolution and function of ubiquitin-like protein-conjugation systems. Nat Cell Biol. 2000;2:E153–157. doi: 10.1038/35019643. [DOI] [PubMed] [Google Scholar]
- 14.Pickart CM. Mechanisms Underlying Ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 15.Huang L, et al. Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science. 1999;286:1321–1326. doi: 10.1126/science.286.5443.1321. [DOI] [PubMed] [Google Scholar]
- 16.Johnston SC, Riddle SM, Cohen RE, Hill CP. Structural basis for the specificity of ubiquitin C-terminal hydrolases. EMBO J. 1999;18:3877–3887. doi: 10.1093/emboj/18.14.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Q, et al. The binding interface between an E2 (UBC9) and a ubiquitin homologue (UBL1) J Biol Chem. 1999;274:16979–16987. doi: 10.1074/jbc.274.24.16979. [DOI] [PubMed] [Google Scholar]
- 18.Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell. 2000;5:865–876. doi: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- 19.Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin- protein ligases. Cell. 2000;102:533–539. doi: 10.1016/s0092-8674(00)00057-x. [DOI] [PubMed] [Google Scholar]
- 20.Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol. 2001;8:833–837. doi: 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- 21.Hamilton KS, et al. Structure of a conjugating enzyme-ubiquitin thiolester intermediate reveals a novel role for the ubiquitin tail. Structure. 2001;9:897–904. doi: 10.1016/s0969-2126(01)00657-8. [DOI] [PubMed] [Google Scholar]
- 22.Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108:345–356. doi: 10.1016/s0092-8674(02)00630-x. [DOI] [PubMed] [Google Scholar]
- 23.Hu M, et al. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041–1054. doi: 10.1016/s0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- 24.VanDemark AP, Hill CP. Structural basis of ubiquitylation. Curr Opin Struct Biol. 2002;12:822–830. doi: 10.1016/s0959-440x(02)00389-5. [DOI] [PubMed] [Google Scholar]
- 25.Verdecia MA, et al. Conformational Flexibility Underlies Ubiquitin Ligation Mediated by the WWP1 HECT Domain E3 Ligase. Mol Cell. 2003;11:249–259. doi: 10.1016/s1097-2765(02)00774-8. [DOI] [PubMed] [Google Scholar]
- 26.Walden H, et al. The Structure of the APPBP1-UBA3-NEDD8-ATP Complex Reveals the Basis for Selective Ubiquitin-like Protein Activation by an E1. Mol Cell. 2003;12:1427–1437. doi: 10.1016/s1097-2765(03)00452-0. [DOI] [PubMed] [Google Scholar]
- 27.Furukawa M, Zhang Y, McCarville JTO, Xiong Y. The CUL1 C-terminal sequence and ROC1 are required for efficient nuclear accumulation, NEDD8 modification, and ubiquitin ligase activity of CUL1. Mol Cell Biol. 2000;20:8185–8197. doi: 10.1128/mcb.20.21.8185-8197.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walden H, Podgorski MS, Schulman BA. Insights into the ubiquitin transfer cascade from the structure of the E1 for NEDD8. Nature. 2003;422:330–334. doi: 10.1038/nature01456. [DOI] [PubMed] [Google Scholar]
- 29.Roussel MF, Cleveland JL, Shurtleff SA, Sherr CJ. Myc rescue of a mutant CSF-1 receptor impaired in mitogenic signalling. Nature. 1991;353:361–363. doi: 10.1038/353361a0. [DOI] [PubMed] [Google Scholar]
- 30.Roussel MF, Theodoras AM, Pagano M, Sherr CJ. Rescue of defective mitogenic signaling by D-type cyclins. Proc Natl Acad Sci USA. 1995;92:6837–6841. doi: 10.1073/pnas.92.15.6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roussel MF, Sherr CJ. Mouse NIH 3T3 cells expressing human colony-stimulating factor 1 (CSF-1) receptors overgrow in serum-free medium containing human CSF-1 as their only growth factor. Proc Natl Acad Sci USA. 1989;86:7924–7927. doi: 10.1073/pnas.86.20.7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lake MW, Wuebbens MM, Rajagopalan KV, Schindelin H. Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB-MoaD complex. Nature. 2001;414:325–329. doi: 10.1038/35104586. [DOI] [PubMed] [Google Scholar]
- 33.Endicott JA, Noble ME, Tucker JA. Cyclin-dependent kinases: inhibition and substrate recognition. Curr Opin Struct Biol. 1999;9:738–744. doi: 10.1016/s0959-440x(99)00038-x. [DOI] [PubMed] [Google Scholar]
- 34.Biondi RM, Nebreda AR. Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem J. 2003;372:1–13. doi: 10.1042/BJ20021641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeda DY, Wohlschlegel JA, Dutta A. A bipartite substrate recognition motif for cyclin-dependent kinases. J Biol Chem. 2001;276:1993–1997. doi: 10.1074/jbc.M005719200. [DOI] [PubMed] [Google Scholar]
- 36.Yashiroda H, Tanaka K. But1 and But2 proteins bind to Uba3, a catalytic subunit of E1 for neddylation, in fission yeast. Biochem Biophys Res Commun. 2003;311:691–695. doi: 10.1016/j.bbrc.2003.10.058. [DOI] [PubMed] [Google Scholar]
- 37.Lyapina S, et al. Promotion of NEDD8-CUL1 conjugate cleavage by COP9 signalosome. Science. 2001;292:1382–1385. doi: 10.1126/science.1059780. [DOI] [PubMed] [Google Scholar]
- 38.Kolman CJ, Toth J, Gonda DK. Identification of a portable determinant of cell cycle function within the carboxyl-terminal domain of the yeast CDC34 (UBC3) ubiquitin conjugating (E2) enzyme. EMBO J. 1992;11:3081–3090. doi: 10.1002/j.1460-2075.1992.tb05380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Silver ET, Gwozd TJ, Ptak C, Goebl M, Ellison MJ. A chimeric ubiquitin conjugating enzyme that combines the cell cycle properties of CDC34 (UBC3) and the DNA repair properties of RAD6 (UBC2): implications for the structure, function and evolution of the E2s. EMBO J. 1992;11:3091–3098. doi: 10.1002/j.1460-2075.1992.tb05381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haldeman MT, Xia G, Kasperek EM, Pickart CM. Structure and function of ubiquitin conjugating enzyme E2-25K: the tail is a core-dependent activity element. Biochemistry. 1997;36:10526–10537. doi: 10.1021/bi970750u. [DOI] [PubMed] [Google Scholar]
- 41.Morrison A, Miller EJ, Prakash L. Domain structure and functional analysis of the carboxyl-terminal polyacidic sequence of the RAD6 protein of Saccharomyces cerevisiae. Mol Cell Biol. 1998;8:1179–1185. doi: 10.1128/mcb.8.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madura K, Dohmen RJ, Varshavsky A. N-recognin/Ubc2 interactions in the N-end rule pathway. J Biol Chem. 1993;268:12046–12054. [PubMed] [Google Scholar]
- 43.Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K. Nedd8 on cullin: building an expressway to protein destruction. Oncogene. 2004;23:1985–1997. doi: 10.1038/sj.onc.1207414. [DOI] [PubMed] [Google Scholar]
- 44.Bencsath KP, Podgorski MS, Pagala VR, Slaughter CA, Schulman BA. Identification of a multifunctional binding site on Ubc9p required for Smt3p conjugation. J Biol Chem. 2002;277:47938–47945. doi: 10.1074/jbc.M207442200. [DOI] [PubMed] [Google Scholar]
- 45.Zheng N, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 46.Bohnsack RN, Haas AL. Conservation in the mechanism of Nedd8 activation by the human AppBp1-Uba3 heterodimer. J Biol Chem. 2003;278:26823–26830. doi: 10.1074/jbc.M303177200. [DOI] [PubMed] [Google Scholar]
- 47.Hannon GJ, et al. MaRX: an approach to genetics in mammalian cells. Science. 1999;283:1129–1130. doi: 10.1126/science.283.5405.1129. [DOI] [PubMed] [Google Scholar]
- 48.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;176:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 49.Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci USA. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brünger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.