

Abstract
A new class of [2]catenanes containing zinc(II)-porphyrin (ZnP) and/or [60]fullerene (C60) as appended groups has been prepared. A complete description of the convergent synthetic approach based on Cu(I) template methodology and “click” 1,3-dipolar cycloaddition chemistry is described. This new electron donor-acceptor catenane family has been subjected to extensive spectroscopic, computational, electrochemical and photophysical studies. 1H NMR spectroscopy and computational analysis have revealed that the ZnP-C60-[2]catenane adopts an extended conformation with the chromophores as far as possible from each other. A detailed photophysical investigation has revealed that upon irradiation, the ZnP singlet excited state initially transfers energy to the (phenanthroline)2-Cu(I) complex core, producing a metal-to-ligand charge transfer (MLCT) excited state, which in turn transfers an electron to the C60 group, generating the ZnP–[Cu(phen)2]2+–C60·− charge separated state. A further charge shift from the [Cu(phen)2]2+ complex to the ZnP subunit, competitive with decay to the ground state, leads to the isoenergetic long distance ZnP·+–[Cu(phen)2]+–C60·− charge separated radical pair state, which slowly decays back to the ground state on the microsecond time scale. The slow rate of back electron transfer indicates that in this interlocked system, as in previously studied covalently linked ZnP-C60 hybrid materials, this process occurs in the Marcus inverted region.
INTRODUCTION
In nature, long-range electron transfer is an essential process, playing a fundamental role in functions indispensable to life such as photosynthesis, respiration, vision and redox mediated enzyme catalysis.1 The importance and complexity of natural photosynthesis have led many researchers to look for ways to study the fundamental chemistry of the charge separation processes involved in the conversion of sunlight into chemical energy in natural photosynthesis.2
Important progress toward the understanding of natural systems has been made in recent years.3 Basically, the incident light is captured by organic pigments and the resulting electronic excitation energy is transduced to the reaction center containing electron donor (D) and acceptor (A) moieties, where a charge separation process occurs to generate a redox potential energy that is used to drive chemical reactions needed to keep the organism alive.3
Although many fundamental features concerning rate and energetic of the electron transfer reactions in natural photosynthesis have been elucidated,2,3 natural systems are extremely complex and many important details of the electron transfer mechanisms still remain to be resolved. Investigation of artificial photosynthetic systems that mimic the behavior of the natural system has emerged as an alternative approach to improve our understanding of the natural light conversion process.4
Many efforts have been undertaken to assemble appropriate D-A groups into a suitably organized molecular topography in order to achieve rapid and efficient ET reactions and generate long-lived charge separated states.5,6 Because of their remarkable spectroscopic and electrochemical properties, as well as their structural relationship to chlorophyll, porphyrinic pigments are the most widely used electron donors in artificial photosynthetic model systems.5e Fullerenes have proven to be superior electron acceptors.5 Due to their low reduction potentials and small reorganization energy, they simultaneously enhance dynamics for ET while inhibiting charge recombination (CR). As a result, a very large number of porphyrin-fullerene systems have been synthesized and their electrochemical and photophysical properties have been extensively investigated.6
Recently, supramolecular concepts have been introduced into the synthesis of porphyrin-fullerene photosynthetic model systems.7 Among the most promising of these are mechanically interlocked electron donor-acceptor systems,8 in which the non-covalent linkage of D and A moieties mimics the natural system more closely than covalently linked D-A systems, which have received the greatest attention to date.6
Catenanes are comprised of two or more interlocked rings, while rotaxanes possess a ring threaded on a rod bearing terminal bulky substituents to prevent dissociation. In such systems, constituent photoactive components are located within a fixed distance without any covalent linkage, allowing the components to undergo submolecular motions by application of an external stimulus, such as light, electrochemical or chemical inputs.9 This molecular topology has been elegantly explored as the dominant principle for construction of molecular motors, shuttles, muscles and information storage devices.10
Rotaxanes incorporating both porphyrin and C60 moieties have been prepared and studied by our group11 and others.8k-p In our first generation systems, modeled on the Cu(I)-templated purely porphyrinic systems elegantly studied by Sauvage and co-workers,8a electronic excitation induces a series of long-range energy and electron transfer processes resulting in the generation of long-lived charge separated radical pair (CSRP) states, i.e., porphyrin·+-C60·−. CSRP lifetimes between 0.59 and 1.17 μs in dichloromethane (DCM) were observed for three first generation rotaxanes in which C60 is covalently linked to the polyether macrocycle, while two ZnP groups serve as termini on the thread.11a,d For rotaxanes in which the groups were switched, i.e., ZnP covalently linked to the macrocycle and C60 moieties now serving as blocking groups on the threads, CSRP lifetimes of 320 ns, 730 ns and 29 us were observed in DCM; in THF the lifetimes were even longer, 890 ns and 32 μs.11b,d The inverse dependence of the rates of charge recombination (CR) on solvent polarity for these interlocked DA materials is evidence that CR is occurring in the Marcus inverted region, as is the case with covalently linked ZnP-C60 hybrid systems.5,6,12
The properties of related DA systems with catenane topology have remained unknown until the present study. The main problem has been that fullerene and porphyrin chromophores are often not chemically compatible with the classical procedures used for the final cyclization step in catenane synthesis.9,10 In particular, introduction of fullerene moieties into the system without damage to the interlocked structure as well as to the porphyrin moiety remained a synthetic challenge.13 As far as we know, only two examples of [2]catenanes bearing a C60 moiety have been reported,14 and the method employed in these studies (involving a π-donor/π-acceptor template) resulted in quite poor yields (5-18%).
The study of the efficiency, energetic and dynamics of ET processes in porphyrin-fullerene-[2]catenanes is of interest for fundamental reasons, including i) better understanding of natural photosynthesis due to the closer resemblance of mechanically interlocked systems vis a vis covalent systems to the topology of the natural photosynthetic reaction center,2a,8a and ii) the longer lifetimes anticipated for charge separated states of catenanes compared to rotaxane analogues. The center-to-center distance between the chromophores is larger in the catenanes than it is in the more flexible rotaxanes, which will affect the crucial electronic coupling parameter in ET dynamics.1,8i,12.
We have found15 that Cu(I)-catalyzed Huisgen-Sharpless-Meldal 1,3-dipolar cycloaddition of azides and alkynes (the CuAAC or “click” reaction)16 coupled with Sauvage’s Cu(I) template synthesis17 is a very effective synthetic approach for the preparation of functionalized macrocycles as well as [2] and [3]catenanes.15a,b,d Taking advantage of the mild conditions of CuAAC reactions, and its tolerance to potentially sensitive groups, including porphyrins and fullerenes,18 we have developed a straightforward strategy to prepare rotaxanes bearing electron donors and C60 in very high yields.15c In the present work, we successfully applied this versatile methodology to the synthesis of the first [2]catenate bearing zinc(II)-porphyrin (ZnP) and C60 moieties as appended groups (compound 1, Figure 1). The new catenate 1 was subjected to extensive spectroscopic, electrochemical, and photophysical studies, along with those of three model [2]catenates (see Figure 1): a [2]catenate without any additional chromophores (2), a [2]catenate bearing only a porphyrin group (3), and a [2]catenate bearing only a C60 moiety (4), all of which were synthesized using the same CuAAC methodology.
Figure 1.

Structures of the new porphyrin-C60-[2]catenates and the model [2]catenates used in the electrochemical and photophysical investigations.
Results and Discussion
1. Molecular Design and Synthesis
The synthetic objectives of the present investigation were inspired by the concept that Cu(I) ions can act as both catalyst and template upon combining 1,3-dipolar cycloaddition CuAAC reaction technology16 with Sauvage’s transition metal-template approach17 for the synthesis of interlocked structures. In the metal template approach, a pair of phenanthroline (phen) ligands are held together thanks to the gathering properties of the Cu(I) ion template, generating a distorted tetrahedral [Cu(phen)2]+ complex, which is subjected to cyclization or stoppering reactions leading to catenanes and rotaxanes, respectively.17 Since Cu(I) ion is also the active species involved in the CuAAC reaction,16 and since catenanes and rotaxanes can be prepared from the same common [Cu(phen)2]+ pseudorotaxane precursor (Chart 1),17 a convenient one-pot procedure using CuAAC reactions in the final stoppering or cyclization step was envisioned for preparation of both types of structures.
Chart 1.

General approach to the synthesis of porphyrin-fullerene interlocked molecules using Cu(I) template synthesis and the CuAAC reaction.
More specifically, we have shown that porphyrin/fullerene rotaxanes15c can be efficiently prepared in high yield by CuAAC reaction between a diazido string-like phen fragment threaded through a phen-macrocycle bearing a C60 moiety and a monoalkynyl-functionalized porphyrin (See Chart 1, path A for a schematic description). This showed that the conditions used for the assembly of these supramolecular systems, and the “click” catalyst we have proposed for the final stoppering reaction, are compatible with porphyrins, fullerenes and the especially sensitive [Cu(phen)2]+ pseudorotaxane precursor. These findings served as a challenge to prepare a porphyrin-C60 [2]catenate using a similar strategy (Chart 1, path B).
The building blocks and precursors needed for the synthesis of catenate 1 by this approach are depicted in Scheme 1. The synthesis began with oligo(ethyleneglycol)-2,9-diphenyl-1,10-phenanthroline derivative.17d Macrocycle 7 was prepared by coupling of 5 and malonic acid 6 using bis(2-oxo-3-oxazolidinyl)phosphonic chloride (BOP-Cl) as the coupling agent. C60 was attached to 7 using the Bingel-Hirsch protocol13 to give macrocycle 8. Compound 5 was converted into ditosylate 9, which was then converted into diazidophenanthroline derivative 10 by reaction with sodium azide. The threading of 10 through the macrocycle 8, using Sauvage’s Cu(I) template protocol,17 quantitatively afforded the C60-linked pseudorotaxane precursor 11, as revealed by 1H NMR spectroscopy.
Scheme 1.

Precursors and synthetic route for preparation of catenate 1.
a) BOP-Cl, Et3N, CH2Cl2, rt, 12 h, 45 % yield; b) C60, DBU, I2, toluene, rt, 24 h, 55 % yield; c) TsCl, Et3N, CH2Cl2, 0°C for 4 h and rt for 20 h, 75 % yield; d) NaN3, DMF, 80°C, 24 h, 93 % yield; e) [Cu(CH3CN)4][PF6], CH2Cl2/CH3CN, rt, 3 h, quantitative; f) BF3OEt2, PPh4Cl, CH2Cl2, rt, 1 h and then 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ), rt, 12 h, 9 % yield; g) Zn(OAc)2, CH2Cl2/CH3OH, reflux, 6 h, quantitative; h) TBAF, THF, rt, 30 min, quantitative; i) CuI, sodium ascorbate, SBP, DBU, H2O/EtOH, rt, 12 h, 57 % yield.
The required meta-diethynylphenyl zinc(II)-porphyrin derivative 14 was prepared by condensation of 3,5-di-tert-butylbenzaldehyde 12 (see Experimental Section for a new and improved synthesis of this material), 3,5-di-(TMS-ethynyl)benzaldehyde 13 and pyrrole, following the Lindsey procedure.19 The free base porphyrin 14a was then metallated using zinc acetate to give 14b. Deprotection of the alkyne group using tetrabutylammonium fluoride (TBAF) gave porphyrin 14 in 9% overall yield, after purification by column chromatography.
The final “double-click” cyclization reaction between the terminal azide groups in phen precursor 11 with porphyrin 14, the crucial step in the preparation of the target catenate, was successfully accomplished following a procedure similar to that developed in our laboratory for the synthesis of the analogous porphyrin-C60 rotaxanes15c (for details, see Experimental). Workup and purification by column chromatography gave target [2]catenate 1 bearing porphyrin and C60 pendant groups as a purple solid in 57% yield.20
The catenate model compounds 2, 3 and 4 (Figure 1) were prepared using the same strategy (see Scheme 2). For the preparation of 2 and 3, diazidophen 10 was threaded through macrocycle 1517d to give pseudorotaxane 16. Subjecting 16 and 1,3-diethynylbenzene or porphyrin 14 to the same CuAAC conditions described for 115c yielded catenates 2 and 3 in 70% and 65% yields, respectively.20 Catenate 4 was obtained in 75% yield from “click” catenation reaction between pseudorotaxane 11 and 1,3-diethynylbenzene, following the same procedure described for 1.15c The reference compounds needed for electrochemical and photophysical studies, namely, malonate-C60 1713 and zinc(II)-tetraphenyl-porphyrin-triazole-benzaldehyde 186j (see Figure 2) were prepared following literature procedures.
Scheme 2.

Precursors and synthetic route for preparation of [2]catenate model compounds.
a) [Cu(CH3CN)4][PF6], CH2Cl2/CH3CN, rt, 3 h, quantitative; b) CuI, sodium ascorbate, SBP, DBU, H2O/EtOH, rt, 12 h.
Figure 2.
Porphyrin and C60 reference compounds used in the electrochemical and photophysical investigations.
2. Structural Characterization
Catenates 1, 2, 3 and 4 were fully characterized by standard spectroscopic techniques. Key evidence supporting the structure assignment to catenate 1 came from MALDI-TOF analysis. The general pattern of the mass spectrum (see Figure 3) is very characteristic of catenanes,21 featuring stepwise fragmentation of each ring from the interlocked system. Thus, one observes the molecular ion peak for 1 at m/z 3215 (M – PF6)+ as well as the peaks for the individual macrocycles at m/z 1805 (ZnP-macrocycle + Cu)+ and m/z 1477 (C60-macrocycle + Cu)+. The absence of fragments between the molecular ion peak and the peaks corresponding to the individual macrocycles provided strong evidence for the proposed structure for 1 in which the ZnP and C60 moieties are covalently linked to the [Cu(phen)2]+-[2]catenate framework.
Figure 3.

MALDI-TOF mass spectrum of catenate 1 (positive mode, α-cyano-4-hydroxycinnamic acid (CCA) matrix). The absence of peaks between the molecular ion peak and the peaks corresponding to the individual macrocycles is characteristic of catenane structures.15,21  = Cu(I) ion;
= Cu(I) ion;  =Zn(II)-porphyrin and
=Zn(II)-porphyrin and  = C60.
= C60.
The 1H NMR spectrum of 1 (Figure 4) exhibits all the peaks expected15,17,22 for a non-symmetrical–[Cu(phen)2]+-[2]catenate. Two sets of phen protons were observed, as the protons of the phen macrocycle bearing ZnP resonate at lower fields than those of the phen fragment on the C60-bearing macrocycle. The expected upfield shifts for the phenyl protons in the two phen fragments entwined around the Cu(I) core (identified as o and m, Figure 4) are observed. This shielding effect is similar to that observed in other metallo-catenanes.17 The triazole ring protons appear at 8.11 ppm, while the adjacent CH2 protons are in the usual region between 4.50-5.00 ppm.15,23 The CH2 groups adjacent to the malonate moiety on the C60-containing macrocycle resonate at 4.56 ppm. The pyrrolic protons in the porphyrin moiety are observed between 8.50-8.80 ppm, while the protons belonging to the oligo(ethyleneglycol) linkers resonate in the expected region of 3.0-4.0 ppm.
Figure 4.

1H NMR spectra (800 MHz, CD3CN, 298 K) of catenate 1. The red peaks correspond to the protons of the zinc-porphyrin moiety. The peaks in green are assigned to the CH2 protons adjacent to the triazole ring and the proton on the triazole ring itself, while the black and the blue peaks are assigned to the protons of the [Cu(phen)2]+ complex core and the oligo(ethyleneglycol) linkers, respectively.
The 1H NMR analysis indicates that catenate 1 adopts an extended conformation, in which the porphyrin and C60 moieties are as far as possible from each other. Molecular modeling studies on catenate 1 (Spartan, PM3 minimization, Figure 5) confirmed this hypothesis.15c In its lowest-energy conformation, the porphyrin and C60 pendant groups reside at the periphery of the catenane backbone, with a center-to-center distance of 2.86 nm. The computed center-to-center distance between the ZnP moiety and the [Cu(phen)2]+ complex was found to be almost double than that of the C60-[Cu(phen)2]+ separation. These differences in interchromophoric distances play an important role in the photoinduced processes in catenate 1, as will be discussed below.
Figure 5.
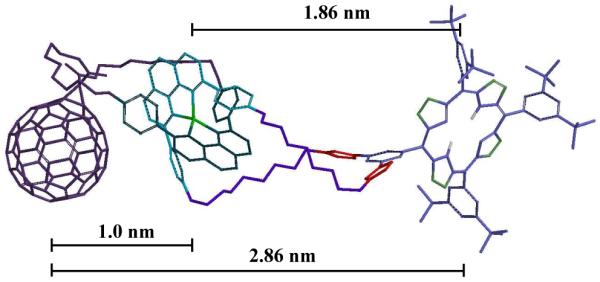
Molecular model of catenate 1. For clarity, the hydrogen atoms have been removed from the structure.
3. Ground State Interactions
3.1. Steady State UV-vis Absorption Spectra
The UV-Vis absorption spectrum of catenate 1 in DCM (not shown) is essentially a linear combination of the spectra of model and reference compounds 2, 17 and 18, suggesting that there is no significant electronic interaction among the ZnP, [Cu(phen)2]+ complex and C60 moieties in the ground state of 1. This conclusion is in agreement with the structural analysis obtained by NMR and computer modeling (see above) which suggests catenate 1 adopts an extended conformation in solution. Thus, the chromophores are well separated from each other, precluding efficient electronic coupling in the ground state. This conclusion is corroborated by the electrochemical data discussed below.
3.2. Electrochemistry
The redox properties of the catenate 1, model compounds 2, 3 and 4 as well as reference compounds 17 and 18 were studied by differential voltammetry experiments in ortho-dichlorobenzene (ODCB) as solvent in the presence of 0.1 M tetrabutylammonium perchlorate [(n-Bu)4NClO4] as supporting electrolyte, using ferrocene/ferricenium as the internal reference. Table 1 shows the electrochemical data obtained.
Table 1.
Electrochemical oxidation and reduction potentials for all compounds studieda
| Oxidation | Reduction | |||||||
|---|---|---|---|---|---|---|---|---|
| Cu+/Cu2+ | ZnP/ZnP·+ | C60/C60· − |
ZnP/ZnP· − | Cu+/Cu0 | ||||
| E0/− | E1−/2− | E2−/3− | E3−/4− | |||||
| Compound | ||||||||
| 1 | +0.18 | +0.18 | −1.12 | −1.48 | --- | −1.96 | −1.92 | −2.30 |
| 2 | +0.16 | --- | --- | --- | --- | --- | --- | −2.30 |
| 3 | +0.20 | +0.20 | --- | --- | --- | --- | −1.92 | −2.30 |
| 4 | +0.16 | --- | −1.12 | −1.48 | −1.96 | −2.54 | --- | −2.30 |
| 17 | --- | --- | −1.12 | −1.48 | −1.96 | −2.54 | --- | --- |
| 18 | --- | +0.24 | --- | --- | --- | --- | −1.92 | --- |
All values (V) are relative to a Fc/Fc+ internal reference. Electrolyte 0.1 M (n-Bu)4NClO4 in ODCB.
The malonate-C60 reference compound 17 was inactive under oxidative conditions. Under reductive conditions, four one-electron steps were observed at −1.12, −1.48, −1.96, and −2.54 V, resembling the trend found for pristine C60.5b,13 However, a sizeable shift to more negative values for the reduction processes in 17 is observed when compared to those of pristine C60 as a consequence of the partial loss of π-conjugation in 17. For triazole-porphyrin reference compound 18, one-electron oxidation and one-electron reduction are observed at +0.24 and −1.92 V, respectively, followed by a second oxidation at +0.54 V.
The anodic scan for [Cu(phen)2]+-catenate model compound 2 reveals an oxidation process at 0.18 V, which has been previously assigned to the one-electron oxidation of the copper center, that is, [Cu(phen)2]+ / [Cu(phen)2]2+.8i,11 In the cathodic scan, only the one-electron reduction of the copper center at −2.30 V is seen, corresponding to the [Cu(phen)2]+ / [Cu(phen)2]0 reduction.
For ZnP–[Cu(phen)2]+ catenate model compound 3 two characteristic oxidation features are discernible in the anodic scan direction. The first oxidation, at +0.20 V, corresponds to one-electron oxidation of both the ZnP group and [Cu(phen)2]+ complex. At more positive potentials, the process at +0.60 V is assigned to the second one-electron oxidation of ZnP moiety. In the cathodic scan, two reduction steps again are observed. A process that qualitatively matches that known for reduction of porphyrin reference 18 is seen at −1.92 V, while a second reduction at −2.30 V, at the same potential as in catenate model 2, is attributed to [Cu(phen)2]+ / [Cu(phen)2]0 reduction. These data indicate lack of significant electronic coupling in 3 between the [Cu(phen)2]+ complex and ZnP moiety in their ground states.
The presence of C60 has no notable impact on the [Cu(phen)2]+ complex centered oxidation and reduction processes in catenate 4, which appear at +0.16 V and −2.30 V, respectively. In the cathodic range, the four one-electron reduction processes of the C60 moiety are observed at −1.12, −1.48, −1.96, and −2.54 V. The lack of appreciable changes upon comparison of the oxidation and reduction potentials of 4 with those of catenate 2 and C60-reference compound 17, attests that the constituents do not electronically interact with each other in the ground state.
Finally, ZnP–[Cu(phen)2]+-C60 catenate 1 reveals similar behavior to catenate models 3 and 4. A first anodic step at +0.20 V corresponds to oxidation of both [Cu(phen)2]+ and ZnP moieties, while the second anodic process at +0.60 V is due to oxidation of the ZnP group. A shift in the one-electron oxidation of the ZnP moiety by nearly 60 mV to more positive potentials is noted upon comparison with that of reference 18 (Table 1). We note a similar trend in the reduction potentials in the model catenates 3 and 4. In particular, the reduction peak at −2.30 V is again assigned to the [Cu(phen)2]+ complex, while reduction of the C60 moiety is seen at −1.12, −1.46, and −1.96 V. The peak corresponding to the latter reduction is broader than the others due to the coalescence of the third one-electron reduction of C60 and the first one-electron reduction of ZnP.
4. Photophysical Properties – Study of Excited State Interactions
To study the electronic interactions between the photoactive constituents upon photoexcitation, emission measurements and transient absorption studies were carried out with catenates 1, 2, 3 and 4 and reference compounds 17 and 18 in solvents of different polarity, including dichloromethane (DCM, Є = 9.2), tetrahydrofuran (THF, Є = 7.6) and benzonitrile (PhCN, Є = 25.0). Table 2 summarizes the spectroscopic data collected in DCM at 298 K.
Table 2.
| Compound | Excited State | λmax (nm) | τ (ns) | Φ | E00 (eV) |
|---|---|---|---|---|---|
| 1 | 1ZnP*-Cu+-C60 | 603 | 0.5 | 1.0 × 10−2 | 2.09 |
| 1ZnP-*Cu+-C60 | 762 | --- | 8.6 × 10−5 | 1.62 | |
| 2 | 3MLCT | 765 | --- | 4.8 × 10−3 | 1.62 |
| 3 | 1ZnP*-Cu+ | 603 | 0.5 | 1.0 × 10−2 | 2.09 |
| ZnP-*Cu+ | 762 | --- | 5.3 × 10−4 | 1.62 | |
| 4 | *Cu+-C60 | 765 | --- | 2.5 × 10−4 | 1.62 |
| Cu+-1C60* | 710 | 0.1 | 2.3 × 10−5 | 1.75 | |
| 17 | 1C60* | 710 | 1.4 | 6.0 × 10−4 | 1.75 |
| 3C60* | 790 | 2.0 × 104 | 0.98 | 1.50c | |
| 18 | 1ZnP* | 600 | 2.3 | 4.0 × 10−2 | 2.09 |
| 3ZnP* | 802 | 4.5 × 104 | 0.88 | 1.55c |
All samples in oxygen free dichloromethane (DCM). Optical densities (ODs) were in the range of 0.12 (C60, 355 nm), 0.14 ([Cu(phen)2]+ complex, 320 nm) and 0.4 (ZnP, 420 nm).
λmax = emission maximum; τ = fluorescence lifetime; Φ = fluorescence quantum yield; E00 = the energy of the corresponding excited state relative to the ground state, calculated from the emission maxima (λmax).
From reference 7d.
4.1. Steady State and Time Resolved Fluorescence
Reference Compounds
The weakest chromophore among all of the photoactive compounds in this work is the malonate-C60 reference compound 17. Excitation of 17 at 355 nm generates a broad emission between 650 and 850 nm. The short wavelength emission maximum at 710 nm is a perfect match to the long wavelength absorption maximum at 695 nm. The low fluorescence quantum yield (ØF) of 17, ~ 6 × 10−4, and the fluorescence lifetime of 1.4 ns are similar to those of pristine C60,5d,e indicating that the major deactivation pathway of the singlet state of 17 is intersystem crossing to the corresponding triplet excited state (vide supra). These features are solvent insensitive as similar findings are noted in THF, DCM, and PhCN. ZnP-reference 18 is the strongest fluorophore, with ØF = 0.04. Its fluorescence spectrum shows maxima at 600 and 650 nm. Following the decay at both of these maxima, the fluorescence lifetime of 18 was found to be 2.3 ns in THF, DCM and PhCN.
Catenate Compounds
Figure 6 compares the emission spectra of the model compounds 2, 3 and 4 and catenate 1, from solutions with equal absorbance. In light of the strong ground state absorption presented by [Cu(phen)2]+ catenate 2 at 320 nm, that wavelength was chosen for photoexcitation. A broad and oxygen sensitive [Cu(phen)2]+ complex MLCT luminescence emerged with a maximum at 765 nm and ØF 4.8 × 10−3, more than an order of magnitude higher than what is typically observed in [Cu(phen)2]+ complex-based systems.24 We ascribe this unexpected increase in fluorescence to the structural rigidity of catenate 2, preventing deactivation processes that might occur in the excited state, such as structural distortion (flattening) or solvent coordination.25
Figure 6.

Fluorescence spectra at 298 K of: A) [Cu(phen)2]+ catenate 2 in DCM, excitation at 320 nm; B) ZnP-[Cu(phen)2]+ catenate 3 in DCM, excitation at 320 nm; C) [Cu(phen)2]+-C60 catenate 4 in DCM, excitation at 320 nm and D) ZnP-[Cu(phen)2]+-C60 catenate 1, red in DCM and black in PhCN, excitation at 420 nm.
We employed ZnP fluorescence as a sensitive probe of catenate model 3. Upon excitation of DCM solutions of 3 and 18 with matched absorptions at 420 nm, the two fluorescence peaks of ZnP at 600 and 650 nm in catenate 3 are quenched by a factor of 4 in DCM when compared to porphyrin reference 18. In addition, the ZnP fluorescence lifetime is 0.5 ns in 3, while it is 2.3 ns in 18. We attribute the ZnP fluorescence quenching to an energy transfer reaction,8a,d,11 since charge transfer reactions between ZnP group and the [Cu(phen)2]2+ complex to yield either ZnP·−-[Cu(phen)2]2+ or ZnP·+-[Cu(phen)2]0 are unlikely to take place, considering the energy levels of 2.12 eV for the former and 2.50 eV for the later. As a complement to selective excitation of the ZnP moiety at 420 nm, 3 was excited at 320 nm, corresponding to the maximum absorption of the [Cu(phen)2]+ complex. A reduced MLCT luminescence quantum yield of 5.3 × 10−4 is attributed to slow transduction of triplet excitation back to ZnP.
For [Cu(phen)2]+-C60 catenate model 4, the characteristically broad and intense [Cu(phen)2]+ MLCT luminescence centered at 765 nm was monitored after excitation at 320 nm. Relative to [Cu(phen)2]+ catenate 2, we noticed for 4 a strong quenching of the luminescence intensity, with ØF = 2.5 × 10−4 (Table 2). Interestingly, the extent of quenching increases with solvent polarity, inferring electronic coupling between the donor and acceptor components, triggering a charge transfer process. A similar trend was seen upon photoexcitation of the C60 moiety in 4 at 355 nm in DCM, where ØF = 2.3 × 10−5 compared to 6.0 × 10−4 for reference compound 17. Complementary time-resolved fluorescence measurements corroborated these results, with lifetimes of 0.1 and 1.4 ns observed for 4 and 17, respectively.
In catenate 1 the strong ZnP fluorescence (ØF = 0.04) was a reliable probe for excited state interactions. As noted in catenate model 3, the fluorescence of 1 is quenched, with ØF = 0.01 in PhCN. The MLCT luminescence of the [Cu(phen)2]+ complex grows in simultaneously with quenching of the ZnP fluorescence, which is clear evidence for energy transduction. This process is barely observable upon excitation at 420 nm since only upon excitation at 320 nm is the MLCT luminescence detectable with significant accuracy. The ØF value of 8.6 × 10−5, which is lower than the values of 4.8 × 10−3 and 5.3 × 10−4 (Table 2) measured for model compounds 2 and 3, respectively, suggests deactivation of the MLCT state via charge transfer. Due to strongly overlapping absorptions, especially between those of the C60 and ZnP moieties, it was not possible to follow the weak fluorescence of C60 as an independent probe in compound 1.
4.2. Transient Absorption Studies
Decisive information about the formation and decay processes following photoexcitation of catenates 1, 2, 3 and 4, and reference compounds 17 and 18 was obtained from transient absorption studies.
Upon excitation of C60-reference 17 in DCM at 387 nm, the lowest vibrational state of the initially generated C60 singlet excited state (absorption at 510 and 920 nm) undergoes quantitative intersystem crossing with a rate constant of 5 × 108 s−1 to the energetically lower lying long lived triplet excited state with maxima at 360 and 720 nm, and a low-energy shoulder around 800 nm. In the absence of molecular oxygen, the C60 triplet excited state has a lifetime of up to 20 μs.
For ZnP reference compound 18 differential absorption changes evolve immediately after the 420 nm laser pulse, characterized by transient bleaching at 420 and 550 nm and broad absorption from 570 to 750 nm in DCM. The singlet excited state features decay at 4 × 108 s−1 to the energetically lower-lying triplet excited state. The newly developing band at 840 nm reflects the diagnostic signature of the ZnP triplet excited state6c with a lifetime of 45 μs. In the presence of molecular oxygen, the ZnP triplet excited state experiences a concentration dependent deactivation process to form singlet oxygen quantitatively.
The model catenate 2 upon excitation at 387 nm in an oxygen free environment such as DCM or PhCN reveals the characteristics of the [Cu(phen)2]+ MLCT excited state.25,26 Figure 7 shows maxima at 540 – 585 and 1000 nm. Minima which evolve in the 440 and 700 nm regions correspond to MLCT absorption, and attest to successful conversion of the [Cu(phen)2]+ complex ground state into the corresponding MLCT excited state. It is widely accepted that the singlet excited MLCT state transforms within a few hundred femtoseconds to the corresponding MLCT triplet excited state.25 Therefore, the transient observed for catenate 2 is assigned to the energetically low lying [Cu(phen)2]+ MLCT triplet state.
Figure 7.

Left: transient absorption spectra (visible and near-infrared) registered upon femtosecond flash photolysis (387 nm, 220 mJ) of [Cu(phen)2]+ catenate 2 in benzonitrile with time delay of 3000 ps at room temperature. Right: time-absorption profiles of the spectra shown on the left at 550 nm, monitoring the MLCT excited state.
On the time scale of the femtosecond experiments (i.e., up to 3 ns) no appreciable decay of the MLCT triplet state is observed. In complementary nanosecond experiments (figure not shown), a similar transient is observed, which is completely quenched in the presence of molecular O2, confirming the triplet excited state character of the transient species. In the absence of molecular O2 the triplet excited state decay is best fitted by a biexponential function to yield lifetimes of 645 ns (95%) and 4.5 μs (5%) in PhCN. In agreement with previous reports,24,25 we assign the former lifetime to that of the [Cu(phen)2]+ triplet excited MLCT state. The latter decay time bears close resemblance to what is seen when probing phenanthroline, and consequently is assigned to be a ligand centered triplet excited state.24,25 Again, as in the fluorescence measurements, the 645 ns lifetime of the triplet excited MLCT state indicates the lack of structural reorganization.
Owing to the strong and dominant absorption of ZnP moiety in catenate 3, visible light excitation at 387 or 420 nm is expected to lead predominantly to the formation of the ZnP singlet excited state. This is confirmed by transient bleaching at 420 and 550 nm and transient absorption from 570 to 750 nm (see Figure 8). This species undergoes a fast decay by energy transfer (see below) in sharp contrast to the slow decay of reference 18 to generate the ZnP triplet excited state. Nevertheless, the ZnP triplet excited state features are also seen when monitoring compound 3 at 840 nm. Concomitant with the ZnP singlet excited state decay of 3 in the 400 to 800 nm range, new features appeared in the 800 to 1200 nm range. A maximum at 1000 nm is a reliable attribute of the triplet excited MLCT state of [Cu(phen)2]+ complex, while in the visible range the ZnP features (i.e., both singlet and triplet excited states) dominate and mask the 580 nm fingerprint of the [Cu(phen)2]+ MLCT triplet excited state. Both triplet species are seen in the complementary nanosecond experiments. The rate of energy transfer from the singlet excited state of ZnP into the lower lying [Cu(phen)2]+ excited state could be followed by the decay at 466 nm, and gave a value of 1.3 × 109 s−1 in PhCN (see Figure 8).
Figure 8.

Left: transient absorption spectra (visible and near-infrared) registered upon femtosecond flash photolysis (420 nm, 150 mJ) of ZnP-[Cu(phen)2]+ catenate 3 in benzonitrile with time delays between 0 and 3000 ps at room temperature – arrow indicates the evolution of the differential changes. Right: time-absorption profiles of the spectra on the left at 466 nm, monitoring the energy transfer process.
Excitation of [Cu(phen)2]+-C60 catenate 4 at 387 nm is mainly directed to the C60 moiety due to its dominant absorption in that range of the spectrum,13 although [Cu(phen)2]+ complex reveals moderate absorption at 387 nm.25-27 Thus, Figure 9 exhibits maxima at 510 and 920 nm as well as at 580 nm, which develop following excitation, in agreement with what has been seen for reference 17 and [Cu(phen)2]+ catenate 2. The nearby [Cu(phen)2]+ complex leads to rapid deactivation of the C60 singlet excited state, with a rate of 2.5 × 1010 s−1, matching the strong quenching of the C60 fluorescence (see Figure 9). As before, the triplet excited [Cu(phen)2]+ MLCT state does not decay appreciably on the 3 ns time scale of these experiments. As the C60 singlet excited state decays a new transition evolves. The absorption features for 4 recorded 1000 (see Figure 9). As before, the triplet excited [Cu(phen)2]+ MLCT state does not decay appreciably on the 3 ns time scale of these experiments. As the C60 singlet excited state decays a new transition evolves. The absorption features for 4 recorded 1000 ps after the laser pulse are clearly different from those of the triplet state of C60 reference 17, which shows a characteristic sharp triplet-triplet maximum around 720 nm.7d In the near-infrared, the diagnostic absorption of the one-electron reduced C60 at 1035 nm, indicating that the deactivation of the C60 singlet excited state of 4 occurs by charge transfer. The C60 radical anion is stable on the 3 ns time scale of our femtosecond experiments.
Figure 9.

Left: transient absorption spectra (visible and near-infrared) registered upon femtosecond flash photolysis (387 nm, 220 mJ) of [Cu(phen)2]+-C60 catenate 4 in DCM with time delays between 0 and 1000 ps at room temperature; the arrow indicates the evolution of the differential changes. Right: time-absorption profiles of the spectra on the left at 930 nm, monitoring the dynamics of charge separation.
In complementary nanosecond experiments (figure not shown), excitation of 4 at 355 nm leads to the same radical ion pair features, indicating formation of [Cu(phen)2]+2-C60·−. A multiwavelength analysis led to a major decay component with a lifetime of 15 ns, which we attribute to charge recombination affording the singlet ground state. A transient with a maximum at 720 nm and a decay lifetime of 4 μs is also observed, which. is most likely the O2-sensitive C60 triplet excited state,5c although contributions from the triplet excited MLCT state cannot be ruled out.
Finally, catenate 1 was probed at 387 and at 420 nm with a fs laser pulse to excite C60 and ZnP moieties, respectively, in DCM, THF, and PhCN. Following photoexcitation at 387 nm we note the successful formation of the singlet excited state of C60 with a strong maximum at 950 nm.7d Instead of seeing the typically slow intersystem crossing noted with 17, the singlet-singlet absorption of 1 decays with accelerated dynamics to yield the fingerprint of the one electron reduced form of C60, namely, absorption at 1040 nm.5c A singlet lifetime of 51 ps for 1 in PhCN was calculated by using a multiwavelength fit of the time profiles, reflecting the charge transfer event. The C60·− species is stable on the femtosecond time scale. Possible transient absorption by [Cu(phen)2]+ MLCT states in the UV-visible region is masked by ZnP-related spectral attributes. These include the ZnP singlet and triplet excited states and the simultaneously formed one-electron oxidized ZnP.
Exclusive ZnP excitation in 1 was accomplished by irradiation at 420 nm. Figure 10 shows that immediately upon excitation, the ZnP singlet excited state signature is seen with maxima at 460, 585, 630, and 725 nm and minima at 560, 605, and 680 nm. The presence of [Cu(phen)2]+ exerts a considerable impact on the decay dynamics, comparing lifetimes of 0.9 and 2.4 ns for 1 and reference 18, respectively. At the conclusion of the ZnP decay, the MLCT excited state of [Cu(phen)2]+ complex evolves as a product of energy transfer rather than charge transfer. No evidence for the one-electron oxidized ZnP, nor for the one-electron reduced ZnP was found in the 600 to 800 nm range, in line with the steady state and time resolved fluorescence experiments. However, the ZnP triplet excited state with a transient maximum at 820 nm is clearly evident, demonstrating competition between energy transfer and intersystem crossing in photoexcited catenate 1.27
Figure 10.

Left: transient absorption spectra (visible and near-infrared) observed upon femtosecond flash photolysis (420 nm, 150 mJ) of ZnP-Cu+-C60 catenate 1 in benzonitrile with time delays between 0 and 3000 ps at room temperature. Right: time-absorption profiles of the spectra on the left at 460 nm, monitoring the kinetics of energy transfer.
To examine the charge recombination (CR) dynamics in 1, solutions were excited with a 6 ns laser pulse at either 355 or 532 nm. In place of transient absorption characteristic of ZnP-Cu2+-C60·− upon 355 nm excitation of ZnP or of 532 nm excitation of [Cu(phen)2]+, the differential absorption spectrum is dominated in the visible region by the one-electron oxidized form of ZnP with a broad band centered at 680 nm and in the near-infrared by a maximum at 1040 nm, which corresponds to the C60·− fingerprint (see Figure 11). The decay of C60·− in the near-infrared (i.e., 900 to 1200 nm) obeys a biexponential rate law with lifetimes of 15 ns and 1.06 μs in PhCN. Based on its similarity with the Cu2+-C60·− radical ion pair state observed for the model compound 4, we ascribe the shorter lifetime to the ZnP-Cu2+-C60·− radical ion pair state, which undergoes charge recombination to give the ground state, competitive with a charge shift to give ZnP·+-Cu+-C60·−. This charge shift must occur within 3 - 10 ns, which corresponds to the time resolution of our femtosecond and nanosecond experiments, respectively.
Figure 11.

Left: transient absorption spectra (visible and near-infrared) observed upon nanosecond flash photolysis (355 nm) of ZnP-Cu+-C60 catenate 1 in benzonitrile with a time delay of 100 ns at room temperature. Right: time-absorption profiles of the spectrum on the left at 1040 nm, monitoring the charge recombination process.
Of particular interest is the presence of the C60 triplet excited state signature absorption at 720 nm when catenate 1 is excited at 355 nm in PhCN.5c This 720 nm transient absorption is a composite of the C60 triplet excited state and the one-electron oxidized form of ZnP. The transient signature absorption for ZnP·+ centered at 680 nm becomes evident only through quenching by molecular O2. A monoexponential fit of the decay at 680 nm generated a lifetime of 1.1 μs in PhCN, in excellent agreement with the longer lived component seen in the decay of the one-electron reduced form of C60. Due to the large spatial separation of the reduced acceptor and oxidized donor, almost 3 nm according to computations (see Figure 5), charge recombination is clearly inhibited.
5. Energetics
Energy transfer (EnT) from the ZnP singlet excited state of catenate 3 (2.09 eV) to give the singlet excited MLCT state and rapid conversion to the triplet excited MLCT state (1.62 eV) proceeds with a rate constant of 1.3 × 109 s−1. This energy transfer is not quantitative (~ 25 %) as the 1ZnP* fluorescence with a lifetime of 2.3 ns is also detected (~ 75 %). The lack of appreciable MLCT emission is rationalized on the basis of rapid triplet-triplet energy transfer from the triplet excited MLCT state (1.62 eV) to the ZnP triplet excited state (1.50 eV). Further support came from experiments that focused on direct excitation of the [Cu(phen)2]+ complex at 320 nm, which revealed strongly quenched MLCT emission. A schematic energy diagram showing the decay pathways upon excitation of 3 at 420 nm is shown in Figure 12a.
Figure 12.

A) Schematic energy level diagrams and decay pathways for catenate 3 upon excitation at 420 nm, and B) catenate 4 upon excitation at 387 or 355 nm.
Excitation of [Cu(phen)2]+-C60 catenate 4 at either 355 or 387 nm 1C60* (1.75 eV).5c Due to the presence of the [Cu(phen)2]+ moiety, the slow intersystem crossing to 3C60* shown by the C60 singlet excited state in 17 is not observed,5c but rather ET occurs to generate the radical ion pair state [Cu(phen)2]2+-C60·− (1.28 eV). Charge transfer (CT) (k = 2.5 × 1010 s−1) occurs 50 times faster than intersystem crossing (k = 7.1 × 108 s−1) to generate the 3C60* (1.50 eV), resulting in a higher efficient CT process for 4. The stabilization of the charge separated state slows down the CR process to 15 ns, which is notably slower than in comparable systems, where lifetimes up to 310 ns are observed.11b One key difference is the closer packing of the two components in 4 leading to stronger electronic coupling of the components, expediting the CR process.
In addition, the energetically lower lying triplet excited MLCT state of the [Cu(phen)2]+ complex (1.62 eV) is also formed in 4 as a product of a nearly instantaneous and highly efficient intersystem crossing. Lower MLCT emission quantum yields in 4 relative to [Cu(phen)2]+ catenate 2 are indicative of a new deactivation pathway when the electron acceptor is present, namely exothermic CT to yield the [Cu(phen)2]2+-C60·− radical ion pair state (1.28 eV), which is much more likely than triplet-triplet energy transfer to populate 3C60* (1.50 eV). Transduction of triplet excited state energy is further hampered by virtue of being a double electron exchange. Nevertheless, the competitive triplet energy transfer is not ruled out entirely upon direct excitation of C60. Evidence for this pathway comes from the fact that the strongly absorbing C60 triplet excited state is seen in the nanosecond experiments. We find that the triplet excited [Cu(phen)2]+ MLCT state is an inferior CT precursor state, as the CT dynamics from the triplet MLCT state are two orders of magnitude slower (between 1.0 - 3.3 × 108 s−1) than CT originating from 1C60* (i.e., 2.5 × 1010 s−1). Based on these observations and the electrochemical data in Table 1, we propose the energy level diagram and pathways depicted in Figure 12b for the decay of catenate 4 upon excitation at either 355 or 387 nm.
The deactivation processes in the full ZnP-Cu+-C60 catenate 1 are basically the composite of those seen for model compound 3 and 4. An additional process available for 1 is the formation initially of ZnP-Cu2+-C60·− (1.28 eV), originating from the [Cu(phen)2]+ triplet excited MLCT state, which subsequently undergoes a charge shift to give the long distance CSRP state ZnP·+-Cu+-C60·− (also estimated at 1.28 eV). Identical and inseparable oxidation potentials of ZnP and [Cu(phen)2]+ complex (see Table 1) lead to isoenergetic CSRP states, for the close ion pair ZnP-Cu2+-C60·− and for the more distantly separated ZnP·+-Cu+-C60·− species. Kinetic evidence for the involvement of both states comes from the CR dynamics, which require biexponential fitting, with lifetimes of 15 ns (k = 6.6 × 107 s−1) and 1.1 μs (k = 9.1 × 105 s−1). The faster decay resembles the dynamics seen for 4 and is assigned to charge recombination in ZnP-Cu2+-C60·−. As a consequence, the lower rate (longer lifetime) is assigned to charge recombination in the long distance CSRP state ZnP·+-Cu+-C60·−, for which strong spectroscopic evidence indicating the formation and decay of the one-electron oxidized form of ZnP and the one-electron reduced form of C60 was obtained. As noted earlier, the center-to-center separation of the positively charge porphyrin and the negatively charged fullerene in this CSRP state is nearly 3 nm.15c Since ZnP-Cu2+-C60·− and ZnP·+-Cu+-C60·− are apparently isoenergetic, the two states are probably in equilibrium, i.e., the charge shift process is reversible. In this context, the pre-exponential factors for fitting the C60·− fingerprint at 1000 nm led to a 3 to 1 ratio for the short- and long-lived species, respectively, indicating that the charge shift occurs within ~ 45 ns (i.e., k ~ 2.2 × 107 s−1).
The lifetimes of the ZnP·+-Cu+-C60·− long distance CSRP state are 1.1 and 1.5 μs in PhCN and THF, respectively, slightly longer than those seen in our first generation rotaxane systems.11a Since the dimensions of catenane 1 are much larger than for the previous studied rotaxanes,11a,d we anticipated this would slow down the CR process to a greater extent in 1 than what was actually observed. Due to the extended structure of catenate 1, the ET processes (i.e., charge separation, charge shift, charge recombination) are likely to be through-space rather than through-bond processes, i.e., the chemical nature of the linkages appears to play only a minor role. Further insight will come from determination of the electronic coupling matrix elements and reorganization energies in these interlocked materials from variable temperature studies, which are planned. The sequence of photoinduced processes, the energy levels determined from spectroscopic and electrochemical data, and the dynamics of the forward and back ET processes upon photoexcitation of catenate 1 are schematically depicted in Figure 13.
Figure 13.

Schematic energy level diagrams, proposed decay pathways and rate constants for ZnP-Cu+-C60 catenate 1 upon excitation at 420 nm. kEnT = energy transfer rate, kET = electron transfer rate, kCR = charge recombination rate.
Summary and Conclusions
Combining the virtues of both “click” chemistry and Sauvage’s Cu(I) template techniques, the synthesis of the first [2]catenate with porphyrin and fullerene subunits was achieved in very good yields. The conformation of the [2]catenate 1 corresponds to an extended structure, with nearly 3 nm center-to-center separation of the ZnP and C60 chromophores. Thus, attractive interactions between the chromophores are not strong enough to bring these groups into close proximity and change the molecular topology of the system, as seems to be the case for the analogous rotaxane.15c A systematic investigation using a variety of techniques revealed that upon excitation of the porphyrin moiety, the long distance ZnP·+–[Cu(phen)2]+-C60·− charge separated radical pair state was formed through a sequence of energy and electron transfer processes, and had a lifetime of 1.1 μs in benzonitrile and 1.5 μs in THF at ambient temperature. In this interlocked system, back electron transfer is clearly occurring in the Marcus inverted region,12 as in previously studied ZnP-C60 covalently linked hybrid materials.6 Minor adjustments in the molecular topology and introduction of different chromophores into the structure might improve the performance of these catenanes to the level required for practical applications. It is worth noting that in a purely porphyrinic [2]catenate in which ZnP acts as the electron donor and AuP+ is the electron acceptor, with the identical [Cu(phen)2]+ central template as in 1, photoinduced ET gave Zn-Cu2+-Au· and not the long distance ZnP·+-Cu+-Au· state, even though the donor-acceptor distance of this porphyrinic catenate is much smaller than in 1.8i Thus, employing C60 as the electron acceptor in the catenate simultaneously provides a driving force for long distance electron transfer while simultaneously retarding the rate of back electron transfer.5,12 The present work illustrates the concept that supramolecular techniques can lead efficiently to highly-complex 3D-nanostructures containing electron donor and acceptor moieties, which upon photoexcitation produce long distance long-lived charge separated states. The use of such materials in light energy conversion systems, specifically photovoltaic devices, will be a subject of future exploration.
EXPERIMENTAL SECTION
1. General Information and Materials
NMR spectra were obtained on either a Bruker AVANCE 400 (400 MHz) or an AVANCE 800 (800 MHz) spectrometer using deuterated solvents as the lock. The spectra were collected at 25 °C and chemical shifts (δ, ppm) were referenced to residual solvent peak (1H, CDCl3 at 7.26 ppm; 13C at 77.2 ppm). In the assignments, the chemical shift (in ppm) is given first, followed, in brackets, by multiplicity (s, singlet; d, doublet; t, triplet; m, multiplet; br, broad), the value of the coupling constants in Hz if applicable, the number of protons implied and finally the assignment. In the 1H NMR assignment (δ), Ho and Hm refer to the hydrogen atoms at the ortho and meta positions, respectively, of the phenyl ring attached to the phenanthroline ring system, whose hydrogen atoms are numbered H3,8, H4,7, H5,6, respectively. Ar is used as an abbreviation for aromatic ring. Mass spectra were obtained on an Agilent 1100 Series Capillary LCMSD Trap XCT Spectrometer in positive or negative-ion mode and ThermoFinnigan PolarisQ ion-trap GCMS Spectrometer. MALDI-TOF mass spectra were recorded in a Bruker OmniFLEX MALDI-TOF MS Spectrometer. This instrument was operated at an accelerating potential of 20 kV in linear mode. The mass spectra represent an average over 256 consecutive laser shots. The mass scale was calibrated using the matrix peaks and the calibration software available from Bruker OmniFLEX. Mentioned m/z values correspond to monoisotopic masses. The compound solutions (10−3 mol/L) were prepared in THF. Matrix compound was purchased from Aldrich and used without further purification. The matrix, α-cyano-4-hydroxy-cinnamic acid (CCA), was dissolved (10 g/L) in a solvent mixture composed of H2O/CH3CN/TFA (25/75/1, v/v). Two microliters of compound solution was mixed with 10 μL of matrix solution. The final solution was deposited onto the sample target and allowed to dry in air. All chemicals were purchased from Sigma-Aldrich and Alfa Aesar and used without further purification. For moisture sensitive reactions, solvents were freshly distilled. Methylene chloride (CH2Cl2) and acetonitrile (CH3CN) were dried over calcium hydride while tetrahydrofuran (THF) was dried using sodium/benzophenone. Anhydrous dimethylformamide (DMF) was used as received. All syntheses were carried out using Schlenk techniques. Moisture sensitive liquids were transferred by canula or syringe. The progress of the reactions was monitored by thin-layer chromatography (TLC) whenever possible. TLC was performed using precoated glass plates (Silica gel 60, 0.25 mm thickness) containing a 254 nm fluorescent indicator. Column chromatography was carried out using Merck Silica gel 60 (0.063-0.200 mm) and neutral alumina (Brockmann I, activated, 150 mesh, 58Å). Compounds 5 and 15 and were synthesized following literature procedures.17d
2. Electrochemical and Photophysical Studies
All solvents used were spectroscopic grade (99.5%) and were purchased from Sigma-Aldrich. The samples were placed in fluorimetric cuvettes with different pathways and, when necessary, purged of oxygen with argon. A single-compartment, three-electrode cell configuration was used in this work. A glassy carbon electrode (3 mm diameter) was used as the working electrode, a platinum wire as the counter, and an Ag wire as the reference electrode. All electrochemical measurements were performed with a E.G.C. Princenton Applied Research model 263A potentiostat/galvanostat. Femtosecond transient absorption studies were performed with 387 and 420 nm laser pulses (1 kHz, 150 fs pulse width) from an amplified Ti:Sapphire laser system (Model CPA 2101, Clark-MXR Inc.). Nanosecond Laser Flash Photolysis experiments were performed with 355 or 532 nm laser pulses from a Quanta-Ray CDR Nd:YAG system (6 ns pulse width) in a front face excitation geometry. Fluorescence lifetimes were measured by using a Fluorolog (Horiba Jobin Yvon). Steady-state fluorescence measurement were performed by using a Fluoromax 3 (Horiba Jobin Yvon). The experiments were performed at room temperature.
3. Synthesis
General Procedure for Synthesis of Catenates15b-d
In flask A, appropriate phen macrocycle (0.041 mmol, 1 equiv) was dissolved in 3 mL of degassed CH2Cl2/CH3CN (7:3, v/v) to which [Cu(CH3CN)4][PF6] (0.015 g, 0.041 mmol) was added under N2 and the solution was stirred at rt for 30 min. The azidophenanthroline ligand 10 (0.028 g, 0.041 mmol) was then added as a solid to flask A and the solution was stirred under N2 at rt for 3 h to generate the corresponding pseudorotaxane precursor. Meanwhile, in the reaction flask, CuI (0.016 g, 0.082 mmol), sodium ascorbate (0.066 g, 0.331 mmol) and sulfonated bathophenathroline (0.098 g, 0.165 mmol) were dissolved in 20 mL of degassed H2O/EtOH (1:1, v/v). The pink suspension was heated at reflux for 2 min and cooled back to rt. The solution in the flask A containing pseudorotaxane precursor was then diluted to 15 mL with degassed CH2Cl2 and added by syringe to the reaction flask. Finally, the suitable 3,5-diethynylphenyl derivative (0.041 mmol), dissolved in 5 mL of degassed CH2Cl2, and 1,8-diaza[5.4.0]-bicycloundec-7-ene (DBU) (0.018 g, 0.124 mmol, 17.7 μL) were added and the resulting mixture was stirred under N2 for 12 h at rt. The crude mixture was neutralized by adding 5 mL of 10% HClaq solution and extracted with CH2Cl2 (3 × 50 mL). The organic phase was washed with water (3 × 100 mL), concentrated to a volume of 10 mL and then stirred for 3 h with a saturated MeOH solution of KPF6 (20 mL) to effect the anion exchange. The solvents were evaporated under reduced pressure, the remaining insoluble solid was extracted with CH2Cl2 (3 × 100 mL) and filtered through paper. The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography (SiO2) using appropriate CH2Cl2/CH3OH mixture for each case as eluent.
Macrocycle 7
Compound 517d (2.00 g, 3.18 mmol) and malonic acid 6 (0.33 g, 3.18 mmol) were dissolved in 100 mL of CH2Cl2. Triethylamine (Et3N) (1.61 g, 2.25 mL, 16 mmol) and bis(2-oxo-3-oxazolidinyl)phosphonic chloride (BOP-Cl) (0.81 g, 3.18 mmol) were added and the reaction mixture was stirred at room temperature for 2 h. Another portion of BOP-Cl (0.81 g, 3.18 mmol) and Et3N (0.80 g, 1.15 mL, 8 mmol) were added and the magnetic stirring was maintained for 12 h at rt to complete the macrocyclization reaction. The crude mixture was neutralized with HClaq (10%), the organic phase was separated, washed with water (3 × 100 mL), dried over Na2SO4, filtered through paper and concentrated under reduced pressure. Final purification was achieved by column chromatography (SiO2) using EtOAc as eluent, affording 7 as a colorless oil (1.00 g, 45% yield). 1H NMR (CDCl3), δ ppm: 8.44 (d, J = 8.8 Hz, 4H, Ho); 8.20 (d, J = 8.5 Hz, 2H, H4 and H7); 8.05 (d, J = 8.5 Hz, 2H, H3 and H8); 7.69 (s, 2H, H5 and H6); 7.14 (d, J = 8.8 Hz, 4H, Hm); 4.34 (t, J = 4.9 Hz, 4H, Ph–O–CH2); 4.26 (t, J = 4.9 Hz, 4H, CH2–O–C=O); 3.90-3.60 (m, 16H, O–CH2–CH2–O); 3.51 (s, 2H, COO–CH2–COO). 13C NMR (CDCl3), δ ppm: CH2 malonic acid group: 207 ppm. Phenanthroline nuclei: 159.5, 156.4, 146.0, 136.9, 132.7, 129.0, 127.6, 125.6, 119.2, 114.8. Oligo(ethyleneglycol) linker: 72.7, 69.7, 69.5, 69.2, 67.6, 66.0. LC-MSD: m/z found 697.55 [M+H]+, calculated 696.27 for C39H40N2O10.
Macrocycle 8
Compound 7 (0.100 g, 0.143 mmol), C60 (0.162 g, 0.225 mmol) and I2 (0.070 g, 0.272 mmol) were dissolved in 160 mL of toluene under magnetic stirring. DBU (0.081 g, 0.080 mL, 0.531 mmol) was added and the reaction mixture was stirred at rt for 24 h. The crude mixture was neutralized with HClaq (10%), the organic phase was separated, washed with water (3 × 100 mL), dried over Na2SO4, filtered through paper and concentrated under reduced pressure. Final purification was achieved by column chromatography (SiO2) using toluene/MeOH (97:3, v/v) as eluent, affording 8 as a brown solid (0.110 g, 55% yield). 1H NMR (CDCl3), δ ppm: 8.36 (d, J = 8.8 Hz, 4H, Ho); 8.17 (d, J = 8.5 Hz, 2H, H4 and H7); 8.00 (d, J = 8.5 Hz, 2H, H3 and H8); 7.67 (s, 2H, H5 and H6); 7.08 (d, J = 8.8 Hz, 4H, Hm); 4.64 (t, J = 4.9 Hz, 4H, Ph–O–CH2); 4.22 (t, J = 4.9 Hz, 4H, CH2–O–C=O); 3.90-3.60 (m, 16H, O–CH2–CH2–O). MALDI-TOF: m/z found 1415.15 [M+H]+, calculated 1414.25 for C99H38N2O10.
Phenanthroline Ditosylate 9
In a three-necked round bottom flask, compound 5 (2.60 g, 7.55 mmol) and triethylamine (5.80 g, 57.50 mmol) were dissolved in 50 mL of dry CH2Cl2 under N2 atmosphere with magnetic stirring. In an addition funnel, p-toluenesulfonyl chloride (p-TsCl) (7.00 g, 36.84 mmol) was dissolved in 50 mL of dry CH2Cl2. The round bottom flask was cooled to 0°C and the p-TsCl solution was added dropwise (over 10 min) under N2 atmosphere with magnetic stirring. After addition, the reaction mixture was kept at 0°C for 4 h and then allowed to warm to rt and stirred for 12 h. The reaction was carefully quenched at 0°C by addition of 10% aqueous HCl. The red organic phase was decanted, washed with water (3 × 100 mL), dried over Na2SO4, filtered through paper and concentrated under reduced pressure. Final purification was achieved by column chromatography (SiO2) using CH2Cl2/MeOH as the eluent (gradient from 0 to 5%, v/v), affording 9 as a red oil (5.30 g, 75% yield). 1H NMR (CDCl3), δ ppm: 8.34 (d, J = 9.0 Hz, 4H, Ho); 8.26 (d, J = 9.0 Hz, 2H, H4 and H7); 8.08 (d, J = 9.0 Hz, 2H, H3 and H8); 7.80 (d, J = 9.0 Hz, 4H, Ho aromatic ring of OTs group); 7.73 (s, 2H, H5 and H6); 7.32 (d, J = 9.0 Hz, 4H, Hm aromatic ring of OTs group); 7.11 (d, J = 9.0 Hz, 4H, Hm); 4.42 (t, J = 4.9 Hz, 4H, Ph–O–CH2); 3.86-3.50 (m, 20H, O–CH2–CH2–O); 2.39 (s, 6H, CH3-OTs group). 13C NMR (CDCl3), δ ppm: phenanthroline nuclei: 159.5, 156.4, 146.0, 136.9, 132.7, 129.0, 127.6, 125.6, 119.2, 114.8. Oligo(ethyleneglycol) linker: 72.7, 69.7, 69.5, 67.6, 66.0, 61.6. Tosyl group: 144.4, 140.3, 130.3, 128.1, 67.4, 21.3. LC-MSD: m/z found 937.43 [M+H]+, calculated 936.30 for C50H52N2O12S2.
Diazidophenanthroline Derivative 10
Caution: Organic azides have been reported in literature as potential explosives. The authors suggest the use of standard PVC blast shield while handling organic azides. Compound 9 (1.00 g, 1.07 mmol) and NaN3 (0.40 g, 6.00 mmol) were dissolved in 50 mL of anhydrous DMF and the reaction mixture was heated at 80 °C for 12 h. The DMF was evaporated under reduced pressure. CH2Cl2 (200 mL) and H2O (50 ml) were added, the organic phase was separated, washed with water (3 × 100 mL), dried over Na2SO4, filtered through paper and concentrated under reduced pressure. Final purification was achieved by flash chromatography (SiO2) using CH2Cl2/MeOH (98:2, v/v) as the eluent, affording 10 as a light yellow solid (0.650 g, 93% yield). 1H NMR (CDCl3), δ ppm: 8.41 (d, J = 8.0 Hz, 4H, Ho); 8.21 (d, J = 8.0 Hz, 2H, H4 and H7); 8.04 (d, J = 8.0 Hz, 2H, H3 and H8); 7.68 (s, 2H, H5 and H6); 7.11 (d, J = 8.0 Hz, 4H, Hm); 4.23 (t, 4H, Ph–O–CH2); 3.90-3.60 (m, 18H, O–CH2–CH2–O); 3.37 (t, 4H, CH2–N3). 13C NMR (CDCl3), δ ppm: phenanthroline nuclei: 159.5, 156.4, 146.0, 136.9, 132.7, 129.0, 127.6, 125.6, 119.2, 114.8. Oligo(ethyleneglycol) linker: 72.7, 69.7, 69.5, 67.6, 66.0. CH2N3: 41.2. IR (KBr) ν cm−1: 2098 (N=N stretch).
3,5-Di-tert-butylbenzaldehyde 12
The following is a new and improved procedure for preparation of this well known compound. A solution of 3,5-di-tert-butyltoluene (25.0 g, 0.122 mol), N-bromosuccinimide (33.0 g, 0.185 mol) and azobisisobutyronitrile (AIBN) (0.900 g, 0.0055 mol) in benzene was heated at reflux under magnetic stirring for 4 h. The reaction mixture was cooled, filtered through paper and the solvents were evaporated under reduced pressure. The residue was dissolved in 70 mL of a solvent mixture composed of EtOH/H2O (1:1) and hexamethylenetetramine (50.0 g, 0.357 mol) was added. The solution was heated at reflux for 4 h. Concentrated HCl was added (21 mL) and heating at reflux was continued for 30 min. The ethanol was removed under reduced pressure, and the remaining aqueous layer was extracted with ether. The ether layer was dried over Na2SO4 and the solvent removed. Recrystallization from EtOH afforded 12 as white crystals (19.10 g, 72% yield). 1H NMR (CDCl3), δ ppm: 10.01 (s, 1H, CHO); 7.72 (d, J = 8.5 Hz, 2H, Ho); 7.71 (m, J = 8.5 Hz,1H, Hp); 1.36 (s, 18H, CH3). 13C NMR (CDCl3), δ ppm: 192 (CHO); 146.9 (C3 and C5); 137.2 (C1); 129.7 (C4); 122.6 (C2 and C6); 34.5 (C–CH3); 31.4 (C–CH3). GC-MS: m/z found 219.02 [M+H]+, calculated 218.17 for C15H22O.
3,5-Di-(trimethylsilylethynyl)benzaldehyde 13
In a round bottomed reaction flask, bis(benzonitrile)-dichloropalladium(II) (0.130 g, 0.325 mmol) and CuI (0.042 g, 0.216 mmol) was dissolved in 11 mL of oxygen-free dioxane under N2 atmosphere at rt. A 2.2 mL aliquot of a 0.27 M solution of triphenylphosphine in dioxane was then added followed by addition of diisopropylamine (1.8 mL, 13 mmol), 3,5-dibromobenzaldehyde (1.42 g, 5.4 mmol) and trimethylsilylacetylene (1.8 mL, 13 mmol). The reaction mixture was stirred under N2 atmosphere and at rt. After ~30 min the solution became warm and a black precipitate formed, turning the reaction mixture nearly solid. The reaction was run for 7 h before being diluted with ethyl acetate and filtered through Celite. The crude product was purified by flash chromatography (SiO2) using hexanes as eluent to afford 13 as a colorless oil (1.53 g, 95% yield). 1H NMR (CDCl3), δ ppm: 9.74 (s, 1H, CHO); 7.65 (d, J = 2.0 Hz, 2H, Ho); 7.56 (m, J = 2.0 Hz, 21, Hp); 0.074 (s, 18H, Si(CH3)3). 13C NMR (CDCl3), δ ppm: 191 (CHO); 142 (C4); 136.3 (C1); 132.6 (C2 and C6); 125.6 (C3 and C5); 102.0 (C≡C–Si(CH3)3); 52.4 (C≡C–Si(CH3)3); 3.3 (C≡C–Si(CH3)3). GC-MS: m/z found 299.0 [M+H]+, calculated 298. 12 for C17H2OSi2.
3,5-Diethynylphenyl-zinc(II) porphyrin 14
Distilled pyrrole (1.49 g, 1.50 mL, 22.5 mmol), 12 (3.30 g, 15.0 mmol), 3,5-di-(trimethylsilylethynyl)benzaldehyde 13 (1.49 g, 5.0 mmol), tetraphenylphosphonium chloride (0.05 g, 0.134 mmol), were dissolved in 200 mL of freshly distilled CH2Cl2 and stirred under N2 at rt for 10 min. BF3.OEt2 (0.265 g, 0.3 mL, 2.5 mmol) was added, the reaction flask covered with aluminium foil and the red solution was stirred at rt for 1 h. DDQ (2,3-dichloro-5,6-dicyanobenzoquinone) (4.0 g, 17.5 mmol) was added and the stirring was maintained for 12 h at rt. Reduction of the volume to ~100 mL under reduced pressure and filtration through celite eliminated undesirable polymeric materials. The porphyrinic products were first separated from the byproducts by flash chromatography using hexanes/CH2Cl2 (1:1, v/v) as eluent. Without any workup, the porphyrin mixture was dissolved in 50 mL of CH2Cl2 and a MeOH saturated solution of zinc acetate dihydrate (0.500 g, in 50 mL) was added and the solution was heated at reflux for 1 h. The mixture was washed with water, dried over Na2SO4, filtered through paper and concentrated under reduced pressure. The crude mixture was then redissolved in 50 mL of THF, tetrabutylammonium fluoride (TBAF) (0.22 g, 0.81 mmol) was added, and the solution was stirred at rt for 30 min to deprotect the alkyne groups. The solvent was evaporated, CH2Cl2 (100 mL) was added and the organic layer was washed with water (3 × 100 mL), dried over Na2SO4, filtered through paper and concentrated under reduced pressure. Final purification was achieved by column chromatography (SiO2), using hexanes/CH2Cl2 (80:20 v/v) as eluent. First product eluted was A4-porphyrin, followed by the desired porphyrin 14 (A3B), which was isolated as a purple solid in 9% yield. Other zinc(II)porphyrins (A2B2 and AB3) were also isolated, while porphyrin B4 was not recovered from the crude mixture. 1H NMR (CDCl3), δ ppm: 9.04 (m, 6H, pyrrolic protons); 8.92 (d, 2H, pyrrolic protons); 8.37 (d, 2H, TMS-Ar-H at orto positions); 8.11 (s, 6H, tert-butyl-Ar-H at orto positions); 8.04 (s, 1H, TMS-Ar-H at para position); 7.82 (s, 3H, s, 6H, tert-butyl-Ar-H at para position); 3.17 (s, 2H, C≡CH); 1.54 (s, 54H, CH3); MALDI-TOF: m/z found 1061.53 [M+H]+, calculated 1060.64 for C72H76N4Zn.
[2]catenate 1. This compound was synthesized from precursor 11 and porphyrin 14 following the general procedure described for preparation of catenates. Final purification was achieved by column chromatography (SiO2) using CH2Cl2/CH3OH (99/1 v/v) as eluent, affording 1 as a purple solid (0.078 g, 57% yield). 1H NMR (CD3CN), δ ppm: 8.98 (s, 1H, Hp of triazole-phenyl group); 8.77 (d, 4H, pyrrolic protons); 8.69 (d, 2H, pyrrolic protons); 8.57 (d, 2H, pyrrolic protons); 8.45 (dd, 4H, H4′, H7′, H4, and H7); 8.11 (s, 2H, H on triazole rings); 7.85 (s, 6H, Ho of ZnP tert-butylphenyl groups); 7.81 (s, 2H, Ho of triazole-phenyl group); 7.80 (s, 2H, H5′ and H6′); 7.79 (s, 2H, H5 and H6); 7.64 (d, 2H, H3′ and H8′); 7.63 (s, 3H, Hp of ZnP-tert-butylphenyl group); 7.36 (d, 2H, H3 and H8); 7.07 (d, 4H, Ho’); 6.74 (d, 4H, Ho); 5.82 (d, 4H, Hm’); 5.67 (d, 4H, Hm); 5.04 (s, 4H, Hb); 4.69 (s, 4H, CH2-triazole groups); 4.80-3.00 (m, oligo(ethyleneglycol) linker); 1.54 (s, 54H, tert-butyl CH3 groups). MALDI-TOF: m/z found 3215.12 [M – PF6]+, calculated, 3215.97 for C207H152N14O16 ZnCu.
[2]catenate 2. This model compound was synthesized from precursor 16 and commercial 1,3-diethynylbenzene following the general procedure described for preparation of catenates. Final purification was achieved by column chromatography (SiO2), using CH2Cl2/CH3OH (98:2 v/v) as eluent, affording 2 as a red solid in 70 % yield. 1H NMR (CDCl3), δ ppm: 8.16 (s, 1H, Hp of triazole-phenyl group); 8.48 (dd, 4H, H4′, H7′, H4, and H7); 8.07 (s, 2H, H on triazole rings); 7.78 (s, 2H, H5′ and H6′); 7.67 (s, 2H, H5 and H6); 7.51 (d, 2H, H3′ and H8′); 7.42 (s, 2H, Ho of triazole-phenyl group); 7.32 (d, 2H, H3 and H8); 7.07 (d, 4H, Ho’); 6.74 (d, 4H, Ho); 5.82 (d, 4H, Hm’); 5.67 (d, 4H, Hm); 4.62 (s, 4H, CH2-triazole groups); 4.80-3.00 (m, oligo(ethyleneglycol) linker). MALDI-TOF: m/z found 1434.05 [M – PF6]+, calculated, 1433.55 for C80H78N10O12Cu.
[2]catenate 3. This model compound was synthesized from precursor 16 and porphyrin 14 following the general procedure described for preparation of catenates. Final purification was achieved by column chromatography (SiO2), using CH2Cl2/CH3OH (98:2 v/v) as eluent, affording 3 as a purple solid in 65 % yield. 1H NMR (CDCl3), δ ppm: 8.96 (s, 1H, Hp of triazole-phenyl group); 8.75 (m, 4H, pyrrolic protons); 8.72 (d, 2H, pyrrolic protons); 8.53 (d, 2H, pyrrolic protons); 8.48 (dd, 4H, H4′, H7′, H4, and H7); 8.11 (s, 2H, H on triazole rings); 7.87 (s, 6H, Ho of ZnP tert-butylphenyl groups); 7.80 (s, 2H, Ho of triazole-phenyl group); 7.82 (s, 2H, H5′ and H6′); 7.77 (s, 2H, H5 and H6); 7.66 (d, 2H, H3′ and H8′); 7.63 (s, 3H, Hp of ZnP-tert-butylphenyl group); 7.32 (d, 2H, H3 and H8); 7.07 (d, 4H, Ho’); 6.74 (d, 4H, Ho); 5.82 (d, 4H, Hm’); 5.67 (d, 4H, Hm); 4.69 (s, 4H, CH2-triazole groups); 4.80-3.00 (m, oligo(ethyleneglycol) linker); 1.54 (s, 54H, tert-butyl CH3 groups). MALDI-TOF: m/z found 2368.75 [M – PF6]+, calculated, 2368.00 for C142H148N14O12ZnCu.
[2]catenate 4. This model compound was synthesized from precursor 11 and 1,3-diethynylbenzene following the general procedure described for preparation of catenates. Final purification was achieved by column chromatography (SiO2), using CH2Cl2/CH3OH (98:1 v/v) as eluent, affording 4 as a brown solid in 75 % yield. 1H NMR (CDCl3), δ ppm: 8.88 (s, 1H, Hp of triazole-phenyl group); 8.42 (dd, 4H, H4′, H7′, H4, and H7); 8.13 (s, 2H, H on triazole rings); 7.81 (s, 2H, Ho of triazole-phenyl group); 7.80 (s, 2H, H5′ and H6′); 7.79 (s, 2H, H5 and H6); 7.64 (d, 2H, H3′ and H8′); 7.36 (d, 2H, H3 and H8); 7.07 (d, 4H, Ho’); 6.74 (d, 4H, Ho); 5.82 (d, 4H, Hm’); 5.67 (d, 4H, Hm); 5.04 (s, 4H, CH2-adjacent to malonate group); 4.69 (s, 4H, CH2-triazole groups); 4.80-3.00 (m, oligo(ethyleneglycol) linker). MALDI-TOF: m/z found 2281.15 [M – PF6]+, calculated, 2281.60 for C145H82N10O16Cu.
ACKNOWLEDGMENTS
Support of the work at NYU by a grant from the National Science Foundation (CHE-0647334) is acknowledged with grateful appreciation. This investigation was conducted in part using an instrumental facility at NYU constructed with support from Research Facilities Improvement Grant Number C06 RR-16572-01 from the National Center for Research Resources, National Institutes of Health. We also would like to thank the Deutsche Forschungsgemeinschaft (SFB 583), FCI and Office of Basic Energy Sciences of the U.S. Department of Energy for financial support.
REFERENCES
- (1).(a) Cannon RD. Electron Transfer Reactions. Butterworths; London, U. K.: 1980. [Google Scholar]; (b) Eberson L. Electron Transfer Reactions in Organic Chemistry. Springer; New York: 1987. [Google Scholar]
- (2).(a) Balzani V, Scandola F. Supramolecular Photochemistry. Horwood; Chichester, U. K.: 1991. [Google Scholar]; (b) Wasielewski MR. Chem. Rev. 1992;92:435–461. [Google Scholar]
- (3).(a) Hader DP, Tevini M. General Photobiology. Pergamon; Elmsford, NY: 1987. [Google Scholar]; (b) Breton J, Vermeglio H, editors. The Photosynthetic Bacterial Reaction Center. Structure and Dynamics. Plenum; New York: 1988. [Google Scholar]; (c) Deisenhofer J, Michel H. Angew. Chem., Int. Ed. Engl. 1989;28:829–847. [Google Scholar]; (d) Feher G, Allen JP, Okamura MY, Rees DC. Nature. 1989;339:111–116. [Google Scholar]; (e) Moser CC, Keske JM, Warncke K, Farid MS, Duttin PL. Nature. 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- (4).(a) Gust D, Moore TA. Science. 1989;244:35–41. doi: 10.1126/science.244.4900.35. [DOI] [PubMed] [Google Scholar]; (b) Gust D, Moore TA, Moore AL. Acc. Chem. Res. 2001;34:40–48. doi: 10.1021/ar9801301. [DOI] [PubMed] [Google Scholar]; (c) Balzani V. Electron Transfer in Chemistry. 1-5. Wiley-VCH; Weinheim, Germany: 2003. [Google Scholar]
- (5).(a) Imahori H, Sakata Y. Adv. Mat. 1997;9:537–546. [Google Scholar]; (b) Echegoyen L, Echegoyen LE. Acc. Chem. Res. 1998;31:593–601. [Google Scholar]; (c) Guldi DM. Chem. Comm. 2000;5:321–327. [Google Scholar]; (d) Prato M, Guldi DM. Acc. Chem. Res. 2000;33:695–703. doi: 10.1021/ar990144m. [DOI] [PubMed] [Google Scholar]; (e) Guldi DM. Chem. Soc. Rev. 2002;31:22–36. doi: 10.1039/b106962b. [DOI] [PubMed] [Google Scholar]; (f) Fukuzumi S, Ohkubo K, Imahori H, Guldi DM. Chem. Eur. J. 2003;9:1585–1593. doi: 10.1002/chem.200390182. [DOI] [PubMed] [Google Scholar]; (g) Figueira-Duarte T, Lloveras V, Vidal-Gancedo J, Gegout A, Delavaux NB, Welter R, Veciana J, Rovira C, Nierengarten JF. Chem. Commum. 2007;42:4345–4347. doi: 10.1039/b707522g. [DOI] [PubMed] [Google Scholar]; (h) Regehly M, Ermilov EA, Helmreich M, Hirsch A, Jux N, Roeder B. J. Phys. Chem. B. 2007;111:998–1006. doi: 10.1021/jp065626b. [DOI] [PubMed] [Google Scholar]; (i) Santos J, Grim B, Islescas BM, Martin N. Chem. Comm. 2008;45:5993–5995. doi: 10.1039/b811358k. [DOI] [PubMed] [Google Scholar]
- (6).For few representative examples, see: Collin J-P, Harriman A, Heitz V, Odobel F, Sauvage J-P. J. Am. Chem. Soc. 1994;116:5679–5690. Schuster DI, Cheng P, Wilson SR, Prokhorenko V, Katterle M, Holzwarth AR, Braslavsky SE, Klihm G, Williams RM, Luo C. J. Am. Chem. Soc. 1999;121:11599–11600. Luo C, Guldi DM, Imahori H, Tamaki K, Sakata Y. J. Am. Chem. Soc. 2000;122:6535–6551. Schuster DI, Cheng P, Jarowski PD, Guldi DM, Luo C, Echegoyen L, Pyo S, Holzwarth AR, Braslavsky SE, Williams RM, Klihm G. J. Am. Chem. Soc. 2004;126:7257–7270. doi: 10.1021/ja038676s. Hagemann O, Jørgensen M, Krebs FC. J. Org. Chem. 2006;71:5546–5559. doi: 10.1021/jo060491r. Vail SA, Schuster DI, Guldi DM, Isosomppi M, Tkachenko N, Lemmetyinen H, Palkar A, Echegoyen L, Chen X, Zhang JZH. J. Phys. Chem. B. 2006;110:14155–14166. doi: 10.1021/jp061844t. Sgobba V, Giancane G, Conoci S, Casilli S, Ricciardi G, Guldi DM, Prato M, Valli L. J. Am. Chem. Soc. 2007;129:3148–3156. doi: 10.1021/ja0655789. Schuster DI, Ke L, Guldi DM, Palkar A, Echegoyen L, Stanisky C, Cross RJ, Niemi M, Tkachenko NV, Lemmetyinen H. J. Am. Chem. Soc. 2007;129:15973–15982. doi: 10.1021/ja074684n. Sarova GH, Hartnagel U, Balbinot D, Sali S, Jux N, Hirsch A, Guldi DM. Chem. Eur. J. 2008;14:3137–3145. doi: 10.1002/chem.200701462. Fazio M, Lee OP, Schuster DI. Org. Lett. 2008;10:4979–4982. doi: 10.1021/ol802053k. Subbaiyan NK, Wijesinghe SA, D’Souza F. J. Am. Chem. Soc. 2009;131:14646–14647. doi: 10.1021/ja9067113.
- (7).For a few representative examples, see: Imahori H, Hagiwara K, Akiyama T, Aoki M, Taniguchi S, Okada T, Shirakawa M, Sakata Y. Chem. Phys. Lett. 1996;263:545–550. Olmstead MM, Costa DA, Maitra K, Noll BC, Philips SL, Van Calcar PM, Balch AL. J. Am. Chem. Soc. 1999;121:7090–7097. Boyd PDW, Hodgson MC, Rickard CEF, Oliver AG, Chaker L, Brothers PJ, Bolskar RD, Tham FS, Reed CA. J. Am. Chem. Soc. 1999;121:10487–10495. Guldi DM, Zerbetto F, Georgakilas V, Prato M. Acc. Chem. Res. 2005;38:38–43. doi: 10.1021/ar040222s. Gayathri SS, Wielopolski M, Perez EM, Fernandez G, Sanchez L, Viruela R, Orti E, Guldi DM, Martin N. Angew. Chem. Int. Ed. 2009;48:815–819. doi: 10.1002/anie.200803984. Kira A, Umeyama T, Matano Y, Yoshida K, Isoda S, Park J, Kang K, Imahori H. J. Am. Chem. Soc. 2009;131:3198–3200. doi: 10.1021/ja8096465. D’Souza F, Subbaiyan NK, Xie Y, Hill JP, Ariga K, Ohkubo K, Fukuzumi S. J. Am. Chem. Soc. 2009;131:16138–16146. doi: 10.1021/ja9048306.
- (8).(a) Armaroli N, Balzani V, Barigelletti F, De Cola L, Flamigni L, Sauvage J-P, Hemmert C. J. Am. Chem. Soc. 1994;116:5211–5217. [Google Scholar]; (b) Amaroli N, Diederich F, Dietrich-Buchecker CO, Flamigni L, Marconi G, Nierengarten J-F, Sauvage J-P. Chem. Eur. J. 1998;4:406–416. [Google Scholar]; (c) Hu Y-Z, van Loyen D, Schwarz O, Bossmann S, Dürr H, Huch V, Veith M. J. Am. Chem. Soc. 1998;120:5822–5823. [Google Scholar]; (d) Cárdenas DJ, Collin J-P, Gaviña P, Sauvage J-P, De Cian A, Fischer J, Armaroli N, Flamigni L, Vicinelli V, Balzani V. J. Am. Chem. Soc. 1999;121:5481–5488. [Google Scholar]; (e) Hu Y-Z, Tsukiji S, Shinkai S, Oishi S, Hamachi I. J. Am. Chem. Soc. 2000;122:241–253. [Google Scholar]; (f) Anderson M, Linke M, Chambron J-C, Davidsson J, Heitz V, Hammarstrom L, Sauvage J-P. J. Am. Chem. Soc. 2002;124:4347–4362. doi: 10.1021/ja0119907. [DOI] [PubMed] [Google Scholar]; (g) Flamigni L, Talarico AM, Serroni S, Puntoriero F, Gunter MJ, Johnston MR, Jeynes TP. Chem. Eur. J. 2003;9:2649–2659. doi: 10.1002/chem.200204502. [DOI] [PubMed] [Google Scholar]; (h) Sasabe H, Kihara N, Furusho Y, Mizuno K, Ogawa A, Takata T. Org. Lett. 2004;6:3957–3960. doi: 10.1021/ol048433k. [DOI] [PubMed] [Google Scholar]; (i) Flamigni L, Talarico AM, Chambron J-C, Heitz V, Linke M, Fujita N, Sauvage J-P. Chem. Eur. J. 2004;10:2689–2699. doi: 10.1002/chem.200305655. [DOI] [PubMed] [Google Scholar]; (j) Sandanayaka ASD, Watanabe N, Ikeshita K-I, Araki Y, Kihara N, Furusho Y, Ito O, Takata T. J. Phys. Chem. B. 2005;109:2516–2525. doi: 10.1021/jp045831w. [DOI] [PubMed] [Google Scholar]; (k) Rajkumar GA, Sandanayaka ASD, Ikeshita K-I, Araki Y, Furusho Y, Takata T, Ito O. J. Phys. Chem. B. 2006;110:6516–6525. doi: 10.1021/jp056699i. [DOI] [PubMed] [Google Scholar]; (l) Saha S, Flood AH, Stoddart JF, Impellizzeri S, Silvi S, Venturi M, Credi A. J. Am. Chem. Soc. 2007;129:12159–12171. doi: 10.1021/ja0724590. [DOI] [PubMed] [Google Scholar]; (m) Mateo-Alonso A, Ehli C, Rahman GMA, Guldi DM, Fioravanti G, Marcaccio M, Paolucci F, Prato M. Angew. Chem., Int. Ed. Engl. 2007;46:3521–3525. doi: 10.1002/anie.200605039. [DOI] [PubMed] [Google Scholar]; (n) Marois J-S, Cantin K, Desmarais A, Morin J-F. Org. Lett. 2008;10:33–36. doi: 10.1021/ol702524f. [DOI] [PubMed] [Google Scholar]; (o) Mateo-Alonso A, Iliopoulos K, Couris S, Prato M. J. Am. Chem. Soc. 2008;130:1534–1535. doi: 10.1021/ja0771235. [DOI] [PubMed] [Google Scholar]; (p) Wang J-Y, Han J-M, Yan J, Ma Y, Pei J. Chem. Eur. J. 2009;15:3585–3594. doi: 10.1002/chem.200802228. [DOI] [PubMed] [Google Scholar]
- (9).Sauvage J-P, Dietrich-Buchecker CO. Molecular Catenanes, Rotaxanes and Knots. Wiley-VCH; Weinheim, Germany: 1999. [Google Scholar]
- (10).(a) Livoreil A, Sauvage J-P, Armaroli N, Balzani V, Flamigni L, Ventura B. J. Am. Chem. Soc. 1997;119:12114–12124. doi: 10.1021/ja9720826. [DOI] [PubMed] [Google Scholar]; (b) Balzani V, Credi A, Venturi M. Molecular Devices and Machines-A Journey into the Nano World. Wiley-VCH; Weinheim, Germany: 2003. [Google Scholar]; (c) Thordarson P, Bijsterveld JA, Rowan AE, Nolte RJM. Nature. 2003;424:915–918. doi: 10.1038/nature01925. [DOI] [PubMed] [Google Scholar]; (d) Badijic JD, Credi V, Silvi S, Stoddart JF. Science. 2004;303:1845–1849. doi: 10.1126/science.1094791. [DOI] [PubMed] [Google Scholar]; (e) Serrel V, Lee C-F, Kay ER, Leigh DA. Nature. 2005;445:523–527. doi: 10.1038/nature05452. [DOI] [PubMed] [Google Scholar]; (f) Green JE, Choi JW, Boukai A, Bunimovich Y, Johnston-Halperin E, Delonno E, Luo Y, Scheriff BA, Xu K, Shin YS, Tseng H-R, Stoddart JF, Heath JR. Nature. 2007;445:414–417. doi: 10.1038/nature05462. [DOI] [PubMed] [Google Scholar]
- (11).(a) Li K, Schuster DI, Guldi DM, Herranz MA, Echegoyen L. J. Am. Chem. Soc. 2004;126:3388–3389. doi: 10.1021/ja039698h. [DOI] [PubMed] [Google Scholar]; (b) Li K, Bracher PJ, Guldi DM, Herranz MA, Echegoyen L, Schuster DI. J. Am. Chem. Soc. 2004;126:9156–9157. doi: 10.1021/ja047548g. [DOI] [PubMed] [Google Scholar]; (c) Schuster DI, Li K, Guldi DM, Ramey J. Org. Lett. 2004;6:1919–1922. doi: 10.1021/ol049540h. [DOI] [PubMed] [Google Scholar]; (d) Schuster DI, Li K, Guldi D. C. R. Chimie. 2006;9:892–908. [Google Scholar]
- (12).(a) Marcus RA. J. Chem. Phys. 1956;24:966–978. [Google Scholar]; (b) Imahori H, Hagiwara K, Akiyama T, Aoki M, Taniguchi S, Okada T, Shirakawa M, Sakata Y. Chem. Phys. Lett. 1996;263:545–550. [Google Scholar]; (c) Guldi DM, Fukuzumi S. In: Fullerenes, From Synthesis to Optoelectronic Properties. Guldi DM, Martin N, editors. Kluwer Academic Publishers; Dordrecht: 2002. pp. 237–265. [Google Scholar]
- (13).Hirsch A, Brettreich M. Fullerenes. Wiley-VCH; Weinheim, Germany: 2005. [Google Scholar]
- (14).(a) Aston PR, Diederich JF, Gomez-Lopez M, Nierengarten J-F, Preece JA, Raymo FM, Stoddart JF. Angew. Chem., Int. Ed. Engl. 1997;36:1448–1451. [Google Scholar]; (b) Nakamura Y, Minami S, Iizuka K, Nishimura J. Angew. Chem., Int. Ed. Engl. 2003;42:3158–3162. doi: 10.1002/anie.200351411. [DOI] [PubMed] [Google Scholar]
- (15).(a) Megiatto JD, Jr., Schuster DI. J. Am. Chem. Soc. 2008;130:12872–12873. doi: 10.1021/ja8050519. [DOI] [PubMed] [Google Scholar]; (b) Megiatto JD, Jr., Schuster DI. Chem. Eur. J. 2009;15:5444–5448. doi: 10.1002/chem.200900536. [DOI] [PubMed] [Google Scholar]; (c) Megiatto JD, Jr., Spencer R, Schuster DI. Org. Lett. 2009;11:4152–4155. doi: 10.1021/ol9016645. [DOI] [PubMed] [Google Scholar]; (d) Megiatto JD, Jr., Schuster DI. New J. Chem. 2009 DOI: 10.1039/b9nj00486f. [Google Scholar]
- (16).(a) Huisgen R. Angew. Chem., Int. Ed. Engl. 1968;7:321–328. [Google Scholar]; (b) Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. Engl. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; (c) Tornøe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- (17).(a) Dietrich-Buchecker CO, Sauvage J-P, Kitzinger JP. Tetrahedron Lett. 1983;24:5095–5098. [Google Scholar]; (b) Dietrich-Buchecker CO, Sauvage J-P. J. Am. Chem. Soc. 1984;106:3043–3044. [Google Scholar]; (c) Dietrich-Buchecker CO, Sauvage J-P. Chem. Rev. 1987;87:795–810. [Google Scholar]; (d) Dietrich-Buchecker CO, Sauvage J-P. Tetrahedron. 1990;46:503–512. [Google Scholar]
- (18).Iehl J, Pereira de Freitas R, Nierengarten J-F. Tetrahedron Lett. 2008;49:4063–4066. [Google Scholar]
- (19).Lindsey JS, Prathapan S, Johnson TE, Wagner RW. Tetrahedron. 1994;50:8941–8946. [Google Scholar]
- (20).All catenated materials reported in this work were isolated as PF6− salts.
- (21).For previous mass spectrometric studies of catenanes and rotaxanes, see: Vetter W, Schill G. Tetrahedron. 1967;23:3079–3093. Vetter W, Logemann E, Schill G. Org. Mass Spectrom. 1977;12:351–355. Bitsch F, Dietrich-Buchecker CO, Khemiss AK, Sauvage J-P, Dorsselaer AV. J. Am. Chem. Soc. 1991;113:4023–4025. Weck M, Mohr B, Sauvage J-P, Grubbs RH. J. Org. Chem. 1999;64:5463–5471. doi: 10.1021/jo990268c.
- (22).The assignment of the protons signals was done by comparison with model compounds and 2D NMR experiments.
- (23).Ornelas C, Aranzaes JR, Cloutet E, Alvez S, Astruc D. Angew. Chem., Int. Ed. Engl. 2007;46:872–877. doi: 10.1002/anie.200602858. [DOI] [PubMed] [Google Scholar]
- (24).Felder D, Nierengarten JF, Barigelletti F, Ventura B, Armarolli N. J. Am. Chem. Soc. 2001;123:6291–6299. doi: 10.1021/ja0043439. [DOI] [PubMed] [Google Scholar]
- (25).Gunaratne T, Rodgers MAJ, Felder D, Nierengarten JF, Accorsi G, Armaroli N. Chem. Comm. 2003;40:3010–3011. doi: 10.1039/b306900a. [DOI] [PubMed] [Google Scholar]
- (26).Armaroli N, Rodgers MAJ, Ceroni P, Balzani V, Dietrich-Buchecker CO, Kern JM, Bailal A, Sauvage JP. Chemical Physical Letters. 1995;241:555–558. [Google Scholar]
- (27).As C60 absorbs at the 420 nm excitation wavelength, we observe a minor contribution of the directly formed ZnP-[Cu(phen)2]2+-C60·− radical pair state.