

Abstract
Parkinson's disease (PD) is second only to Alzheimer's disease as the most common devastating human neurodegenerative disorder. Despite intense investigation, no interdictive therapy is available for PD. We investigated whether simvastatin, a Food and Drug Administration-approved cholesterol-lowering drug, could protect against nigrostriatal degeneration after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxication to model PD in mice. First, MPP+ induced the activation of p21ras and nuclear factor-κB (NF-κB) in mouse microglial cells. Inhibition of MPP+-induced activation of NF-κB by Δp21ras, a dominant-negative mutant of p21ras, supported the involvement of p21ras in MPP+-induced microglial activation of NF-κB. Interestingly, simvastatin attenuated activation of both p21ras and NF-κB in MPP+-stimulated microglial cells. Consistently, we found a very rapid activation of p21ras in vivo in the substantia nigra pars compacta of MPTP-intoxicated mice. However, after oral administration, simvastatin entered into the nigra, reduced nigral activation of p21ras, attenuated nigral activation of NF-κB, inhibited nigral expression of proinflammatory molecules, and suppressed nigral activation of glial cells. These findings paralleled dopaminergic neuronal protection, normalized striatal neurotransmitters, and improved motor functions in MPTP-intoxicated mice. Similarly, pravastatin, another cholesterol-lowering drug, suppressed microglial inflammatory responses and protected dopaminergic neurons in MPTP-intoxicated mice, but at levels less than simvastatin. Furthermore, both the statins administered 2 d after initiation of the disease were still capable of inhibiting the demise of dopaminergic neurons and concomitant loss of neurotransmitters, suggesting that statins are capable of slowing down the progression of neuronal loss in the MPTP mouse model. Therefore, we conclude that statins may be of therapeutic benefit for PD patients.
Introduction
Parkinson's disease (PD) is the most common human neurodegenerative disorder affecting movement, balance, flexibility, and coordination. Disease symptoms are commonly associated with advancing age. Clinically, PD is characterized by tremor, bradykinesia, rigidity, and postural instability (Olanow and Tatton, 1999; Vila and Przedborski, 2004). Pathologically, it is indicated by gliosis and progressive degeneration of the dopaminergic neurons associated with the presence of intracytoplasmic inclusions (Lewy bodies) in the substantia nigra pars compacta (SNpc) (Dauer and Przedborski, 2003).
Recent studies support an important role for inflammation in nigrostriatal degeneration (Dauer and Przedborski, 2003; Gao et al., 2003; Teismann et al., 2003). First, microglial activation is evident in close proximity to damaged or dying dopaminergic neurons. Second, the CSF level of NO2− (nitrite), a metabolite of NO, and the CNS level of inducible nitric oxide synthase (iNOS) are higher in patients with PD in comparison with a group of patients without dopaminergic dysfunction (Qureshi et al., 1995; Hunot et al., 1996). Consistently, the ablation of iNOS in mutant mice significantly attenuates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxicity (Dehmer et al., 2000). Third, a variety of proinflammatory cytokines including tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), IL-6, eicosanoids, and other immune neurotoxins are found in either CSF or affected brain regions in PD (Mogi et al., 1994; Bessler et al., 1999; Nagatsu et al., 2000). Fourth, transgenic mice carrying homozygous mutant alleles for both the TNF receptors, but not the individual receptors, are completely protected against MPTP (Sriram et al., 2006). Finally, we have demonstrated that nuclear factor-κB (NF-κB), a transcription factor required for the transcription of most of the proinflammatory molecules, is activated in the SNpc of PD patients and MPTP-intoxicated mice and that selective inhibition of NF-κB in mice by NBD (NEMO-binding domain) peptides protect dopaminergic neurons from MPTP toxicity (Ghosh et al., 2007). According to Hunot et al. (1997), NF-κB-dependent apoptogenic transduction pathway in dopaminergic neurons may play a role in neuronal death in PD. Recently, it has been demonstrated that Nurr1, a nuclear orphan receptor, suppresses glial activation by docking to NF-κB–p65 on target inflammatory gene promoters and protects dopaminergic neurons in vivo in the nigra (Saijo et al., 2009). Together, controlling inflammation may be an important step to protect dopaminergic neurons in PD and its animal model.
Previously, we (Pahan et al., 2000) have demonstrated that small G-protein p21ras plays an important role in the expression of proinflammatory molecules and that lovastatin, a Food and Drug Administration (FDA)-approved cholesterol-lowering drug, inhibits the expression of proinflammatory molecules in astroglia, microglia, and macrophages (Pahan et al., 1997). Here, we examined the antiinflammatory efficacy of simvastatin against parkinsonian neurotoxin MPTP/MPP+. We demonstrate that p21ras plays an important role in MPP+-induced activation of NF-κB in mouse microglial cells and that simvastatin inhibits the activation of p21ras and the activation of NF-κB in MPP+-stimulated microglial cells. We also demonstrate that activation of p21ras was induced rapidly in vivo in the SNpc of MPTP-intoxicated mice and that statins inhibited nigral activation of p21ras, suppressed nigral expression of proinflammatory molecules, and exhibited significant protection of the nigrostriatal axis after MPTP intoxication. These results provide a mode of action of statins against parkinsonian neurotoxin and open an option for treating PD patients with FDA-approved cholesterol-lowering drugs.
Materials and Methods
Animals and MPTP intoxication.
Six- to 8-week-old C57BL/6 mice were purchased from Harlan. Animal maintaining and experiments were in accordance with National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of Rush University Medical Center. For MPTP intoxication, mice received four intraperitoneal injections of MPTP-HCl (18 mg/kg of free base; Sigma-Aldrich) in saline at 2 h intervals (Ghosh et al., 2007). Control animals received only saline.
Human brain tissue.
Autopsy brain tissues from four male PD patients and four control subjects were obtained from the Rush PD Center Brain Bank. PD patients and control subjects had similar mean age at death (PD, 74 + 3; control, 79 + 18 years). The mean postmortem interval for PD and controls were 4.1 + 0.8 and 10.9 + 1.1 h, respectively.
Drugs and antibodies.
Simvastatin, pravastatin, and rabbit anti-mouse iNOS were obtained from Calbiochem. Rabbit and goat anti-NF-κB p65 and goat anti-glial fibrillary acidic protein (GFAP) were purchased from Santa Cruz Biotechnology. Rat anti-mouse CD11b and mouse anti-human CD11b were purchased from Abcam and Serotec, respectively. Cy2- and Cy5-conjugated antibodies were obtained from Jackson ImmunoResearch Laboratories.
Treatment of mice with statins.
Based on the known dosage (80 mg/d) of an adult human patient with hypercholesterolemia for statins, we treated mice with a dose of 1 mg · kg body weight−1 · d−1 statins via gavage. Whereas pravastatin was solubilized in water, simvastatin powder was mixed with 0.5% methylcellulose. Mice were treated with 100 μl of either simvastatin or pravastatin by gavage either 3 h or 2 d after the last injection of MPTP. Previously, Przedborski and colleagues (Tieu et al., 2003) showed that sufficient amount of MPTP could be converted into MPP+ within 90 min of the last injection of MPTP. Therefore, to avoid any possible influence of statins on entry and conversion of MPTP into MPP+ in the midbrain, oral treatment began a minimum of 3 h after the last injection of MPTP. Control MPTP mice received only 100 μl of vehicle (0.5% methylcellulose) every day.
Activation of p21ras.
Activation of p21ras was monitored as described previously (Pahan et al., 2000) with the following modifications. Briefly, after 6 and 12 h of MPTP insult, ventral midbrain was dissected out and frozen immediately on dry ice. The p21ras-binding domain (RBD) of the p21ras effector kinase Raf1 has been shown to bind specifically to the GTP-bound (active) form of p21ras proteins. Therefore, using an assay kit from Millipore, ventral midbrain tissues were homogenized with lysis buffer containing inhibitors of different proteases and kinases followed by immuno-pulldown of active p21ras using Raf–RBD–glutathione S-transferase (GST) beads. Then the amount of activated p21ras was determined in GST beads by a Western blot using a p21ras-specific antibody.
Transcriptional activity of NF-κB.
Transcriptional activity of NF-κB was assayed as described by us in several studies (Jana et al., 2001; Liu et al., 2002). Briefly, cells plated at 50–60% confluence in 12-well plates were transfected with 0.25 μg of pBIIX-Luc (an NF-κB-dependent reporter construct) and 12.5 ng of pRL-TK (a plasmid encoding Renilla luciferase, used as transfection efficiency control; Promega) using LipofectAMINE Plus (Invitrogen). After 24 h of transfection, cells were stimulated with MPP+ for an additional 6 h, and firefly and Renilla luciferase activities were recorded in a TD-20/20 Luminometer (Turner Designs) by analyzing total cell extract according to standard instructions provided in the Dual Luciferase kit (Promega). Relative luciferase activity of cell extracts was typically represented as follows: (firefly luciferase value/Renilla luciferase value) × 10−3.
Semiquantitative reverse transcription-PCR analysis for proinflammatory molecules (iNOS, IL-1β, and TNF-α) and glial cell markers.
Total RNA was isolated from ventral midbrain using Ultraspec-II RNA reagent (Biotecx Laboratories) following the manufacturer's protocol. To remove any contaminating genomic DNA, total RNA was digested with DNase. Reverse transcription (RT)-PCR was performed as described previously (Roy et al., 2006; Ghosh et al., 2007; Jana et al., 2007) using a RT-PCR kit from Clontech. Briefly, 1 μg of total RNA was reverse transcribed using oligo-dT12-18 as primer and Moloney murine leukemia virus reverse transcriptase (Clontech) in a 20 μl reaction mixture. The resulting cDNA was amplified using Titanium TaqDNA polymerase and the following primers, electrophoresed on a 1.5% agarose gels, and visualized by ethidium bromide staining: iNOS, sense, 5′-CCCTTCCGAAGTTTCTGGCAGCAGC-3′; antisense, 5′-GGCTGTCAGAGCCTCGTGGCTTTGG-3′; IL-1β, sense, 5′-CTCCATGAGCTTTGTACAAGG-3′; antisense, 5′-TGCTGATGTACCAGTTGGGG-3′; TNF-α, sense, 5′-TTCTGTCTACTGAACTTCGGGGTGATCGGTCC-3′; antisense, 5′-GTATGAGATAGCAAATCGGCTGACGGTGTGGG-3′; CD11b, sense, 5′-GTGAGGATTCCTACGGGACCCAGGT-3′; antisense, 5′-GGCGTACTTCACAGGCAGCTCCAAC-3′; GFAP, sense, 5′-GGCGCTCAATGCTGGCTTCA-3′; antisense, 5′-TCTGCCTCCAGCCTCAGGTT-3′; GAPDH, sense, 5′-GGTGAAGGTCGGTGTGAACG-3′; antisense, 5′-TTGGCTCCACCCTTCAAGTG-3′. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to ascertain that an equivalent amount of cDNA was synthesized from different samples. The relative expression of proinflammatory molecules or glial cell markers was measured after scanning the bands.
Real-time PCR analysis.
It was performed in the ABI-Prism 7700 sequence detection system (Applied Biosystems) as described previously (Roy et al., 2006; Ghosh et al., 2007; Jana et al., 2007) using TaqMan Universal Master mix and optimized concentrations of FAM-labeled probes and primers. All primers and FAM-labeled probes for mouse iNOS, cytokines, CD11b, GFAP, and GAPDH were obtained from Applied Biosystems. The mRNA expression of iNOS, cytokines, and glial markers was normalized to the level of GAPDH mRNA. Data were processed by the ABI Sequence Detection System 1.6 software.
Immunohistochemistry and quantitative morphology.
Seven days after MPTP intoxication, mice were killed, and their brains were fixed, embedded, and processed for tyrosine hydroxylase (TH) and thionin staining as described previously (Benner et al., 2004; Ghosh et al., 2007). Total numbers of TH- and Nissl-stained neurons in SNpc were counted stereologically with STEREO INVESTIGATOR software (MicroBrightField) by using an optical fractionator (Benner et al., 2004; Ghosh et al., 2007). Quantitation of striatal TH immunostaining was performed as described previously (Benner et al., 2004; Ghosh et al., 2007). Optical density measurements were obtained by digital image analysis (Scion). Striatal TH optical density reflected dopaminergic fiber innervation. For immunofluorescence staining on fresh frozen sections, rat anti-mouse CD11b (1:100), goat anti-mouse GFAP (1:100), rabbit anti-NF-κB p65 (1:100), goat anti-NF-κB p65 (1:100), rabbit anti-NF-κB p50 (1:100), and rabbit anti-mouse iNOS (1:250) were used. The samples were mounted and observed under a Bio-Rad MRC1024ES confocal laser-scanning microscope.
HPLC analysis for measurement of striatal dopamine and its metabolite levels.
Striatal level of dopamine (DA), 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) was quantified as described previously (Benner et al., 2004; Roy et al., 2006; Ghosh et al., 2007). Briefly, mice were killed by cervical dislocation after 7 d of MPTP intoxication, and their striata were collected and immediately frozen in dry ice and stored at −80°C until analysis. On the day of the analysis, tissues were sonicated in 0.2 m perchloric acid containing isoproterenol, and the resulting homogenates were centrifuged at 20,000 × g for 15 min at 4°C. After pH adjustment and filtration, 10 μl of supernatant was injected onto a Eicompak SC-3ODS column (Complete Stand-Alone HPLC- ECD System EiCOMHTEC-500 from JM Science) and analyzed following manufacturer's protocol.
HPLC analysis for quantifying simvastatin and pravastatin in nigra.
Substantia nigra were isolated from each brain, weighed, and immediately placed in −70°C for overnight. Next day, the samples were homogenized in 1 ml of chloroform/methanol/perchloric acid (2:1:0.05), and the homogenate was centrifuged at 10,000 × g for 10 min at room temperature. Ibuprofen was added as internal standard before homogenization. The organic layer (upper) was taken out carefully and dried up in a Centrivap concentrator (Labconco), followed by resuspension in 50 μl of acetonitrile. Ten microliter sample was then analyzed in Waters 2695 separation module HPLC system using the Phenomenex Luna C18 separation column (250 × 4.6 mm). Samples were analyzed with an isocratic gradient that consisted of acetonitrile/0.01 m phosphoric acid (1:1), at the flow rate of 0.5 ml/min at room temperature. The amount of statins was quantified by monitoring different concentrations of standards (0.1, 1, 2, 5, and 10 μg/ml).
Behavioral analysis.
Two types of behavioral experiments were conducted. This included open-field experiment for locomotor activity and rotorod experiment for feet movement as described previously (Ghosh et al., 2007). Locomotor activity was measured after 7 d of the last dose of MPTP injection in Digiscan Monitor (Omnitech Electronics). This Digiscan Monitor records stereotypy and rearing, behaviors that are directly controlled by striatum, as well as other basic locomotion parameters, such as horizontal activity, total distance traveled, number of movements, movement time, rest time, mean distance, mean time, center time, etc. Before any insult or treatment, mice were placed inside the Digiscan Infrared Activity Monitor for 10 min daily and on rotorod for 10 min daily for 3 consecutive days to train them and record their baseline values. Briefly, animals were removed directly from their cages and gently placed nose first into a specified corner of the open-field apparatus, and after release, data acquisition began every 5 min interval. DIGISCAN software was used to analyze and store horizontal and vertical activity data, which were monitored automatically by infrared beams. In rotorod, the feet movement of the mice was observed at different speeds. To eliminate stress and fatigues, mice were given a 5 min rest interval. Then, 7 d after the last dose of MPTP, open-field assays and rotorod tests were performed twice at 6 h interval on each mouse separately (Ghosh et al., 2007). For each mouse, impairment as well as improvement in locomotor activity was decided after comparing with baseline values.
Statistics.
All values are expressed as means ± SEM. Differences among means were analyzed by one- or two-way ANOVA considering time, dose, or treatment as the independent factor. The one-way ANOVA was performed while analyzing dose-dependent effect of MPP+ on the transcriptional activity of NF-κB, dose-dependent effect of simvastatin on MPP+-induced activation of NF-κB, or time-dependent activation of p21ras after MPTP intoxication in vivo in the nigra. However, two-way ANOVA was used to analyze the effect of simvastatin on MPP+-induced time-dependent activation of p21ras in microglia or to analyze the effect of simvastatin and pravastatin on rotorod activity in MPTP-intoxicated mice at different rpm. In other cases, Student's t test was used to compare outcome between two groups (e.g., control vs PD, control vs MPTP, MPTP vs simvastatin, MPTP vs pravastatin, etc.).
Results
MPP+ induces the activation of microglial NF-κB through p21ras
Microglial activation plays an important role in the pathogenesis of PD as well as other neurodegenerative disorders (Dauer and Przedborski, 2003; Gao et al., 2003; Benner et al., 2004; Roy et al., 2006; Ghosh et al., 2007). Some features of PD are modeled in MPTP-intoxicated animals. The neurotoxic effect of MPTP depends on its conversion into MPP+. In glial cells, monoamine oxidase B (MAO-B) converts MPTP to MPP+, which then activates glial cells (Dauer and Przedborski, 2003; Teismann et al., 2003). Recently, we found that MPP+ induces the activation of NF-κB in glial cells (Ghosh et al., 2007). However, mechanisms by which MPP+ induces the activation of NF-κB in glial cells are poorly understood. Previously, we showed that bacterial lipopolysaccharides (LPSs) induce the activation of NF-κB in astroglia via p21ras (Pahan et al., 2000). Therefore, we investigated whether MPP+ might also induce the activation of NF-κB in glial cells via p21ras. First, we examined whether MPP+ alone was capable of activating p21ras in microglial cells. Activation of p21ras was monitored by immunoprecipitation of active p21ras using Raf–RBD–GST beads. As evident from Figure 1, A, top panel, and B, MPP+ alone significantly (p < 0.0001) induced the activation of p21ras in mouse BV-2 microglial cells with a time course showing maximal activation at 15 min of stimulation. However, p21ras activation was not observed after 30 min of stimulation by MPP+ (Fig. 1A,B). Next, we examined whether p21ras was involved in MPP+-induced activation of NF-κB in microglial cells. As expected, MPP+ markedly induced the transcriptional activity of NF-κB (F(3,8) = 59.03; p < 0.0001). After multiple comparisons using Bonferroni adjustment, it has been found that 1 μm (MPP+) was the most effective dose in inducing the activation of NF-κB in microglial cells (Fig. 1C). We used a dominant-negative mutant of p21ras (Δp21ras) in which the Ser residue at position 17 was changed to Asn (S17N). This mutant binds preferentially to GDP and acts as the dominant inhibitor of p21ras function presumably by blocking access to exchange factors (Qiu et al., 1995; Garnovskaya et al., 1996). Previously, we demonstrated that this Δp21ras inhibits LPS-induced GTP loading of p21ras and the expression of iNOS in astroglia (Pahan et al., 2000). Inhibition of MPP+-induced activation of NF-κB by Δp21ras compared with an empty vector (Fig. 1D) clearly suggests that MPP+ requires p21ras to induce the activation of NF-κB in microglial cells.
Figure 1.

MPP+ induces the activation of p21ras and NF-κB in mouse BV-2 microglial cells via simvastatin-sensitive pathway. A, Cells were stimulated with 1 μm MPP+ under serum-free condition. At different time points, activation of p21ras was monitored (top panel) as described in Materials and Methods. In another set, cells pretreated with 10 μm simvastatin for 6 h were challenged with 1 μm MPP+ under serum-free condition followed by monitoring activation of p21ras at different time points (bottom panel). B, Bands from three different experiments were quantified and activation of Ras is shown as percentage of control. ap < 0.0001 versus control (0 min). C, Cells were cotransfected with 0.25 μg of pBIIX-Luc and 12.5 ng of pRL-TK. After 24 h of transfection, cells were stimulated with different concentrations of MPP+ under serum-free condition. After 6 h of stimulation, firefly and Renilla luciferase activities were obtained by analyzing total cell extract. Data are mean + SEM of three separate experiments. bp < 0.0001 versus control (0 μm). D, Cells were cotransfected with 0.125 μg of pBIIX-Luc, 0.125 μg of either an empty vector or Δp21ras, and 12.5 ng of pRL-TK. After 24 h of transfection, cells were stimulated with MPP+ under serum-free condition. After 6 h of stimulation, firefly and Renilla luciferase activities were analyzed. E, Cells were cotransfected with 0.25 μg of pBIIX-Luc and 12.5 ng of pRL-TK. After 24 h of transfection, cells were treated with different concentrations of simvastatin for 6 h followed by stimulation with MPP+. After 6 h of stimulation, firefly and Renilla luciferase activities were analyzed. Data are mean + SD of three separate experiments. cp < 0.001 versus MPP+.
Simvastatin inhibits MPP+-induced activation of p21ras and NF-κB in microglial cells
Next, we investigated the effect of simvastatin on the activation of p21ras and NF-κB in MPP+-stimulated microglial cells. Cells preincubated with 10 μm simvastatin for 6 h were stimulated by MPP+ followed by assay of p21ras at different minutes of stimulation. It is clearly evident from Figure 1, A, bottom panel, and B, that simvastatin markedly inhibited MPP+-induced activation of p21ras. Multiple-comparison analysis showed that simvastatin inhibited the activation of p21ras at different minute of MPP+ stimulation (2 min: F(1,4) = 151.84, p = 0.0002; 5 min: F(1,4) = 162.78, p = 0.0002; 10 min: F(1,4) = 99.31, p = 0.0006; 15 min: F(1,4) = 137.55, p = 0.0003). Accordingly, we also observed significant (F(3,8) = 45.15; p < 0.0001) inhibition of MPP+-induced activation of NF-κB by simvastatin in microglial cells (Fig. 1E). Multiple comparisons using Bonferroni adjustment shows that, at a dose of 2 μm, simvastatin was not significantly efficient in suppressing MPP+-induced activation of NF-κB (Fig. 1E). However, significant (p < 0.0001) inhibition of NF-κB activation by simvastatin was found at 5 and 10 μm (Fig. 1E). These results suggest that simvastatin is capable of inhibiting MPP+-induced activation of p21ras and NF-κB in microglial cells.
Activation of p21ras in ventral midbrain of MPTP-intoxicated mice
To investigate the role of induced activation of p21ras in the loss of invaluable dopaminergic neurons in MPTP mouse model, first we examined whether the activation of p21ras was induced in the SNpc of MPTP-intoxicated mice. It is clearly evident from Figure 2, A and B, that MPTP was a marked inducer of p21ras activation (F(2,6) = 82.28; p < 0.0001) in vivo in the nigra. Multiple-comparison analysis using Bonferroni adjustment revealed maximum nigral activation (p < 0.0001) of p21ras at 6 h of MPTP intoxication (Fig. 2A,B). The magnitude of p21ras activation decreased after 12 h of MPTP challenge (Fig. 2A,B). In contrast, saline-treated animals showed no or little evidence for p21ras activation.
Figure 2.
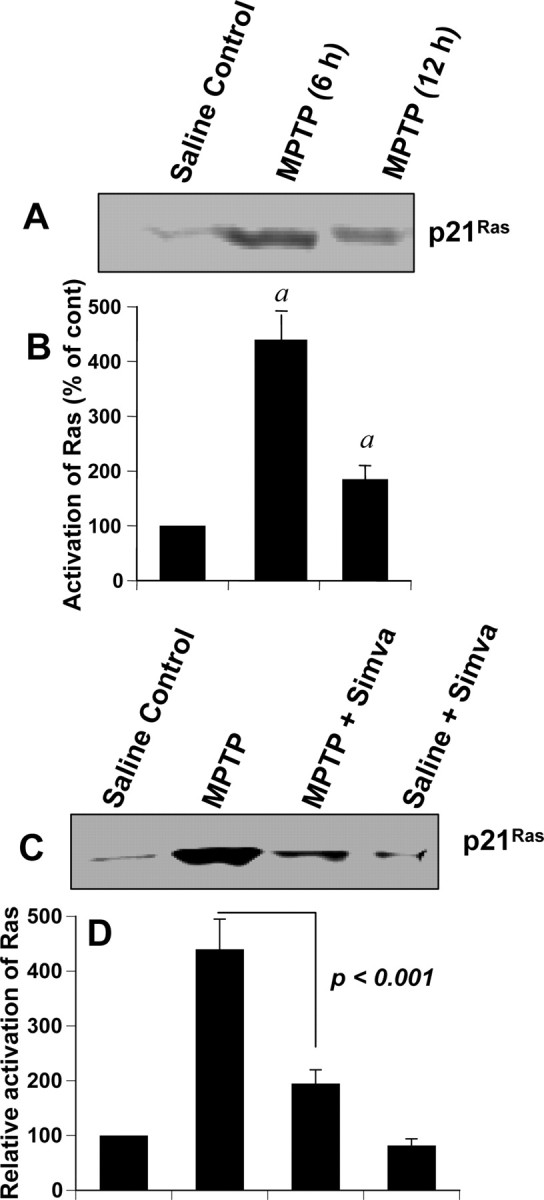
Activation of p21ras in ventral midbrain of MPTP-intoxicated mice is simvastatin sensitive. A, Six and 12 h after the last injection of MPTP, activation of p21ras was monitored in ventral midbrain tissues. Saline was used as control. The experiment was repeated four times, each time on one animal in each group. B, Bands from four different mice were quantified, and activation of Ras is shown as percentage of control. ap < 0.001 versus saline control. C, Mice were treated with simvastatin (1 mg · kg body weight−1 · d−1) via gavage from 24 h before MPTP injection. Six hours after the last injection of MPTP, activation of p21ras was monitored in ventral midbrain tissues. The experiment was repeated four times each time using one animal in each group. D, Bands from four different mice were quantified and activation of p21ras is shown as percentage of control. Error bars indicate SEM.
Because simvastatin suppressed MPP+-induced activation of p21ras in mouse microglial cells (Fig. 1A), we examined whether simvastatin was capable of attenuating the activation of p21ras in vivo in the SNpc of MPTP-intoxicated mice. Mice were treated with simvastatin (1 mg · kg body weight−1 · d−1) through gavage from 1 d before the MPTP intoxication, and the activation of p21ras was monitored 6 h after the last injection of MPTP. As seen in Figure 2, C and D, simvastatin significantly inhibited the activation of p21ras in vivo in the midbrain of MPTP-intoxicated mice.
Simvastatin inhibited the activation of NF-κB in ventral midbrain of MPTP-intoxicated mice
Immunofluorescence analysis of PD midbrain tissue sections demonstrated NF-κB p65 protein (Fig. 3A). When NF-κB p65-positive cells were counted in four nigral sections (two images per slide) from each of four different PD and age-matched control brains in an Olympus IX81 fluorescence microscope using the MicroSuite imaging software, p65 was found to be increased significantly (p < 0.0001) in PD brains compared with age-matched controls. The p65 colocalized with CD11b- and GFAP-positive microglia and astrocytes (Fig. 3A). Because simvastatin inhibited the activation of p21ras in vivo in the SNpc of MPTP-intoxicated mice (Fig. 2) and MPP+ induced the activation of NF-κB in glial cells via p21ras, we examined whether simvastatin was capable of suppressing the activation of NF-κB in vivo in the SNpc of MPTP-intoxicated mice. It is clearly evident from Figure 3B that MPTP intoxication markedly induced the expression of RelA p65 in SNpc compared with saline treatment. Double-label immunofluorescence analysis indicates that p65 was principally expressed in CD11b-positive microglia and GFAP-positive astroglia (Fig. 3B). Next, mice were treated with simvastatin (1 mg · kg body weight−1 · d−1) via gavage from 3 h after the last injection of MPTP, and the activation of NF-κB was examined 24 h after the last injection of MPTP. To understand whether the effect is specific to only simvastatin, we included another statin drug, pravastatin, in the study. However, before doing that, we examined whether pravastatin was capable of entering into the CNS of MPTP-intoxicated mice because according to previous reports (Quion and Jones, 1994; Vuletic et al., 2006), pravastatin does not cross the blood–brain barrier (BBB). Therefore, normal and MPTP-intoxicated mice were fed pravastatin and simvastatin and 24 h after the last injection of MPTP, nigral samples were analyzed for simvastatin and pravastatin by HPLC using ibuprofen as an internal standard. As reported previously (Quion and Jones, 1994; Vuletic et al., 2006), we could not detect pravastatin in the nigra of pravastatin-fed normal mice (data not shown). However, we were able to detect simvastatin in the nigra of simvastatin-fed normal mice (data not shown). In contrast, as evident from Figure 4, both pravastatin (Fig. 4B) and simvastatin (Fig. 4C) were able to enter into the nigra of MPTP-insulted mice. However, nigral level of simvastatin (1.37 + 0.27 ng/mg tissue) was slightly higher than that of pravastatin (1.12 + 0.22 ng/mg tissue). As expected, we did not notice any peak for statin in the nigra of MPTP-intoxicated mice (Fig. 4A). Together, although pravastatin is not capable of entering into the nigra of normal mice, it readily enters into the nigra of MPTP-challenged mice.
Figure 3.
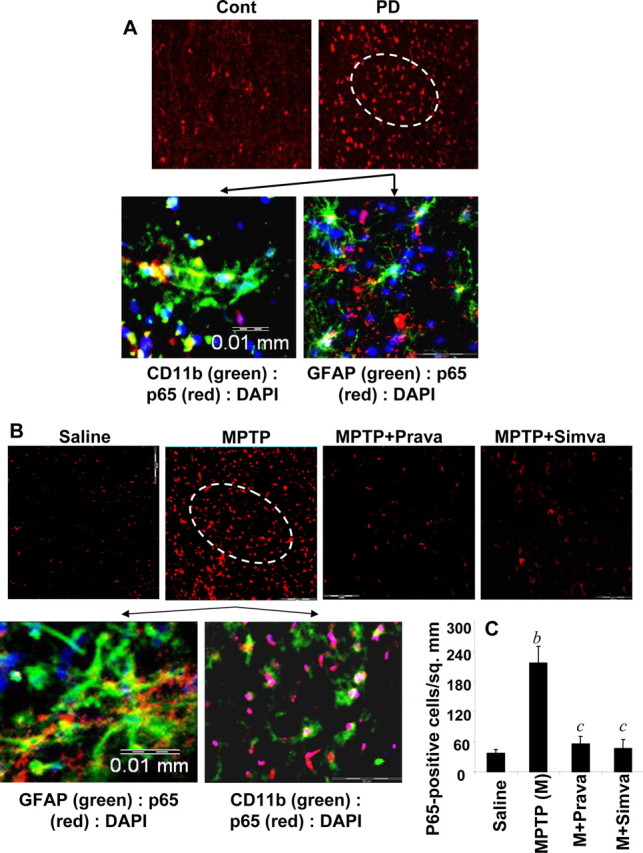
Activation of NF-κB in ventral midbrain of MPTP-intoxicated mice is statin sensitive. A, Midbrain sections of PD patients and age-matched controls were immunostained for p65 (low magnification). Sections of PD patients were also double-labeled for p65 and glial cell markers (GFAP and CD11b). Results represent three independent experiments. B, Mice were treated with pravastatin and simvastatin (1 mg · kg body weight−1 · d−1) from 3 h after the last injection of MPTP. Twenty-four hours after the last injection of MPTP, ventral midbrain sections were immunostained for p65 (low magnification). Midbrain sections of MPTP-intoxicated mice were also double-labeled for p65 and glial cell markers (GFAP for astrocytes and CD11b for microglia). Results represent three independent experiments. C, NF-κB p65-positive cells were counted in four nigral sections (2 images per slide) from each of four mice. bp < 0.0001 versus saline-control; cp < 0.0001 versus MPTP. Error bars indicate SEM.
Figure 4.

Entry of statins into the nigra of MPTP-intoxicated mice. Mice were treated with pravastatin and simvastatin (1 mg · kg body weight−1 · d−1) from 24 h before MPTP injection. Twenty-four hours after the last injection of MPTP, nigra was dissected out and concentrations of pravastatin and simvastatin were analyzed in nigral samples by HPLC using ibuprofen as an internal standard. A, MPTP. B, MPTP plus pravastatin. C, MPTP plus simvastatin.
Next, we investigated the effect of simvastatin and pravastatin on the activation of NF-κB in vivo in the nigra. As evident from Figure 3, B and C, both simvastatin and pravastatin markedly inhibited the level of p65 in vivo in the midbrain of MPTP-intoxicated mice. These results suggest that statins are capable of inhibiting the activation of NF-κB in vivo in the SNpc of MPTP-intoxicated mice.
Statins inhibit the expression of proinflammatory molecules in vivo in the midbrain of MPTP-intoxicated mice
Inflammation plays a role in the loss of dopaminergic neurons in PD and its animal model (Mogi et al., 1994; Hunot et al., 1996; Bessler et al., 1999; Dehmer et al., 2000; Nagatsu et al., 2000; Wu et al., 2002; Benner et al., 2004; Sriram et al., 2006). Consistently, we performed immunofluorescence assays on PD midbrain sections to examine for the expression of iNOS. Figure 5A shows that the expression of iNOS protein was much more in midbrain sections of PD compared with age-matched controls and that iNOS colocalized with CD11b-positive microglia and GFAP-positive astroglia. When iNOS-positive cells were counted in four nigral sections (two images per slide) from each of four different PD and age-matched control brains in an Olympus IX81 fluorescence microscope using the MicroSuite imaging software, iNOS was found to be increased significantly (p < 0.0001) in PD brains compared with age-matched controls. Because simvastatin inhibited the activation of p21ras and NF-κB in vivo in the midbrain of MPTP-intoxicated mice, we examined whether statin drugs were able to suppress the expression of iNOS in vivo in the SNpc of MPTP-insulted mice. Immunofluorescence analysis for iNOS in ventral midbrain sections shows that MPTP intoxication led to marked increase in nigral iNOS protein expression and that iNOS colocalized with GFAP-positive astroglia and CD11b-positive microglia (Fig. 5B). However, consistent to their inhibitory effect on the activation of NF-κB, both simvastatin and pravastatin suppressed MPTP-induced expression of iNOS protein (Fig. 5B,C). As shown by semiquantitative RT-PCR (Fig. 6A) and quantitative real-time PCR (Fig. 6B) experiments, MPTP intoxication led to marked increase in mRNA expression of iNOS, IL-1β, and TNF-α in the SNpc. However, both simvastatin and pravastatin strongly inhibited MPTP-induced expression of these proinflammatory molecules in vivo in the SNpc (Fig. 6A,B). These results suggest statins can suppress the expression of proinflammatory molecules in vivo in the SNpc of MPTP-intoxicated mice.
Figure 5.
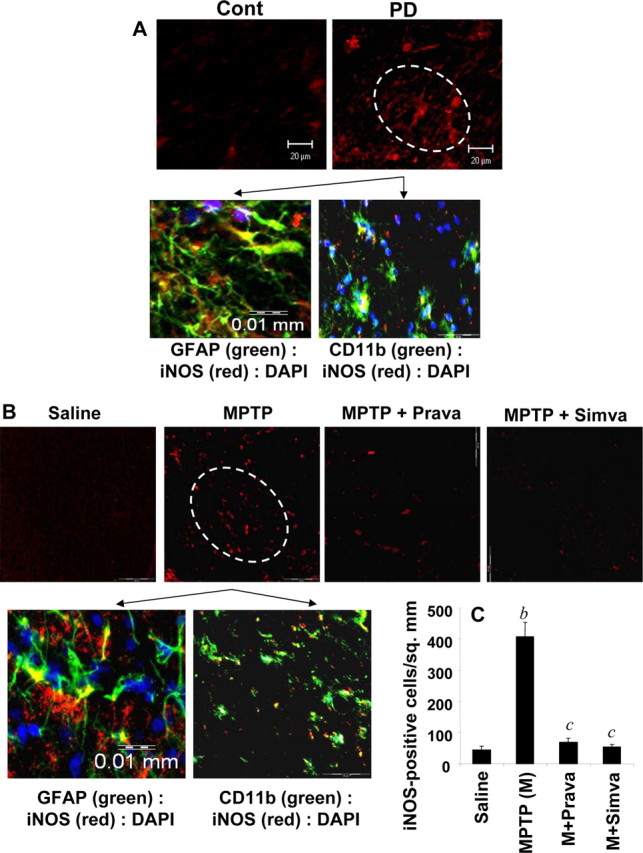
Expression of iNOS in ventral midbrain of MPTP-intoxicated mice is statin sensitive. A, Midbrain sections of PD patients and age-matched controls were immunostained for iNOS (low magnification). Sections of PD patients were also double-labeled for iNOS and glial cell markers (GFAP and CD11b). Results represent three independent experiments. B, Mice were treated with pravastatin and simvastatin (1 mg · kg body weight−1 · d−1) from 3 h after the last injection of MPTP. Twenty-four hours after the last injection of MPTP, ventral midbrain sections were immunostained for iNOS (low magnification). Midbrain sections of MPTP-intoxicated mice were also double-labeled for iNOS and glial cell markers (GFAP for astrocytes and CD11b for microglia). Results represent three independent experiments. C, Cells positive for iNOS were counted in four nigral sections (2 images per slide) from each of four mice. bp < 0.0001 versus saline-control; cp < 0.0001 versus MPTP. Error bars indicate SEM.
Figure 6.
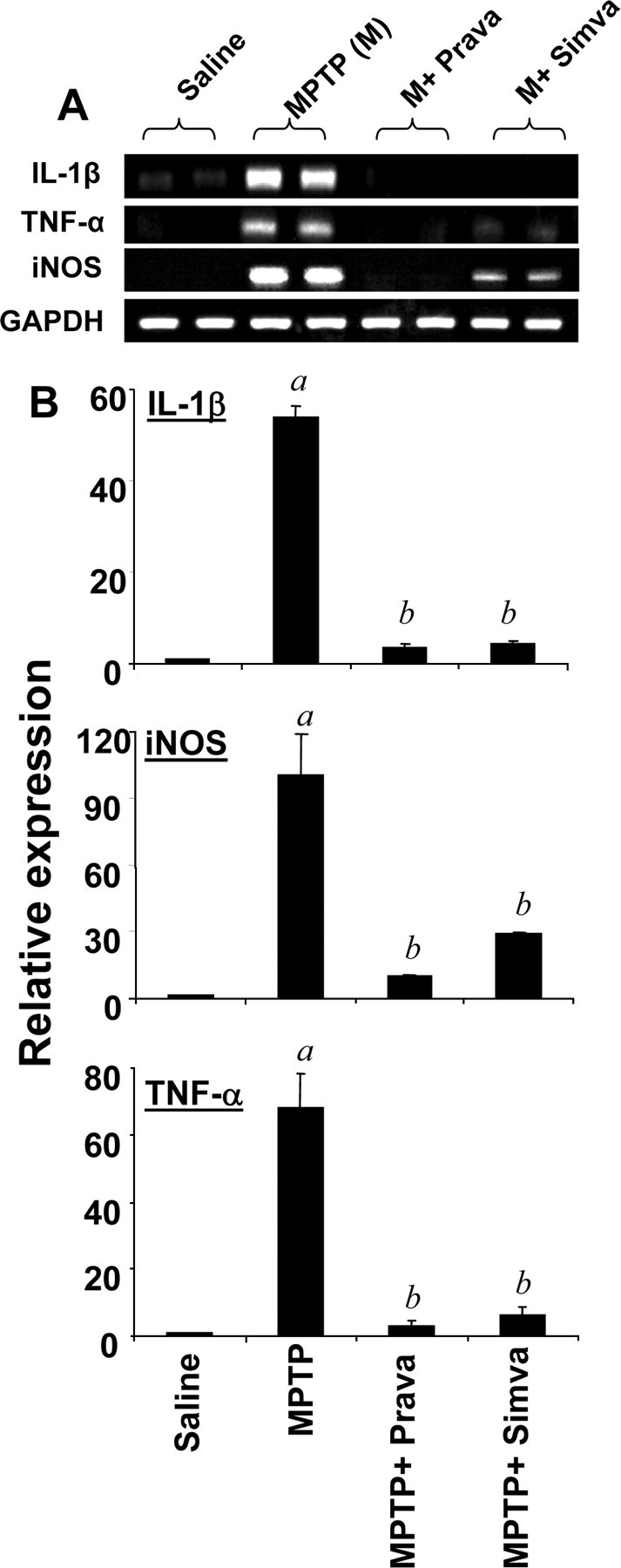
Simvastatin and pravastatin inhibit the expression of iNOS and proinflammatory cytokines in ventral midbrain of MPTP-intoxicated mice. Mice receiving simvastatin and pravastatin (1 mg · kg body weight−1 · d−1) from 3 h after the last injection of MPTP were killed 24 h after the last injection of MPTP. The mRNA expression of iNOS, IL-1β, and TNF-α was analyzed by semiquantitative RT-PCR (A) and quantitative real-time PCR (B). Data are means + SEM of five mice per group. ap < 0.0001 versus saline group; bp < 0.0001 versus the MPTP group.
Statins inhibit the activation of glial cells in vivo in the midbrain of MPTP-intoxicated mice
Recently, activation of glial cells is being considered as a pathological hallmark in PD and other neurodegenerative disorders (González-Scarano and Baltuch, 1999; Wu et al., 2002; Dauer and Przedborski, 2003; Teismann et al., 2003; Benner et al., 2004; Ghosh et al., 2007). Increased expression of CD11b, the β-integrin marker of microglia, represents microglial activation during neurodegenerative inflammation (González-Scarano and Baltuch, 1999). Similarly, on activation, astrocytes also express enhanced level of GFAP, which is considered to be a marker protein for astrogliosis (Eng and Ghirnikar, 1994; Brahmachari et al., 2006). Consistent with these findings, immunofluorescence analysis of midbrain sections in Figure 7A shows that levels of GFAP and CD11b protein were much more in midbrain sections of PD compared with age-matched controls. When GFAP- and CD11b-positive cells were counted in four nigral sections (two images per slide) from each of four different PD and age-matched control brains in an Olympus IX81 fluorescence microscope using the MicroSuite imaging software, GFAP and CD11b were found to be increased significantly (p < 0.0001) in PD brains compared with age-matched controls. We investigated whether simvastatin and pravastatin could attenuate MPTP-induced activation of glial cells in vivo in the SNpc of mice. As evident from immunofluorescence analysis of CD11b and GFAP in ventral midbrain sections (Fig. 7B), MPTP intoxication led to marked increase in nigral CD11b and GFAP protein expression. However, treatment of MPTP-intoxicated mice with simvastatin and pravastatin led to the inhibition of GFAP and CD11b protein expression (Fig. 7B). It is clearly evident from semiquantitative RT-PCR in Figure 8A and real-time PCR in Figure 8B that MPTP intoxication led to marked increase in mRNA expression of both CD11b and GFAP in the SNpc. However, similar to the inhibition of proinflammatory molecules, both simvastatin and pravastatin suppressed MPTP-induced expression of CD11b and GFAP in vivo in the SNpc (Fig. 8A,B). Simvastatin was more potent than pravastatin in inhibiting the expression of GFAP mRNA in vivo in the SNpc (Fig. 8). These results suggest statins are capable of attenuating the expression of proinflammatory molecules in vivo in the SNpc of MPTP-intoxicated mice.
Figure 7.

Increased expression of CD11b and GFAP in ventral midbrain of MPTP-intoxicated mice is statin sensitive. A, Midbrain sections of PD patients and age-matched controls were immunostained for GFAP and CD11b. B, Mice were treated with pravastatin and simvastatin (1 mg · kg body weight−1 · d−1) from 3 h after the last injection of MPTP. Twenty-four hours after the last injection of MPTP, ventral midbrain sections were immunostained for GFAP and CD11b. Results represent analysis of four nigral sections (2 images per slide) from each of four mice.
Figure 8.
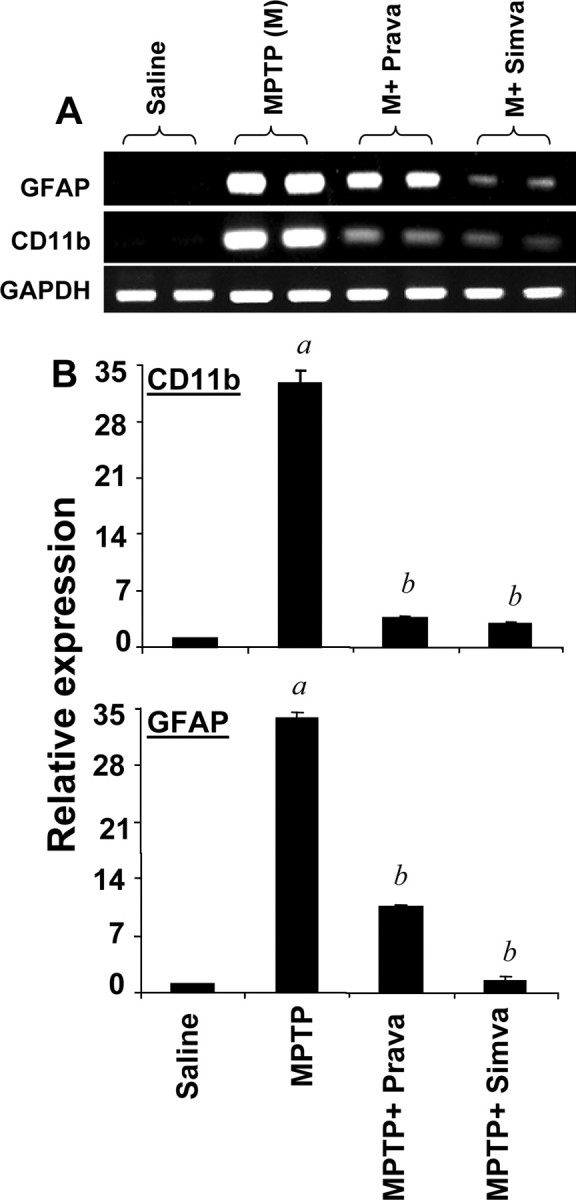
Simvastatin and pravastatin inhibit mRNA expression of GFAP and CD11b in ventral midbrain of MPTP-intoxicated mice. Mice receiving simvastatin and pravastatin (1 mg · kg body weight−1 · d−1) from 3 h after the last injection of MPTP were killed 24 h after the last injection of MPTP. The mRNA expression of GFAP and CD11b was analyzed by semiquantitative RT-PCR (A) and quantitative real-time PCR (B). Data are means + SEM of five mice per group. ap < 0.0001 versus saline group; bp < 0.0001 versus the MPTP group.
Statins protect against MPTP-induced neurodegeneration
Mice were treated with simvastatin and pravastatin (1 mg · kg body weight−1 · d−1) via gavage from 3 h after the last injection of MPTP. Seven days after the last injection of MPTP, animals were processed for quantification of dopaminergic cell bodies in the SNpc and of projecting dopaminergic fibers in the striatum using TH immunostaining. MPTP intoxication led to ∼73% loss of SNpc TH-positive neurons (Fig. 9A,B) and 70% reduction of striatal TH ODs (Fig. 9A,C) compared with saline-injected controls. However, in MPTP-injected mice treated with simvastatin and pravastatin, less reduction in SNpc TH-positive neurons and striatal TH ODs was observed (Fig. 9A–C). Simvastatin was slightly more efficient than pravastatin in protecting TH-positive neurons and fibers against MPTP toxicity (Fig. 9A–C).
Figure 9.

Simvastatin and pravastatin protect dopaminergic neurons in MPTP-intoxicated mice. Mice receiving simvastatin and pravastatin (1 mg · kg body weight−1 · d−1) from 3 h after the last injection of MPTP were killed 7 d after the last injection of MPTP followed by TH immunostaining of SNpc (top panel) and striatum (bottom panel) (A), counting of TH-positive neurons in SNpc (B), quantification of TH-positive fibers in striatum (C), and assay of neurotransmitters in striatum (D). To understand whether statin alone was toxic for the nigrostriatum, groups of mice received two different doses of simvastatin (1 and 40 mg · kg body weight−1 · d−1). After 7 d, the concentration of dopamine was assayed in striatum (E). Data are means + SEM of eight mice per group.
Next, to determine whether statins protect against biochemical deficits caused by MPTP, we quantified levels of DA and two of its metabolites, DOPAC and HVA, in striata 7 d after the MPTP treatment. As evident from Figure 9D, MPTP intoxication led to ∼78% decrease in striatal DA compared with striata of saline-injected mice. In contrast, MPTP-intoxicated animals that received simvastatin and pravastatin showed only 21–26% decrease in striatal dopamine (Fig. 9D).
Although we observed protection of the nigrostriatum by simvastatin in MPTP-intoxicated mice, Kreisler et al. (2007) have shown that simvastatin is deleterious for the nigrostriatum. The major difference between the work by Kreisler et al. and our current study is that they treated mice (intraperitoneally) with simvastatin at a dose of 40 mg · kg body weight−1 · d−1, a dose that is 40 times higher than that used by us. We also used the same dose of simvastatin to examine the truth behind its protective effect. Consistent to that observed by Kreisler et al. (2007), simvastatin alone at this high dose significantly led to a decrease in striatal dopamine (Fig. 9E). In contrast, simvastatin at a dose of 1 mg · kg body weight−1 · d−1 had no inhibitory effect on striatal dopamine (Fig. 9E). Together, these results suggest that simvastatin is protective for the nigrostriatum at a FDA-approved therapeutic dose (1 mg · kg body weight−1 · d−1 equivalent to 80 mg · adult−1 · d−1) and that simvastatin is toxic for the nigrostriatum at a very high dose.
Is inhibition of p21ras farnesylation alone sufficient to protect the nigrostriatum against MPTP toxicity?
In addition to inhibiting farnesylation of p21ras, simvastatin and other statins exhibit many other biological functions (Pahan, 2006), which could be responsible for the protection of nigrostriatum from MPTP neurotoxicity. Therefore, we investigated whether inhibition of p21ras alone was sufficient for the protection of the nigrostriatum. Farnesyltransferase (FPT) inhibitor II is a potent and fairly selective inhibitor of farnesylation of p21ras (Leftheris et al., 1996). MPTP-intoxicated mice received FPT inhibitor II via intraperitoneal injection at a dose of 10 mg · kg body weight−1 · d−1 from 3 h after the last injection of MPTP. After 7 d of MPTP intoxication, level of neurotransmitters was monitored in the striatum. FPT inhibitor II alone was able to reverse the loss of dopamine by >70% in MPTP-intoxicated mice (Fig. 10), suggesting that suppression of p21ras farnesylation alone is sufficient to protect the nigrostriatum in MPTP-intoxicated mice.
Figure 10.

Effect of FPT inhibitor II on the loss of dopamine in MPTP-intoxicated mice. Mice receiving FPT inhibitor II (10 mg · kg body weight−1 · d−1) from 3 h after the last injection of MPTP were killed 7 d after the last injection of MPTP followed by assay of dopamine in striatum (DA). Data are means + SEM of eight mice per group.
Statins improve locomotor functions in MPTP-intoxicated mice
The ultimate therapeutic goal of neuroprotection is to decrease functional impairment. Therefore, to examine whether statins protect not only against structural and neurotransmitter damage but also against functional deficits caused by MPTP, we monitored locomotor and open-field activities. As reported previously (Tillerson et al., 2002; Ghosh et al., 2007), MPTP injection caused marked decrease in rotorod performance (Fig. 11A), horizontal activity (Fig. 11B), movement time (Fig. 11C), number of movements (Fig. 11D), stereotypy counts (Fig. 11E), rearing (Fig. 11F), and total distance (Fig. 11H). However, MPTP increased the rest time (Fig. 11G). Interestingly, both simvastatin and pravastatin significantly improved MPTP-induced hypolocomotion (Fig. 11A–H). Although both simvastatin and pravastatin improved rotorod performance, pairwise comparison at each rpm reveals that simvastatin was more potent than pravastatin in enhancing this locomotor activity at 16 rpm (F(1,21) = 15.52; p = 0.0008) and 18 rpm (F(1,21) = 34.21; p < 0.0001).
Figure 11.

Simvastatin and pravastatin improve motor functions in MPTP-intoxicated mice. Mice receiving simvastatin and pravastatin (1 mg · kg body weight−1 · d−1) from 3 h after the last injection of MPTP were tested for motor functions [rotorod (A); horizontal activity (B); movement time (C); number of movements (D); stereotypy counts (E); rearing (F); rest time (G); total distance (H)] 7 d after the last injection of MPTP. Data are means + SEM of eight mice per group. b,c,e,f,hp < 0.001 versus MPTP. d,gp < 0.05 versus MPTP. For rotorod, we performed two-way ANOVA analysis and observed significant improvement in rotorod performance by simvastatin and pravastatin at 16 rpm (F(2,21) = 72.35; p < 0.0001), 18 rpm (F(2,21) = 170.41; p < 0.0001), and 22 rpm (F(2,21) = 108.94; p < 0.0001). Error bars indicate SEM.
Do statins halt the disease progression?
Usually after the diagnosis of the disease, PD patients are treated with neuroprotective drugs. Therefore, we investigated whether statins administered 2 d after initiation of the disease were still capable of inhibiting the demise of TH-positive neurons and concomitant loss of neurotransmitters. As evident from TH immunostaining in Figure 12A, stereological counting of TH-positive neurons in Figure 12B and quantitation of TH-positive fibers in the striatum in Figure 12C, both simvastatin and pravastatin protected TH-positive neurons in the SNpc and TH-positive fibers in the striatum from MPTP toxicity. To establish whether this protection of nigrostriatal neurons was correlated with striatal neurotransmitter levels, we analyzed striatal tissues for DA, DOPAC, and HVA by HPLC. MPTP intoxication sharply reduced (∼78% decrease) striatal DA compared with striata of saline-injected mice (Fig. 12D). However, MPTP-intoxicated animals that received pravastatin from 2 d after the initiation of the disease showed a ∼52% decrease in striatal dopamine (Fig. 12D). MPTP-intoxicated mice that received simvastatin from 2 d after the initiation of the disease showed a ∼30% decrease in striatal dopamine (Fig. 12D). These results suggest that statins are capable of slowing down the progression of neuronal loss in the MPTP mouse model.
Figure 12.

Simvastatin and pravastatin suppress disease progression in the MPTP mouse model. Mice receiving simvastatin and pravastatin (1 mg · kg body weight−1 · d−1) from 2 d after the last injection of MPTP were killed 7 d after the last injection of MPTP followed by TH immunostaining of SNpc (top panel) and striatum (bottom panel) (A), counting of TH-positive neurons in SNpc (B), quantification of TH-positive fibers in striatum (C), and assay of neurotransmitters in striatum (D). Data are means + SEM of eight mice per group.
In addition, the neurotoxic effect of MPTP depends on several key toxicokinetic steps such as its conversion into MPP+ in glial cells by MAO-B and the uptake of MPP+ by dopaminergic neurons (Dauer and Przedborski, 2003), suggesting that statins may confer protection by interfering with the conversion of MPTP to MPP+ and/or the uptake of MPP+ by dopaminergic neurons. However, in this disease progression model, in which the treatment began from 2 d after the last injection of MPTP, statins also showed its efficacy nullifying this possibility.
Discussion
Understanding the mechanism of the disease process of PD and development of effective neuroprotective therapeutic approach to halt the disease progression are of paramount importance. The MPTP mouse model is particularly useful in testing new therapeutic intervention in PD. Because of the facts that inflammation plays a role in the loss of dopaminergic neurons in PD and its MPTP mouse model and that statins, FDA-approved cholesterol-lowering drugs, inhibit the expression of proinflammatory molecules, we decided to investigate the efficacy of statins in protecting nigrostriatal neurons in the MPTP mouse model of PD.
Several lines of evidence presented in this manuscript clearly establish that statins are capable of protecting dopaminergic neurons from parkinsonian toxicity. Our conclusion is based on the following. First, MPTP intoxication led to the induction of various proinflammatory molecules within the nigra of MPTP-intoxicated mice. However, treatment of mice with simvastatin and pravastatin resulted in attenuation of expression of proinflammatory molecules. Second, as expected, MPTP intoxication led to marked increase in gliosis as evident from increased expression of CD11b and GFAP in the SNpc. However, both simvastatin and pravastatin were capable of suppressing the expression of CD11b and GFAP in vivo in the SNpc. Third, as observed in PD, nigrostriatal neurons disappeared and the level of neurotransmitters decreased in MPTP-intoxicated mice. But treatment with simvastatin and pravastatin protected TH-positive dopaminergic neurons from MPTP toxicity and restored the level of neurotransmitters. Fourth, simvastatin and pravastatin also ameliorated functional impairment in MPTP-intoxicated mice. Fifth, simvastatin and pravastatin administered 2 d after initiation of the disease still suppressed the demise of TH-positive neurons and concomitant loss of neurotransmitters, suggesting that statins have the capability of attenuating disease progression. Although pravastatin is not capable of entering into the CNS of normal mice (Quion and Jones, 1994), this statin drug enters into the nigra of MPTP-intoxicated mice. It is most probably attributable to the fact that MPTP challenge increases permeability through the BBB (Zhao et al., 2007; Brochard et al., 2009).
Wolozin et al. (2007) have reported that simvastatin is associated with a reduction in the incidence of dementia and PD. According to Wahner et al. (2008), all statins are inversely associated with PD except for pravastatin. A possible explanation for pravastation being the exception in this study may be its inability in crossing the BBB. However, a neuroprotective effect of statins in MPTP mouse model remains controversial. For example, Kreisler et al. (2007) have shown that both simvastatin and atorvastatin have deleterious effects on the nigrostriatum in MPTP-intoxicated mice. In contrast, Selley (2005) has reported that simvastatin prevents MPTP-induced striatal dopamine depletion and protein tyrosine nitration in mice. These studies used a concentration of simvastatin that is 10–40 times higher than the regular human dose and treatment began 5 or 7 d before MPTP intoxication. When we repeated their experiment, we also found that simvastatin at a high dose (40 mg · kg body weight−1 · d−1) was deleterious for the nigrostriatum. Therefore, although simvastatin at an FDA-approved therapeutic dose is protective, at a higher dose, this drug is toxic for the nigrostriatum.
Statins are the most widely used cholesterol-lowering drugs throughout the world. These drugs inhibit HMG-CoA reductase, the regulatory enzyme in the cholesterol biosynthesis pathway, and thereby lower the level of cholesterol in patients with hypercholesterolemia. Previously, Pahan et al. (1997) showed that lovastatin inhibits LPS-induced expression of proinflammatory molecules (iNOS, IL-1β, TNF-α, and IL-6) in glial cells and macrophages possibly through the inhibition of farnesylation/activation of p21ras, the central molecule in intracellular signal transduction. Subsequently, we also demonstrated that overexpression of Δp21ras, a dominant-negative mutant of p21ras, inhibits the expression of proinflammatory molecules in astroglia (Pahan et al., 2000), suggesting the involvement of p21ras in glial expression of proinflammatory molecules. The p21ras proto-oncogene proteins, a family of GTP-binding proteins, function by binding to the cytoplasmic surface of the plasma membrane. This membrane localization of p21ras involves prenylation of cysteine in CAAX motif present at the C terminus, proteolytic removal of AAX tripeptide, and then carboxymethylation of C-terminal cysteine (Hancock et al., 1991). Statins inhibit the mevalonate pathway, lowering the level of farnesyl pyrophosphate required for the farnesylation of p21ras.
Being a highly lipophilic molecule, MPTP readily crosses the BBB. It has been shown that, after systemic administration, MPTP reaches the brain within minutes (Markey et al., 1984). Once in the brain, MPTP is converted to MPDP+ and MPP+. The latter one is believed to be responsible for neurotoxicity (Dauer and Przedborski, 2003). Mechanisms by which MPP+ insult leads to glial activation and neuronal death are poorly understood. Here, we demonstrate that MPP+ alone is capable of activating p21ras in mouse microglial cells within minutes. Because the activation of NF-κB is necessary for the transcription of different proinflammatory molecules (Hayden and Ghosh, 2008) and MPP+ also induces the activation of NF-κB in glial cells (Ghosh et al., 2007), we examined whether p21ras was involved in MPP+-induced activation of NF-κB. Interestingly, Δp21ras attenuated MPP+-induced activation of NF-κB in microglia, indicating that MPP+ induces the activation of NF-κB in microglia via p21ras. Therefore, inhibition of MPP+-induced activation of p21ras and NF-κB in microglial cells by simvastatin suggests that simvastatin attenuates MPP+-induced activation of NF-κB via inhibition of p21ras (Fig. 13). This has been also supported by our in vivo data in which MPTP intoxication led to rapid activation of p21ras within the SNpc and oral treatment of MPTP-intoxicated mice with simvastatin suppressed nigral activation of p21ras. Although we observed marked activation of p21ras by MPP+/MPTP, we do not know a mechanism by which MPP+ couples to the activation of p21ras. On farnesylation, activated p21ras goes to the plasma membrane from where it transmits downstream signals. Therefore, rapid activation of p21ras by MPP+ in microglia suggests that either MPP+ binds to any putative tyrosine kinase receptor for the activation of p21ras or MPP+ enters into the cell to activate and mobilize guanine nucleotide exchange factors such as, son of sevenless homologs 1 and 2 (SOS-1 and SOS-2), p21ras protein-specific guanine nucleotide-releasing factor 1, etc. Currently, experiments are underway in our laboratory to investigate such possibilities.
Figure 13.
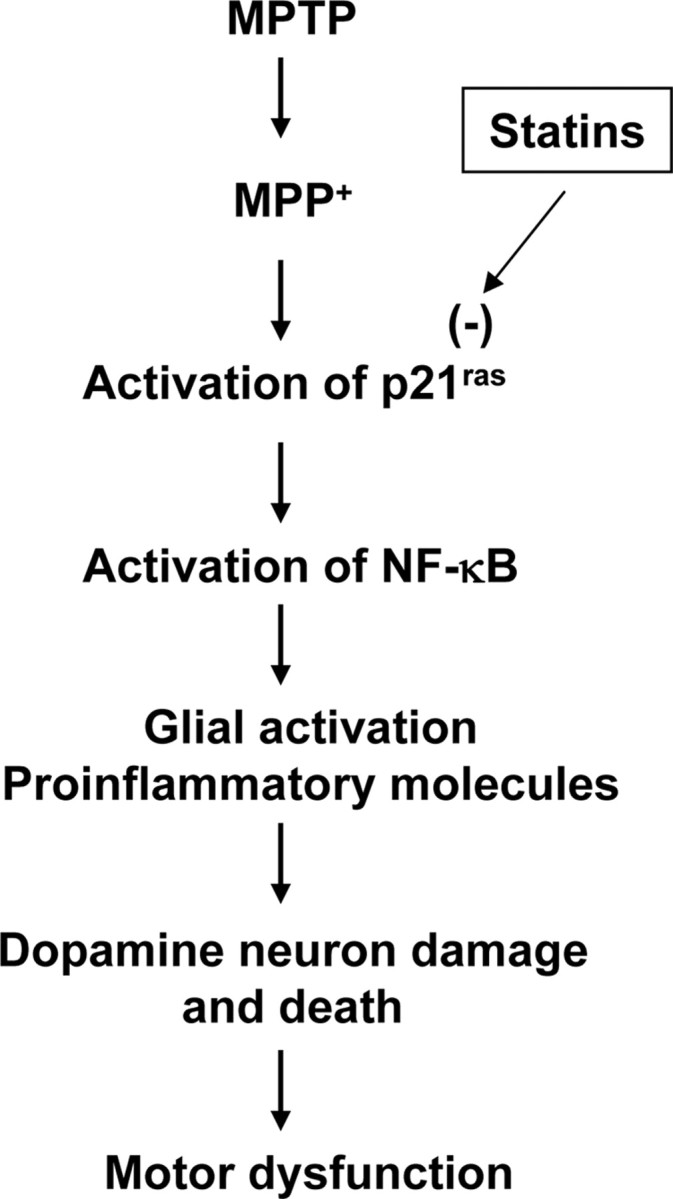
Schematic presentation of possible hypothesis.
Once NF-κB is activated on MPP+/MPTP insult, in collaboration with other proinflammatory transcription factors, it drives the transcription of several proinflammatory molecules including iNOS, TNF-α, and IL-1β in both microglia and astroglia (Liu et al., 2002; Dasgupta et al., 2003; Hayden and Ghosh, 2008), which have been shown to play an important role in the loss of dopaminergic neurons in the MPTP mouse model (Liberatore et al., 1999; Dehmer et al., 2000; Nagatsu et al., 2000; Sriram et al., 2006). Recently, we demonstrated that once NO is produced via NF-κB-dependent pathway, it upregulates the expression of GFAP in astroglia and the expression of CD11b in microglia through NF-κB-independent but GC (guanylate cyclase)–cGMP–PKG (protein kinase G)-dependent pathway (Brahmachari et al., 2006; Roy et al., 2006). Because glial production of proinflammatory molecules and gliosis are important features of PD pathology, it appears that NF-κB activation regulates these pathological features either directly or indirectly and that statins protect dopaminergic neurons in MPTP-intoxicated mice via suppression of (p21ras–NF-κB)-dependent pathological steps. Restoration of striatal dopamine content in MPTP-intoxicated mice by FPT inhibitor II, a cell-permeable potent inhibitor of p21ras farnesylation, suggests that inhibition of p21ras alone is sufficient for the protection of the nigrostriatum from MPTP. Although inhibition of p21ras by statins and FPT inhibitor II led to the protection of the nigrostriatum in MPTP-lesioned mice, one study by Heumann et al. (2000) shows that transgenic activation of p21ras in neurons promotes hypertrophy and protects from MPTP-induced degeneration. However, this study used RasV12, the oncogenic form of p21ras, which always transduces robust activation of the classical MAP kinase [Raf–MEK (mitogen-activated protein kinase kinase)–ERK (extracellular signal-regulated kinase)] pathway making the normal cell vulnerable to abnormal growth.
Protective effects in animal models of PD have been obtained with various molecules including GDNF (glial cell line-derived neurotrophic factor), neurturin, BDNF, TGF-β, and bFGF (basic fibroblast growth factor) (Date et al., 1993; Flanders et al., 1998; Kordower et al., 2000). However, these peptides do not readily diffuse across the BBB or ventricular lining and have limited or unstable bioavailability and some toxicity (Kordower et al., 1999). However, statins enter into the CNS (Fig. 4) (Saheki et al., 1994) and protect the nigrostriatum against MPTP insult (Figs. 9, 12). Although statins could have multiple effects leading to neuroprotection such as stimulation of heat shock protein expression, suppression of matrix metalloproteinase, activation of Akt, inhibition of apoptosis, etc. (Delanty et al., 2001; Pahan, 2006), here we have demonstrated that p21ras is activated very early in vivo in the SNpc of MPTP-intoxicated mice and that statins suppress the activation of p21ras and block the activation of NF-κB in the SNpc, inhibit nigral expression of proinflammatory molecules and the activation glial cells, protect the loss of dopaminergic neurons, and improve the behavioral functions in MPTP-intoxicated mice (summarized in Fig. 13). These results suggest that statins may be used for therapeutic intervention in PD.
Footnotes
This work was supported by National Institutes of Health Grants NS39940, NS48923, and T32 AG000269, and The Michael J. Fox Foundation for Parkinson's Research. We thank Dr. Bichun Ouyang (Department of Neurological Sciences, Rush University Medical Center) for her help with statistical analysis and Dr. Karie Scrogin of Loyola University Medical Center in Chicago for providing acetonitrile.
References
- Benner EJ, Mosley RL, Destache CJ, Lewis TB, Jackson-Lewis V, Gorantla S, Nemachek C, Green SR, Przedborski S, Gendelman HE. Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:9435–9440. doi: 10.1073/pnas.0400569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessler H, Djaldetti R, Salman H, Bergman M, Djaldetti M. IL-1 beta, IL-2, IL-6 and TNF-alpha production by peripheral blood mononuclear cells from patients with Parkinson's disease. Biomed Pharmacother. 1999;53:141–145. doi: 10.1016/S0753-3322(99)80079-1. [DOI] [PubMed] [Google Scholar]
- Brahmachari S, Fung YK, Pahan K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci. 2006;26:4930–4939. doi: 10.1523/JNEUROSCI.5480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Jana M, Liu X, Pahan K. Role of very-late antigen-4 (VLA-4) in myelin basic protein-primed T cell contact-induced expression of proinflammatory cytokines in microglial cells. J Biol Chem. 2003;278:22424–22431. doi: 10.1074/jbc.M301789200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date I, Yoshimoto Y, Imaoka T, Miyoshi Y, Gohda Y, Furuta T, Asari S, Ohmoto T. Enhanced recovery of the nigrostriatal dopaminergic system in MPTP-treated mice following intrastriatal injection of basic fibroblast growth factor in relation to aging. Brain Res. 1993;621:150–154. doi: 10.1016/0006-8993(93)90312-b. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB. Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J Neurochem. 2000;74:2213–2216. doi: 10.1046/j.1471-4159.2000.0742213.x. [DOI] [PubMed] [Google Scholar]
- Delanty N, Vaughan CJ, Sheehy N. Statins and neuroprotection. Expert Opin Investig Drugs. 2001;10:1847–1853. doi: 10.1517/13543784.10.10.1847. [DOI] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS. GFAP and astrogliosis. Brain Pathol. 1994;4:229–237. doi: 10.1111/j.1750-3639.1994.tb00838.x. [DOI] [PubMed] [Google Scholar]
- Flanders KC, Ren RF, Lippa CF. Transforming growth factor-betas in neurodegenerative disease. Prog Neurobiol. 1998;54:71–85. doi: 10.1016/s0301-0082(97)00066-x. [DOI] [PubMed] [Google Scholar]
- Gao HM, Liu B, Zhang W, Hong JS. Novel anti-inflammatory therapy for Parkinson's disease. Trends Pharmacol Sci. 2003;24:395–401. doi: 10.1016/S0165-6147(03)00176-7. [DOI] [PubMed] [Google Scholar]
- Garnovskaya MN, van Biesen T, Hawe B, Casañas Ramos S, Lefkowitz RJ, Raymond JR. Ras-dependent activation of fibroblast mitogen-activated protein kinase by 5-HT1A receptor via a G protein beta gamma-subunit-initiated pathway. Biochemistry. 1996;35:13716–13722. doi: 10.1021/bi961764n. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, Ghosh S, Mosley RL, Gendelman HE, Pahan K. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2007;104:18754–18759. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- Hancock JF, Cadwallader K, Marshall CJ. Methylation and proteolysis are essential for efficient membrane binding of prenylated p21K-ras(B) EMBO J. 1991;10:641–646. doi: 10.1002/j.1460-2075.1991.tb07992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Heumann R, Goemans C, Bartsch D, Lingenhöhl K, Waldmeier PC, Hengerer B, Allegrini PR, Schellander K, Wagner EF, Arendt T, Kamdem RH, Obst-Pernberg K, Narz F, Wahle P, Berns H. Transgenic activation of Ras in neurons promotes hypertrophy and protects from lesion-induced degeneration. J Cell Biol. 2000;151:1537–1548. doi: 10.1083/jcb.151.7.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunot S, Boissière F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y, Hirsch EC. Nitric oxide synthase and neuronal vulnerability in Parkinson's disease. Neuroscience. 1996;72:355–363. doi: 10.1016/0306-4522(95)00578-1. [DOI] [PubMed] [Google Scholar]
- Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, Faucheux BA, Agid Y, Hirsch EC. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with Parkinson disease. Proc Natl Acad Sci U S A. 1997;94:7531–7536. doi: 10.1073/pnas.94.14.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana M, Liu X, Koka S, Ghosh S, Petro TM, Pahan K. Ligation of CD40 stimulates the induction of nitric-oxide synthase in microglial cells. J Biol Chem. 2001;276:44527–44533. doi: 10.1074/jbc.M106771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana M, Jana A, Liu X, Ghosh S, Pahan K. Involvement of phosphatidylinositol 3-kinase-mediated up-regulation of IκB alpha in anti-inflammatory effect of gemfibrozil in microglia. J Immunol. 2007;179:4142–4152. doi: 10.4049/jimmunol.179.6.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Palfi S, Chen EY, Ma SY, Sendera T, Cochran EJ, Cochran EJ, Mufson EJ, Penn R, Goetz CG, Comella CD. Clinicopathological findings following intraventricular glial-derived neurotrophic factor treatment in a patient with Parkinson's disease. Ann Neurol. 1999;46:419–424. doi: 10.1002/1531-8249(199909)46:3<419::aid-ana21>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Emborg ME, Bloch J, Ma SY, Chu Y, Leventhal L, McBride J, Chen EY, Palfi S, Roitberg BZ, Brown WD, Holden JE, Pyzalski R, Taylor MD, Carvey P, Ling Z, Trono D, Hantraye P, Déglon N, Aebischer P. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson's disease. Science. 2000;290:767–773. doi: 10.1126/science.290.5492.767. [DOI] [PubMed] [Google Scholar]
- Kreisler A, Gelé P, Wiart JF, Lhermitte M, Destée A, Bordet R. Lipid-lowering drugs in the MPTP mouse model of Parkinson's disease: fenofibrate has a neuroprotective effect, whereas bezafibrate and HMG-CoA reductase inhibitors do not. Brain Res. 2007;1135:77–84. doi: 10.1016/j.brainres.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Leftheris K, Kline T, Vite GD, Cho YH, Bhide RS, Patel DV, Patel MM, Schmidt RJ, Weller HN, Andahazy ML, Carboni JM, Gullo-Brown JL, Lee FY, Ricca C, Rose WC, Yan N, Barbacid M, Hunt JT, Meyers CA, Seizinger BR, et al. Development of highly potent inhibitors of Ras farnesyltransferase possessing cellular and in vivo activity. J Med Chem. 1996;39:224–236. doi: 10.1021/jm950642a. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K. Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem. 2002;277:39312–39319. doi: 10.1074/jbc.M205107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markey SP, Johannessen JN, Chiueh CC, Burns RS, Herkenham MA. Intraneuronal generation of a pyridinium metabolite may cause drug-induced parkinsonism. Nature. 1984;311:464–467. doi: 10.1038/311464a0. [DOI] [PubMed] [Google Scholar]
- Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180:147–150. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson's disease. J Neural Transm Suppl. 2000:277–290. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- Pahan K. Lipid-lowering drugs. Cell Mol Life Sci. 2006;63:1165–1178. doi: 10.1007/s00018-005-5406-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Namboodiri AM, Singh I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K, Liu X, McKinney MJ, Wood C, Sheikh FG, Raymond JR. Expression of a dominant-negative mutant of p21ras inhibits induction of nitric oxide synthase and activation of nuclear factor-kappaB in primary astrocytes. J Neurochem. 2000;74:2288–2295. doi: 10.1046/j.1471-4159.2000.0742288.x. [DOI] [PubMed] [Google Scholar]
- Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature. 1995;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- Quion JA, Jones PH. Clinical pharmacokinetics of pravastatin. Clin Pharmacokinet. 1994;27:94–103. doi: 10.2165/00003088-199427020-00002. [DOI] [PubMed] [Google Scholar]
- Qureshi GA, Baig S, Bednar I, Södersten P, Forsberg G, Siden A. Increased cerebrospinal fluid concentration of nitrite in Parkinson's disease. Neuroreport. 1995;6:1642–1644. doi: 10.1097/00001756-199508000-00013. [DOI] [PubMed] [Google Scholar]
- Roy A, Fung YK, Liu X, Pahan K. Up-regulation of microglial CD11b expression by nitric oxide. J Biol Chem. 2006;281:14971–14980. doi: 10.1074/jbc.M600236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saheki A, Terasaki T, Tamai I, Tsuji A. In vivo and in vitro blood-brain barrier transport of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors. Pharm Res. 1994;11:305–311. doi: 10.1023/a:1018975928974. [DOI] [PubMed] [Google Scholar]
- Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selley ML. Simvastatin prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain Res. 2005;1037:1–6. doi: 10.1016/j.brainres.2004.02.083. [DOI] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: role of TNF-alpha. FASEB J. 2006;20:670–682. doi: 10.1096/fj.05-5106com. [DOI] [PubMed] [Google Scholar]
- Teismann P, Tieu K, Cohen O, Choi DK, Wu DC, Marks D, Vila M, Jackson-Lewis V, Przedborski S. Pathogenic role of glial cells in Parkinson's disease. Mov Disord. 2003;18:121–129. doi: 10.1002/mds.10332. [DOI] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson-Lewis V, Ramasamy R, Przedborski S. d-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest. 2003;112:892–901. doi: 10.1172/JCI18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillerson JL, Caudle WM, Reverón ME, Miller GW. Detection of behavioral impairments correlated to neurochemical deficits in mice treated with moderate doses of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Exp Neurol. 2002;178:80–90. doi: 10.1006/exnr.2002.8021. [DOI] [PubMed] [Google Scholar]
- Vila M, Przedborski S. Genetic clues to the pathogenesis of Parkinson's disease. Nat Med. 2004;10(Suppl):S58–S62. doi: 10.1038/nm1068. [DOI] [PubMed] [Google Scholar]
- Vuletic S, Riekse RG, Marcovina SM, Peskind ER, Hazzard WR, Albers JJ. Statins of different brain penetrability differentially affect CSF PLTP activity. Dement Geriatr Cogn Disord. 2006;22:392–398. doi: 10.1159/000095679. [DOI] [PubMed] [Google Scholar]
- Wahner AD, Bronstein JM, Bordelon YM, Ritz B. Statin use and the risk of Parkinson disease. Neurology. 2008;70:1418–1422. doi: 10.1212/01.wnl.0000286942.14552.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, Wang SW, Li NC, Lee A, Lee TA, Kazis LE. Simvastatin is associated with a reduced incidence of dementia and Parkinson's disease. BMC Med. 2007;5:20. doi: 10.1186/1741-7015-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Ling Z, Newman MB, Bhatia A, Carvey PM. TNF-alpha knockout and minocycline treatment attenuates blood-brain barrier leakage in MPTP-treated mice. Neurobiol Dis. 2007;26:36–46. doi: 10.1016/j.nbd.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]