

Abstract
Upon activation, microglia and astrocytes produce a number of proinflammatory molecules that participate in the pathophysiology of several neurodegenerative disorders. This study explores the anti-inflammatory property of cinnamon metabolite sodium benzoate (NaB) in microglia and astrocytes. NaB, but not sodium formate, was found to inhibit LPS-induced expression of inducible NO synthase (iNOS), proinflammatory cytokines (TNF-α and IL-1β) and surface markers (CD11b, CD11c, and CD68) in mouse microglia. Similarly, NaB also inhibited fibrillar amyloid β (Aβ)-, prion peptide-, double-stranded RNA (polyinosinic-polycytidylic acid)-, HIV-1 Tat-, 1-methyl-4-phenylpyridinium+-, IL-1β-, and IL-12 p402-induced microglial expression of iNOS. In addition to microglia, NaB also suppressed the expression of iNOS in mouse peritoneal macrophages and primary human astrocytes. Inhibition of NF-κB activation by NaB suggests that NaB exerts its anti-inflammatory effect through the inhibition of NF-κB. Although NaB reduced the level of cholesterol in vivo in mice, reversal of the inhibitory effect of NaB on iNOS expression, and NF-κB activation by hydroxymethylglutaryl-CoA, mevalonate, and farnesyl pyrophosphate, but not cholesterol and ubiquinone, suggests that depletion of intermediates, but not end products, of the mevalonate pathway is involved in the antiinflammatory effect of NaB. Furthermore, we demonstrate that an inhibitor of p21ras farnesyl protein transferase suppressed the expression of iNOS, that activation of p21ras alone was sufficient to induce the expression of iNOS, and that NaB suppressed the activation of p21ras in microglia. These results highlight a novel anti-inflammatory role of NaB via modulation of the mevalonate pathway and p21ras.
Activation of glial cells (microglia and astroglia) has been implicated in the pathogenesis of a variety of neurodegenerative diseases, including Alzheimer's disease (AD), 3 Parkinson's disease, Creutzfeld-Jacob disease, HIV-associated dementia (HAD), stroke, and multiple sclerosis (MS) (1, 2). It has been found that activated microglia and astroglia accumulate at sites of injury or plaques in neurodegenerative CNS (1–7). Although activated microglia scavenge dead cells from the CNS and secrete different neurotrophic factors for neuronal survival, it is believed that severe activation causes various autoimmune responses leading to neuronal death and brain injury (1–7). During activation, microglia and astroglia express various genes related to inflammation, such as proinflammatory cytokines, proinflammatory enzymes, and proinflammatory adhesion molecules (1–9). Therefore, characterization of signaling pathways required for the activation of glial cells is an active area of investigation since compounds capable of antagonizing such signaling steps may have therapeutic effect in neurodegenerative disorders.
Cinnamon contains three major compounds (cinnamaldehyde, cinnamyl acetate and cinnamyl alcohol), which are converted into cinnamic acid by oxidation and hydrolysis, respectively. In the liver, this cinnamic acid is β-oxidized to benzoate (10) that exists as sodium salt (sodium benzoate; NaB) or benzoyl-CoA. It has been reported that minor amount of NaB is also excreted in the urine of humans (11, 12). NaB is of medical importance because it is a component of Ucephan, a Food and Drug Administration (FDA)-approved drug used in the treatment for hepatic metabolic defects associated with hyperammonemia such as urea cycle disorder in children (13, 14). It is also widely used as a preservative in broad range of foods and cosmetic products (15). It is nontoxic and can be administered as a solution in drinking water. It has been reported that 2% solution of NaB in drinking water is safe for lifelong treatment in mice without any noticeable side effects (16). Because Ayurvedic as well as Yunani medicines have been using cinnamon as vital medicines for inflammatory diseases like arthritis for centuries, we were prompted to test the effect of NaB on the expression of proinflammatory molecules in glial cells.
Here we provide the first evidence that NaB attenuates the expression of inducible NO synthase (iNOS) and proinflammatory cytokines in microglia, astrocytes, and macrophages. Although NaB reduced the level of cholesterol in vivo in mice, it was not the cause behind the anti-inflammatory activity of NaB. Alternatively, hydroxymethylglutaryl-CoA (HMG-CoA), mevalonate, and farnesylpyrophosphate reversed NaB-mediated inhibition of iNOS, indicating the involvement of intermediates, but not the end product, of the mevalonate pathway in the anti-inflammatory effect of NaB. Consistently, inhibition of the expression of iNOS and the production of NO by farnesylpyrophosphate transferase inhibitor, suppression of p21ras activation by NaB and induction of iNOS by the activated p21ras alone suggest that NaB exerts its anti-inflammatory effect in glial cells via modulating farnesylation and activation of p21ras. Our findings raise a possibility that NaB, a component of a prescribed drug for human urea cycle disorder and a widely used food preservative, may find further application in neuroinflammatory and neurodegenerative disorders.
Materials and Methods
Reagents
NaB, sodium formate (NaFO), LPS (Escherichia coli), polyinosinic-polycytidylic acid (poly(IC)), and 1-methyl-4-phenylpyridinium (MPP)+ were purchased from Sigma-Aldrich. FBS, HBSS, trypsin, and DMEM/F-12 were from Mediatech. HIV-1 coat protein gp120 (expressed in Chinese hamster ovary cells; strain HIV-1 MN) was obtained from U.S. Biological. Prion peptides and human Aβ peptides 1–42 were obtained from Bachem Bioscience. Recombinant mouse IL-1β and IL-12 p40 homodimer (p402) were obtained from R&D Systems. 125I-Labeled protein A and [α-32P]dCTP (3000 Ci/mmol) were purchased from PerkinElmer Life Sciences. Pravastatin and Abs against human and mouse iNOS were purchased from Calbiochem.
Isolation of primary mouse microglia
Microglia were isolated from mixed glial cultures according to the procedure of Giulian and Baker (17). Briefly, on days 7–9, the mixed glial cultures were washed three times with DMEM/F-12 and shaken at 240 rpm for 2 h at 37°C on a rotary shaker. The floating cells were washed and seeded on to plastic tissue culture flasks and incubated at 37°C for 1 h. The attached cells were seeded onto new plates for further studies. From 90 to 95% of this preparation was found to be positive for Mac-1 surface Ag. Mouse BV-2 microglial cells (gift from Dr. Virginia Bocchini, University of Perugia, Perugia, Italy) were also maintained and induced as indicated above.
Isolation of primary human astroglia
Primary human astroglia were prepared as described by us in many studies (18–20). All of the experimental protocols were reviewed and approved by the Institutional Review Board of the Rush University Medical Center. Briefly, 11- to 17-wk-old fetal brains obtained from the Human Embryology Laboratory (University of Washington, Seattle, WA) were dissociated by trituration and trypsinization. On the ninth day, these mixed glial cultures were placed on a rotary shaker at 240 rpm at 37°C for 2 h to remove loosely attached microglia. Then on the 11th day, flasks were shaken again at 190 rpm at 37°C for 18 h to remove oligodendroglia. The attached cells remaining were primarily astrocytes. These cells were trypsinized and sub-cultured in complete medium at 37°C with 5% CO2 in air to yield more viable and healthy cells (18–20). More than 98% of these cells obtained by this method were positive for glial fibrillary acidic protein (GFAP), a marker for astrocytes.
Isolation of mouse peritoneal macrophages
Resident macrophages were obtained from mouse by peritoneal lavage with sterile RPMI 1640 containing 1% FBS and 100 μg/ml gentamicin as described earlier (21).
Assay for NO synthesis
Synthesis of NO was determined by assay of culture supernatant for nitrite, a stable reaction product of NO with molecular oxygen, using Griess reagent (21–24), except that nitrate in culture supernatants was first reduced to nitrite with nitrate reductase and NADPH (Sigma-Aldrich) for 1 h at 37°C before the assay. Protein was measured by the procedure of Bradford (25).
Immunoblot analysis for iNOS
Immunoblot analysis for iNOS was conducted as described earlier (22, 23, 26). Briefly, cells homogenates were electrophoresed, proteins were transferred onto a nitrocellulose membrane, and the iNOS band was visualized by immunoblotting with Abs against human iNOS.
Northern blot analysis
Analysis was conducted as described earlier (21, 26). Briefly, total RNA was isolated using Ultraspec-II RNA reagent (Biotecx Laboratories) according to the manufacturer's protocol. For Northern blot analyses, 20 μg of total RNA were electrophoresed on 1.2% denaturing formaldehyde-agarose gels, electrotransferred to Hybond-nylon membrane (Amersham Biosciences) and hybridized at 68°C with 32P-labeled cDNA probe. After hybridization, filters were washed two or three times in solution I (2× SSC, 0.05% SDS) for 1 h at room temperature followed by solution II (0.1× SSC, 0.1% SDS) at 50°C for another hour. The membranes were then dried and exposed to x-ray films (Eastman Kodak). The same amount of RNA was hybridized with probe for GAPDH.
Semiquantitative RT-PCR analysis
To remove any contaminating genomic DNA, total RNA was digested with DNase. Semiquantitative RT-PCR was conducted as described earlier (27, 28) using a RT-PCR kit from Clontech. Briefly, 1 μg of total RNA was reverse transcribed using oligo(dT)12–18 as primer and Moloney murine leukemia virus reverse transcriptase (Clontech). The resulting cDNA was appropriately diluted and then cDNA amplified. Amplified products were electrophoresed on a 1.8% agarose gels and visualized by ethidium bromide staining.
iNOS: sense, 5′-CCCTTCCGAAGTTTCTGGCAGCAGC-3′; antisense, 5′-GGCTGTCAGAGCCTCGTGGCTTTGG-3′. IL-1β: sense, 5′-CTCCAT GAGCTTTGTACAAGG-3′; antisense, 5′-TGCTGATGTACCAGTTGGG G-3′. IL-6: sense, 5′-GACAACTTTGGCATTGTGG-3′; antisense, 5′-ATGC AGGGATGATGTTCTG-3′. TLR4: sense, 5′-CAGAAATTCCTGCAGTGG GT-3′; antisense, 5′-GTGAAGGCAGAGGTGAAAGC-3′. TLR3: sense, 5′-ACAACGTAGCTGACTGCAGC-3′; antisense, 5′-GAGTTCTGTCAGGT TCGTGC-3′. B7-1: sense, 5′-AAGTACCTGGGCCGCACGAGC-3′; antisense, 5′-GCCACACACCATCCGGGAATG-3′. B7-2: sense, 5′-GGGCCTGGTCCTTTCAGACCG-3′; antisense, 5′-CCTCTGACAC GTGAGCATCTC-3′. CD11b: sense, 5′-CAGATCAACAATGTGACC GTATGGG-3′; antisense, 5′-CATCATGTCCTTGTACTGCCGCTTG-3′. MHC-II: sense, 5′-AGCCTCTGTGGAGGTGAAGA-3′; antisense, 5′-GGTTGACGAAGAAGCTGGTC-3′. CD11c: sense, 5′-CAGTCT GGCAGATGTGGCTA-3′; antisense, 5′-CACCTGCTCCTGACACTC AA-3′. CD68: sense, 5′-GACCTACATCAGAGCCCGAG-3′; antisense, 5′-AGAGGGGCTGGTAGGTTGAT-3′. GAPDH: sense, 5′-GG TGAAGGTCGGTGTGAACG-3′; antisense, 5′-TTGGCTCCACCCTT CAAGTG-3′.
Real-time PCR analysis
It was performed using the ABI-Prism7700 sequence detection system (Applied Biosystems) as described earlier (27, 28). The mRNA expressions of respective genes were normalized to the level of GAPDH mRNA. Data were processed by the ABI Sequence Detection System 1.6 software and analyzed by ANOVA.
Assay of iNOS promoter-driven reporter activity
Cells plated at 50–60% confluence in six-well plates were cotransfected with 1 μg of phiNOS(7.2)Luc and 50 ng of pRL-TK (a plasmid encoding Renilla luciferase, used as transfection efficiency control; Promega) by LipofectAMINE Plus (Invitrogen) as described in several studies (20, 26). Twenty-four hours after transfection, cells were treated with different stimuli for 12 h. Firefly and R. luciferase activities were obtained by analyzing total cell extract according to standard instructions provided in the Dual Luciferase Kit (Promega) in a TD-20/20 Luminometer (Turner Designs). Relative luciferase activity of cell extracts was typically represented as the ratio of firefly luciferase value to the R. luciferase value × 10−3.
Preparation of nuclear extracts and EMSA
Nuclear extracts were prepared from microglial cells as described previously (7, 19, 21). Nuclear extracts were used for EMSA using 32P-end-labeled double-stranded (NF-κB; 5′-AGTTGAGGGGACTTTCCCAGGC-3′; Promega) oligonucleotides. Double-stranded mutated (NF-κB, 5′-AG TTGAGGCGACTTTCCCAGGC-3′; Santa Cruz Biotechnology) oligonucleotides were used to verify the specificity of NF-κB binding to DNA.
Assay of transcriptional activities of NF-κB
Cells plated at 70–80% confluence in 12-well plates were cotransfected with 0.25 μg of either PBIIX-Luc (an NF-κB-dependent reporter construct) and 12.5 ng of pRL-TK using LipofectAMINE Plus (22, 23). After 24 h of transfection, cells were treated with different stimuli for 6 h. Firefly and R. luciferase activities were obtained as described above.
Assay of cytokines by ELISA
Microglial cells preincubated with NaB for 6 h were stimulated with LPS. After 24 h of stimulation, supernatants were collected to assay TNF-α and IL-1β by high-sensitivity ELISA kits (BD Biosciences).
Immunofluorescence analysis
It was performed as described earlier (19, 28). Briefly, coverslips containing 100–200 cells/mm2 were fixed with 4% paraformaldehyde followed by treatment with cold ethanol and two rinses in PBS. Samples were blocked with 3% BSA in PBS-Tween 20 (PBST) for 30 min and incubated in PBST containing 1% BSA and goat anti-CD11b (1/50), rabbit anti-iNOS (1/200), or goat anti-GFAP (1/50). After three washes in PBST (15 min each), slides were further incubated with Cy2 (Jackson ImmunoResearch Laboratories). For negative controls, a set of culture slides was incubated under similar conditions without the primary Abs. The samples were mounted and observed under a Bio-Rad MRC1024ES confocal laser-scanning microscope.
Assay of cholesterol in serum
Total cholesterol was quantified in serum by using an Amplex Red Cholesterol Assay kit from Invitrogen. Briefly, cholesterol was oxidized by cholesterol oxidase to yield H2O2, which then reacted with 10-acetyl-3,7-dihydroxyphenoxazine (Amplex Red). In the presence of HRP, this Amplex Red-H2O2 complex produced highly fluorescent resorufin, which was detected by fluorometry.
Statistics
Statistical comparisons were made using one-way ANOVA followed by Student's t test.
Results
NaB attenuates the expression of iNOS and proinflammatory cytokines in LPS-stimulated mouse microglia
Activated microglia are known to produce excessive amount of NO having the potential of damaging neurons and oligodendrocytes. Because cinnamon has been used as a natural supplement for normal human health for centuries, we investigated the effect of NaB, a major metabolite of cinnamon, on the expression of iNOS in microglia. LPS is a prototype inducer of various proinflammatory molecules in different cell types including mouse microglia. Therefore, BV-2 microglial cells preincubated with different doses of NaB and sodium formate (NaFO), the negative control for NaB, for 6 h were stimulated with LPS under serum-free condition. Although at lower concentrations (50 and 100 μM), NaB was not effective in inhibiting the production of NO (data not shown), at higher concentrations, NaB markedly suppressed LPS-induced production of NO in microglial cells as evident from estimation of total NO (nitrite and nitrate; Fig. 1A). Significant inhibition of LPS-induced NO production was observed at 500 μM NaB, and maximum inhibition was noted at 1 mM or higher concentration (Fig. 1A). Alternatively, NaFO had no effect on LPS-induced production of NO (Fig. 1A). However, a 1- or 2-h preincubation of microglial cells with NaB was not enough for this molecule to exhibit its inhibitory effect on LPS-induced NO production (data not shown). To understand the mechanism further, we investigated the effect of NaB on the mRNA expression of iNOS in microglial cells. It is clearly evident from RT-PCR in Fig. 1B and real-time PCR in Fig. 1C that NaB, but not NaFO, inhibited LPS-induced mRNA expression of iNOS in BV-2 microglial cells. It is well known that LPS requires TLR4 to transduce its signal and subsequently to induce iNOS. Therefore, there is a possibility that NaB might inhibit the expression of TLR4, and as a result decreased expression of iNOS is observed. To examine this possibility, we investigated the effect of NaB on the expression of TLR4. The RT-PCR results in Fig. 1D show that NaB had no effect on the expression of TLR4 in LPS-treated microglial cells, clearly suggesting that NaB-mediated inhibition of iNOS is not due to any inhibition of expression of TLR4.
FIGURE 1.

Dose-dependent inhibition of LPS-induced NO production by NaB, but not NaFO, in mouse microglia. Mouse BV-2 microglial cells were treated with different concentrations of NaB and NaFO for 6 h followed by stimulation with LPS under serum-free condition. A, After 24 h of stimulation, concentrations of total NO (nitrate plus nitrite) were measured in supernatants. After 5 h of stimulation, the expression of iNOS mRNA was monitored by semiquantitative RT-PCR (B) and real-time PCR (C). D, The expression of TLR4 was monitored by semiquantitative RT-PCR. Mouse peritoneal macrophages isolated by peritoneal lavage were treated with NaB (1 mM) or NaFO (1 mM) for 5 h followed by stimulation with LPS. After 6 h of stimulation, the mRNA expression of iNOS was monitored by semiquantitative RT-PCR (E) and real-time PCR (F). Primary microglia isolated from 7- to 9-day-old mouse pups were treated with different concentrations of NaB and NaFO for 6 h followed by stimulation with LPS under serum-free conditions. After 24 h of stimulation, concentration of nitrite (G) was measured in supernatants. H, Cell viability was assayed by MTT. Results are means ± SD of three different experiments. b, p < 0.05 vs LPS; a, p < 0.001 vs LPS.
Next, to investigate whether NaB suppresses the induction of NO production in primary cells, we used peritoneal macrophages and primary microglia. As evident from semiquantitative RT-PCR in Fig. 1E and quantitative PCR in Fig. 1F, NaB, but not NaFO, markedly suppressed the expression of iNOS mRNA in LPS-stimulated peritoneal macrophages. Similar to BV-2 microglial cells, NaB dose-dependently suppressed LPS-induced production of NO in primary mouse microglia (Fig. 1G). On the other hand, NaFO had no effect on LPS-induced production of nitrite (Fig. 1G). MTT results show that neither NaB nor NaFO was toxic to microglia at any of the concentrations tested (Fig. 1H), suggesting that the inhibitory effect of NaB on microglial expression of iNOS was not due to any change in cell viability.
In addition to producing NO, activated microglia also secrete various proinflammatory cytokines. Therefore, we examined whether NaB was capable of suppressing the expression of proinflammatory cytokines in BV-2 microglial cells. Similar to the inhibition of iNOS, NaB dose-dependently inhibited the production of TNF-α and IL-1β protein (Fig. 2A) and the expression of TNF-α and IL-1β mRNA (Fig. 2, B and C). On the other hand, NaFO had no effect on the expression of these proinflammatory cytokines.
FIGURE 2.

NaB, but not NaFO, inhibits LPS-induced expression of proinflammatory cytokines in mouse BV-2 microglial cells. Cells were treated with different concentrations of NaB and NaFO for 6 h followed by stimulation with 1 μg/ml LPS under serum-free condition. A, After 24 h of stimulation, concentrations of TNF-α and IL-1β were measured in supernatants by ELISA. Results are means ± SD of three different experiments. b, p < 0.05 vs LPS-TNF; a, p < 0.001 vs LPS-TNF; c, p < 0.05 vs LPS-IL-1; d, p < 0.001 vs LPS-IL-1. After 5 h of stimulation, the mRNA expression of TNF-α and IL-1β was monitored by semiquantitative RT-PCR (B) and real-time PCR (C). a, p < 0.001 vs LPS.
NaB inhibits Aβ-, prion peptide (PrP)-, dsRNA (poly(IC))-, HIV-1 gp120-, MPP+-, IL-1β-, and IL-12 p402-induced expression of iNOS in microglial cells
Activated microglia are considered to play an important role in various pathological conditions associated with viral encephalopathy, AD, MS, Parkinson's disease, HAD, Creutzfeldt-Jakob disease, etc. (1). Because NaB inhibited LPS-induced expression of proinflammatory molecules in microglia, we were prompted to investigate whether NaB was also capable of negating the expression of iNOS in microglial cells stimulated with etiological reagents of various neurological disorders. BV-2 microglial cells were challenged with fibrillar Aβ peptides (etiological reagent for AD), fibrillar PrP peptides (etiological reagent for prion diseases), dsRNA in the form of poly(IC) (one of the etiological reagents for viral encephalopathy), HIV-1 gp120 (one of the etiological reagents for HAD), IL-12 p402 (one of the etiological reagent for MS), and MPP+ (Parkinsonian toxin). It is clearly evident from Fig. 3 that Aβ peptides (Fig. 3A), gp120 (Fig. 3B), MPP+ (Fig. 3C), PrP (Fig. 3D), poly(IC) (Fig. 3E), IL-12 p402 (Fig. 3F), and IL-1β (Fig. 3G) induced the expression of iNOS mRNA in microglial cells. However, NaB knocked down Aβ-, gp120-, MPP+-, PrP-, poly(IC)-, p402-, and IL-1b-induced microglial expression of iNOS (Fig. 3). On the other hand, NaFO had no such inhibitory effect (Fig. 3) suggesting the specificity of the effect. We further examined the effect of NaB on the expression of TLR3, the prototype receptor of poly(IC). It is clearly evident from the Fig. 3E (middle) that NaB had no effect on the mRNA level of TLR3, indicating that inhibition of iNOS by NaB in poly(IC)-stimulated cells is not due to any suppression of TLR3.
FIGURE 3.

Effect of NaB and NaFO on Aβ-, HIV-1 gp120-, MPP+-, PrP-, poly(IC)-, IL-1β p402, and IL-12 p402-induced expression of iNOS in mouse BV-2 microglial cells. Cells preincubated with NaB (1 mM) or NaFO (1 mM) for 6 h were stimulated with 1 μM Aβ1–42 (A), 200 pg/ml gp120 (B), 1 μM MPP+ (C), 1 μg/ml PrP (D), 100 μg/ml poly(IC) (E), 20 ng/ml p402 (F), or 10 ng/ml IL-1β (G) under serum-free condition. After 5 h of stimulation, the expression of iNOS mRNA (A–G) and TLR3 mRNA (E) was analyzed in microglial cells by semiquantitative RT-PCR. Results represent three independent experiments.
NaB inhibits the expression of microglial surface markers, MHC class II, and costimulatory molecules in LPS- or MPP+-stimulated mouse microglial cells
During severe activation, microglia not only secrete various neurotoxic molecules but also express different proteins and surface markers. Among different surface markers, CD11b is the most potential one with immense biological significance (29–31). It is reported that in various neuroinflammatory diseases, the increased CD11b expression corresponds to the severity of microglial activation (1). We examined whether NaB could abrogate increased expression of various surface molecules in microglia. As expected, LPS increased the expression of CD11b, CD11c, and CD68 in BV-2 microglial cells (Fig. 4A). We also used MPP+ (Parkinsonian neurotoxin) to stimulate CD11b in microglia. Double-label immunofluorescence analysis of CD11b and iNOS in mouse primary microglia shows that MPP+ stimulation increased the expression of CD11b and iNOS and that NaB attenuated LPS-mediated CD11b and iNOS expression (Fig. 4B). We further examined the surface expression of CD11b and CD68 by FACS. It is evident from Fig. 4 that NaB, but not NaFO, markedly inhibited the surface expression of CD11b (Fig. 4C) and CD68 (Fig. 4D) in LPS-stimulated microglial cells. Taken together, these studies suggest that NaB is capable of suppressing the activation of microglia. Activated microglia also express MHC class II and costimulatory molecules like B7-1 and B7-2, which are critical for Ag presentation. As evident from Fig. 4A, LPS increased the expression of class-II MHC and the costimulatory molecules B7-1 and B7-2 (Fig. 4A). However, NaB, but not NaFO, markedly suppressed the expression of these activation markers and costimulatory molecules in microglial cells (Fig. 4A).
FIGURE 4.

NaB, but not NaFO, inhibits the expression of surface markers, MHC class II, and costimulatory molecules in activated microglia. BV-2 microglial cells were treated with NaB and NaFO for 6 h followed by stimulation with 1 μg/ml LPS under serum-free conditions. A, After 24 h of stimulation, the expressions of CD11b, CD11c, CD68, B7-2, B7-1, and MHC class II were monitored by semiquantitative RT-PCR. B, Primary mouse microglia were treated with NaB followed by stimulation with 1 μM MPP+. After 24 h of stimulation, levels of CD11b and iNOS were monitored by double-label immunofluorescence. Cells were incubated with appropriately diluted FITC-conjugated CD11b (C) or CD68 (D) followed by FACS. The percentage of cells in the indicated quadrant has been mentioned. Results are means ± SD of three different trials.
NaB inhibits the activation of NF-κB in mouse BV-2 microglial cells
LPS, proinflammatory cytokines (TNF-α and IL-1β) and other stimuli (HIV-1 gp120, IL-12 p402, Aβ, PrP, etc.) are known to induce iNOS expression via activation of NF-κB (19–24, 26, 27). Because NaB attenuated the expression of iNOS in mouse microglia, we examined the effect of NaB on the activation of NF-κB. Activation of NF-κB was monitored by both DNA binding and transcriptional activity of NF-κB. As expected, treatment of BV-2 microglial cells with LPS resulted in the induction of DNA-binding activity of NF-κB (Fig. 5A). However, NaB inhibited LPS-induced DNA-binding activity of NF-κB in microglial cells (Fig. 5A). We then tested the effect of NaB on LPS-induced transcriptional activity of NF-κB. Consistent to the effect of NaB on the DNA binding activity of NF-κB, NaB also suppressed NF-κB-dependent transcription of luciferase in a dose-dependent manner in LPS-stimulated cells (Fig. 5B). NaFO had no effect on the LPS-induced transcriptional activity of NF-κB in microglial cells (Fig. 5B). We next examined the effect of NaB on other proinflammatory stimuli-induced transcriptional activity of NF-κB. The wild-type IκB kinase-γ (IKK-γ) NEMO binding domain peptide (wtNBD), a specific inhibitor of induced NF-κB activation, was used as a positive control. As expected, NaB markedly suppressed NF-κB-dependent transcription of luciferase in microglial cells stimulated by Aβ, IL-1β, gp120, PrP, and IL-12 p402 (Fig. 5, C–G) These results suggest that NaB attenuates the expression of iNOS by suppressing the activation of NF-κB.
FIGURE 5.

NaB attenuates activation of NF-κB in mouse BV-2 microglial cells. A, Cells were treated with different concentrations of NaB for 6 h followed by stimulation with 1 μg/ml LPS under serum-free conditions. After 1 h of stimulation, the DNA-binding activity of NF-κB was monitored. B, Cells plated in 12-well plates were cotransfected with 0.25 μg of PBIIX-Luc (an NF-κB-dependent reporter construct) and 12.5 ng of pRL-TK. Twenty-four hours after transfection, cells received different concentrations of NaB and NaFO. After 6 h of incubation, cells were stimulated with LPS for 6 h under serum-free condition. Firefly (ff-Luc) and Renilla luciferase (r-Luc) activities were obtained by analyzing the total cell extract. C–H, Twenty-four hours after transfection, cells received NaB (1 mM); 1 h before stimulation, cells were treated with wtNBD (10 μM). After 6 h of incubation with NaB, cell were stimulated with LPS (1 μg/ml; C), IL-1β (10 ng/ml; D), PrP (1 μg/ml; E), Aβ (1 μM; F), gp120 (200 pg/ml; G), or p402 (20 ng/ml; H). Firefly and Renilla luciferase activities were obtained by analyzing the total cell extract. Results are means ± SD of three different experiments. a, p < 0.001 vs control; b, p < 0.05 vs LPS; c, p < 0.001 vs LPS.
NaB suppresses the expression of iNOS and GFAP in primary human astrocytes
Next we examined whether NaB could suppress the expression of iNOS in human brain cells. Astroglia are the major glial cells in the CNS, and astroglial activation also plays a role in various neurodegenerative disorders. Therefore, we investigated the effect of NaB on the expression of iNOS in primary human astroglia. Earlier, we found that IL-1β is the only cytokine that induces iNOS in primary astroglia (20). Consistently, IL-1β induced the production of nitrite (Fig. 6A) and the expression of iNOS protein (Fig. 6B) in primary astroglia isolated from human fetal brains. NaB markedly inhibited IL-1β-induced production of NO (Fig. 6A) and the expression of iNOS protein (Fig. 6B) in human fetal astroglia.
FIGURE 6.

NaB inhibits IL-1β-induced expression of iNOS and the activation of human iNOS promoter in primary human astrocytes. Cells pretreated with different concentrations of NaB for 6 h were stimulated with 20 ng/ml IL-1β. After 48 h of stimulation, the level of nitrite was measured by Griess reagent (A) and the level of iNOS protein was monitored by Western blot (B). Actin was run as control. a, p < 0.001 vs IL-1β. C, Cells plated at 70–80% confluence in 12-well plates were cotransfected with 0.25 μg of phiNOS(7.2)Luc and 12.5 ng of pRL-TK using the Lipofectamine-Plus (Invitrogen). Twenty-four hours after transfection, cells received NaB and NaFO. After 6 h of incubation, cells were stimulated with IL-1β (20 ng/ml) for 12 h under serum-free condition. Firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were obtained by analyzing the total cell extract. Data are means ± SD of three different experiments. a, p < 0.001 vs control; b, p < 0.001 vs IL-1β. Primary human astroglia were treated with NaB (1 mM) and NaFO (1 mM) for 6 h followed by stimulation with IL-1β (20 ng/ml). After 24 h of stimulation, mRNA expression of GFAP was examined by semiquantitative RT-PCR (E), and protein expression of GFAP and iNOS was monitored by double-label immunofluorescence (D). Cells pretreated with appropriate concentrations of NaB or NaFO for 6 h or wtNBD (10 μM) alone or along with NaB for 1 h were stimulated with 20 ng/ml IL-1β under serum-free conditions. After 48 h of stimulation, the level of iNOS and GFAP proteins was monitored by Western blot (F). β-Actin was run as control Results represent three independent experiments.
To understand the effect of NaB on the transcription of the iNOS gene, primary human astrocytes were transfected with phiNOS(7.2)Luc, a construct containing the human iNOS promoter fused to the luciferase gene, and activation of this promoter was measured after stimulating the cells with IL-1β in the presence or absence of NaB. As we found earlier (20, 23), IL-1β induced iNOS promoter-driven luciferase activity by ∼4-fold (Fig. 6C). Consistent with the effect of NaB on the expression of iNOS, NaB itself had no effect on iNOS promoter-driven luciferase activity but it significantly inhibited iNOS promoter-driven luciferase activity in IL-1β-stimulated cells (Fig. 6C), suggesting that NaB inhibits IL-1β-induced production of NO and the expression of iNOS by inhibiting the activation of iNOS promoter.
Increased expression of glial fibrillary acidic protein (GFAP) represents astroglial activation and gliosis during neurodegeneration. Because NaB decreased the expression of iNOS in human astroglia, we investigated whether this drug was capable of inhibiting the increased expression of GFAP in primary human astroglia. As expected, IL-1β markedly increased the mRNA expression of GFAP in astroglia (Fig. 6E). However, NaB, but not NaFO, suppressed IL-1β-induced astroglial mRNA expression of GFAP (Fig. 6E). Immunofluorescence analysis in Fig. 6D also indicates that IL-1β stimulation increased the level of GFAP and iNOS compared with control and that NaB markedly attenuated IL-1β-mediated up-regulation of GFAP and iNOS in human astrocytes. Western blot analysis in Fig. 6F further substantiates our findings and also indicates that NaB has no additive effect when used in combination with wtNBD, suggesting that NaB-mediated inhibition of proinflammatory markers is solely due to the inhibition of NF-κB.
Intermediates of the mevalonate pathway reverses the inhibitory effect of NaB on the expression of iNOS and the activation of NF-κB in mouse BV-2 microglial cells
The requirement of at least 6 h of preincubation of cells with NaB to see its anti-inflammatory effect suggests that metabolite(s) sensitive to NaB may be involved in the process. Earlier, Pahan et al. (32) have demonstrated that intermediates of the mevalonate pathway play a role in the expression of iNOS and proinflammatory cytokines in glial cells. The end product of the mevalonate pathway is cholesterol; therefore, we investigated whether NaB had any effect on the level of cholesterol in vivo in mice. After 7 days of treatment, NaB reduced the level of cholesterol in serum of mice by ∼28%, and this reduction was comparable (∼30%) with that by the so-called cholesterol-lowering drug pravastatin (Fig. 7). Alternatively, NaFO had no effect on serum level of cholesterol (Fig. 7), indicating the specificity of the effect. These results are important because they suggest that NaB may be used to lower cholesterol in patients with hypercholesterolemia.
FIGURE 7.

Effect of NaB on serum level of cholesterol in male C57BL/6 mice. Mice (6–8 wk old) were treated with NaB (100 mg/kg body weight), NaFO (100 mg/kg body weight), and pravastatin (1 mg/kg body weight) separately via gavage for 7 days followed by quantification of cholesterol in serum using a simple fluorometric method. Results represent means ± SD of five mice per group (n = 5). b, p < 0.05 vs control.
Next we examined the role of different members of the mevalonate pathway in the anti-inflammatory effect of NaB. HMG-CoA, mevalonate, and farnesyl pyrophosphate abrogated the inhibitory effect of NaB on the expression of iNOS mRNA (Fig. 8A), the production of NO (Fig. 8B), and the activation of NF-κB (Fig. 8C) in microglial cells. Alternatively, cholesterol (the end product of the mevalonate pathway) and coenzyme Q (an unrelated lipid molecule) had no effect on NaB-mediated inhibition of iNOS mRNA (Fig. 8A), NO production (Fig. 8B), and the activation of NF-κB (Fig. 8C). These results suggest that depletion of intermediary products rather than end products of the mevalonate pathway is responsible for the observed anti-inflammatory effect of NaB.
FIGURE 8.

Intermediates of the mevalonate pathway negate the inhibitory effect of NaB on the expression of iNOS and the activation of NF-κB in mouse BV-2 microglial cells. Cells were treated with NaB in the presence or absence of different concentrations of HMG-CoA, mevalonate, FPP, cholesterol, and ubiquinone for 6 h followed by stimulation with LPS. A, After 5 h of stimulation, the expression of iNOS mRNA was monitored by RT-PCR. B, After 24 h of stimulation, the level of nitrite was measured in supernatants using Griess reagent. Results are means ± SD of three independent experiments. a, p < 0.001 vs LPS; b, p < 0.001 vs LPS+NaB. C, Cells were cotransfected with 0.25 μg of PBIIX-Luc and 12.5 ng of pRL-TK. Twenty-four hours after transfection, cells were incubated with NaB in the presence or absence of HMG-CoA, mevalonate, FPP, cholesterol, and ubiquinone. After 6 h of incubation, cells were stimulated with LPS for 6 h followed by assay of firefly (ff-Luc) and Renilla (r-Luc) luciferase activities. Results are means ± SD of three different experiments. a, p < 0.001 vs LPS; b, p < 0.001 vs LPS plus NaB.
An inhibitor of p21ras farnesyl protein transferase (FPT inhibitor II) suppresses LPS-induced expression of iNOS mRNA and activation of NF-κB in mouse BV-2 microglial cells
Inhibition of LPS-induced expression of iNOS and activation of NF-κB by NaB and its reversal by farnesyl pyrophosphate (FPP) and other upstream members of the mevalonate pathway (HMG-CoA and mevalonate), but not by the end product of the same pathway, suggest a possible involvement of the farnesylation reaction in the activation of NF-κB and the expression of iNOS. Because farnesylation is a necessary step for the activation of p21ras, we examined the effect of FPT inhibitor II on LPS-induced expression of iNOS and activation of NF-κB in microglial cells. FPT inhibitor II selectively inhibits p21ras farnesyl protein transferase with the IC50 of 75 nM. In whole cells, however, a higher concentration (25–250 μM) of FPT inhibitor II is required to inhibit farnesylation of p21ras by 90% (33). Therefore, BV-2 cells were pretreated with 100, 200, and 300 μM FPT inhibitor II for 2 h followed by stimulation with LPS. It is clearly evident from Fig. 9 that FPT inhibitor II dose-dependently suppressed LPS-induced expression of iNOS mRNA (Fig. 9A) and activation of NF-κB (Fig. 9B) in microglial cells. These results suggest that p21ras farnesylation plays an important role in the activation of NF-κB and the expression of iNOS in LPS-stimulated microglia.
FIGURE 9.

Effect of FPT inhibitor II (FPTI II) on the expression of iNOS and the activation of NF-κB in mouse BV-2 microglial cells. A, Cells treated with different concentrations of FPT inhibitor II for 2 h were stimulated by LPS. After 5 h of stimulation, the expression of iNOS mRNA was monitored by RT-PCR. B, Cells were cotransfected with 0.25 μg of PBIIX-Luc and 12.5 ng of pRL-TK. Twenty-four hours after transfection, cells were incubated with different concentrations of FPT inhibitor II for 2 h followed by stimulation with LPS. After 6 h of stimulation, activities of firefly (ff-Luc) and Renilla (r-Luc) luciferase were monitored. Results are mean ± S.D. of three different experiments. a, p < 0.001 vs LPS; b, p < 0.05 vs LPS. C–E, Cells treated with appropriate concentrations of FPT inhibitor II (200 μM) for 2 h (C), FPP (200 μM; D), or NaB (1 mM; E) alone for 6 h or along with wtNBD (10 μM; 1 h before stimulation) were stimulated by LPS. After 5 h of stimulation, the expression of iNOS mRNA was monitored by RT-PCR. F, Cells treated with different concentrations of NaB for 6 h were stimulated by IFN-γ (12.5 U/ml). After 5 h of stimulation, the expression of iNOS mRNA was monitored by RT-PCR. The results represent three independent experiments.
To exclude any possibility that FPTI-II inhibits iNOS independent of NF-κB, we examined the effect of wtNBD and FPT inhibitor II, alone or together, on the mRNA expression of iNOS. It is evident from Fig. 9C that FPT inhibitor II did not show any additive inhibitory effect on the expression of iNOS in wtNBD-treated cells, suggesting that inhibition of iNOS by FPT inhibitor II is purely due to the suppression of NF-κB activation. Furthermore, inability of FPP to reverse the inhibitory effect of wtNBD on iNOS as evident from Fig. 9D clearly indicates that farnesylation of p21ras is an upstream event of NF-κB activation and thus whether NF-κB is directly inhibited, inhibition of iNOS is not reversed by FPP. The semiquantitative RT-PCR studies in Fig. 9E again shows that NaB has no additive inhibitory effect on iNOS over wtNBD, further substantiating the fact that NaB-mediated inhibition of iNOS in microglial cells solely involves suppression of NF-κB activation. To further confirm, we investigated the effect of NaB on the expression of IFN-γ-induced expression of iNOS in BV2 microglial cells. IFN-γ induces iNOS via activation of STATs, which is independent of NF-κB. It is evident from Fig. 9F that NaB had no significant effect on IFN-γ-induced expression of iNOS mRNA in microglial cells even at a dose as high as 2 mM. These results suggest that NaB is only able to inhibit iNOS if induced by activation of NF-κB and that the observed inhibition of iNOS by NaB is purely due to suppression of NF-κB through the depletion of mevalonate intermediates.
NaB suppresses the activation of p21ras in mouse BV-2 microglial cells
Because p21ras farnesylation is involved in the activation of NF-κB and the expression of iNOS and NaB-mediated inhibition of these events were reversed by FPP, we examined the whether NaB suppressed the activation of p21ras. Earlier, we have seen activation of p21ras in human astroglioma cells within 2–4 min of LPS stimulation (34). Therefore, at different times (2, 3, and 5 min) of stimulation by LPS, microglial cells were analyzed for the activation of p21ras. Although we did not see p21ras activation at 2 min of LPS stimulation, marked activation was observed at 3 and 5 min of stimulation (Fig. 10A). Therefore, we examined the effect of NaB on the activation of p21ras at 3 min of LPS stimulation. It is clearly evident from Fig. 10B that NaB, but not NaFO, markedly inhibited LPS-induced activation of p21ras in microglial cells. These results suggest that NaB attenuates the expression of proinflammatory molecules in glial cells probably by suppressing the activation of p21ras.
FIGURE 10.

Effect of NaB on the activation of p21ras in mouse BV-2 microglial cells. A, Cells were stimulated with LPS under serum-free conditions. At different time points, activation of p21ras was monitored. B, In another set, cells pretreated with 1 mM NaB/NaFO for 6 h were challenged with LPS under serum-free conditions followed by monitoring activation of p21ras at 3 min of stimulation. Results represent two independent experiments.
Activation of p21ras alone is sufficient to induce the expression of iNOS and the activation of NF-κB in mice BV-2 microglial cells
Our results that FPT inhibitor II inhibited microglial expression of iNOS and that NaB suppressed microglial activation of p21ras and the expression of iNOS prompted us to examine whether activation of p21ras alone was sufficient for the expression of iNOS in microglial cells. Activation of p21ras in BV-2 glial cells was achieved by the expression of RasV12, a constitutively active mutant of p21ras. It is clear from Fig. 11A that the expression of RasV12 alone markedly induced the production of NO in microglial cells whereas the expression of empty vector was unable to induce NO production. Inhibition of RasV12-induced production of NO by arginase, an enzyme that degrades the substrate (l-arginine) of NOS, and N-methyl-L-arginine acetate, a competitive inhibitor of NOS activity, suggests that the induction of NO production in RasV12-transfected cells is dependent on NOS-mediated arginine metabolism (data not shown). To understand the mechanism of induction of NO production, we analyzed the status of iNOS protein and mRNA in RasV12-transfected cells. Western blot analysis with Abs against murine macrophage iNOS and Northern blot analysis for iNOS mRNA clearly showed that RasV12 induced the expression of iNOS protein (Fig. 11B) and mRNA (Fig. 11C), suggesting that signal(s) provided by the activation of p21ras alone is sufficient to induce the expression of iNOS.
FIGURE 11.
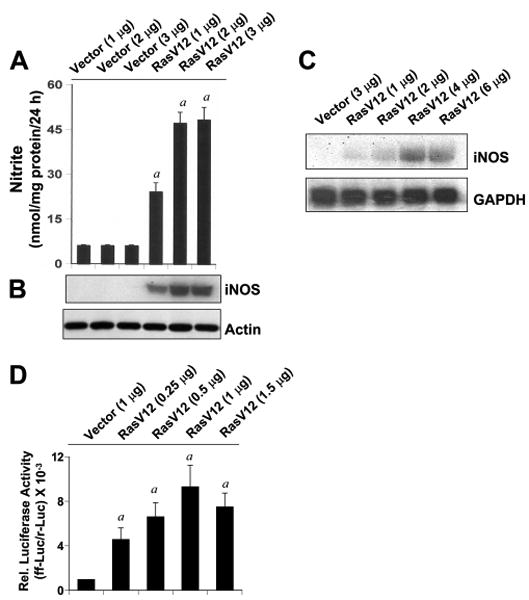
Activation of p21ras alone induces the expression of iNOS and the activation of NF-κB in mouse BV-2 microglial cells. Cells were transfected with different concentrations of an empty vector and RasV12 (constitutively active mutant of Ras). Twenty-four hours after transfection, cells were incubated in serum-free medium for 24 h followed by assay of nitrite concentration in supernatant (A) and monitoring the level of iNOS protein in cells by Western blot (B). Actin was run as control. a, p < 0.001 vs IL-1β. C, Cells plated at 70–80% confluence in 12-well plates were cotransfected with 0.25 μg of phiNOS(7.2)Luc and 12.5 ng of pRL-TK using the Lipofectamine-Plus (Invitrogen). Twenty-four hours after transfection, cells received NaB and NaFO. After 6 h of incubation, cells were stimulated with IL-1β (20 ng/ml) for 12 h under serum-free condition. Firefly (ff-Luc) and Renilla (r-Luc) luciferase activities were obtained by analyzing the total cell extract as described in Materials and Methods. Data are means ± SD of three different experiments. a, p < 0.001 vs control; b, p < 0.001 vs IL-1β.
Because the activation of NF-κB is important for the induction of iNOS (19–23, 26–28), to understand the basis of iNOS expression by RasV12, we also investigated whether Ras alone was sufficient for the activation of NF-κB. Activation of NF-κB was monitored by transcriptional activity of NF-κB using the expression of luciferase from a reporter construct, PBIIX-Luc. Cells were cotransfected with PBIIX-Luc and RasV12 followed by incubation in serum-free medium. As evident from Fig. 11D, RasV12 markedly induced NF-κB-dependent transcription of luciferase, whereas the empty vector was unable to induce the transcriptional activity of NF-κB.
Discussion
Although microglial activation has an important repairing function through scavenging of unwanted bodies in the CNS and activation of astrocytes may have important beneficial effects in the recovery of injured CNS by actively monitoring and controlling the extracellular water, pH, and ion homeostasis, once microglia and astroglia are activated in the neurodegenerating microenvironment, activation always goes beyond control, and eventually detrimental effects of glial activation override its beneficial effects. Activated glia produce NO, a number of proinflammatory cytokines, reactive oxygen species, etc., in excessive amounts for a prolonged time period that ultimately damage neurons and oligodendrocytes. Therefore, understanding mechanisms that regulate microglial and astroglial activation is an important area of investigation.
Cinnamon, the brown bark of the cinnamon tree, is a commonly used spice and flavoring material for desert, candies, chocolate, etc. It has a long history as a medicine as well. Medieval physicians used cinnamon in medicines to treat a variety of disorders including arthritis, coughing, hoarseness, and sore throats. In addition to containing manganese, dietary fiber, iron, and calcium, cinnamon contains three major compounds, cinnamaldehyde, cinnamyl acetate, and cinnamyl alcohol. After intake, these three active compounds are converted into cinnamic acid by oxidation and hydrolysis, respectively. Then cinnamic acid is β-oxidized to benzoate in the liver. This benzoate exists as sodium salt (NaB) or benzoyl-CoA. This NaB is a widely used food preservative due to its antimicrobial properties. Earlier, we have demonstrated that NaB modifies T cells at multiple steps and protects experimental allergic encephalomyelitis, an animal model of MS (35). Recently, one study by Cao et al. (36) demonstrates that cinnamon polyphenol extract increases the expression of tristetraprolin, an antiinflammatory molecule, more rapidly than those of proinflammatory cytokines in macrophages. Several lines of evidence presented in this study clearly support the conclusion that the cinnamon metabolite NaB attenuates the activation of mouse microglia and human astroglia. Our conclusion is based on the following observations. First, LPS, a prototype inducer of inflammation in many cell types including microglia, induced the expression of iNOS and proinflammatory cytokines and up-regulated the expression of various surface markers in mouse microglia. However, NaB attenuated LPS-induced expression of proinflammatory molecules without altering cell survival suggesting that this attenuation is not due to any cell death. This inhibition was also specific as NaFO, a compound structurally similar to NaB but without having the benzene moiety, had no effect. Second, we extended the study beyond LPS and examined whether NaB was capable of suppressing microglial expression of iNOS induced by other neurotoxins and etiological reagents of various neurodegenerative disorders. It is important that NaB, but not NaFO, attenuated microglial expression of iNOS mRNA induced by various neurotoxins such as Aβ (related to AD), poly(IC) (related to viral neuropathy), HIV-1 gp120 (related to HAD), MPP+ (related to Parkinson's disease), PrP (related to prion disorders), IL-1β (related to neuroinflammation), and IL-12 p402 (related to MS). Third, IL-1β is critical for inducing iNOS and activating primary human astrocytes (20). NaB also suppressed the production of NO, the expression of iNOS, the activation human iNOS promoter, and the up-regulation of GFAP in human astrocytes. Because these proinflammatory molecules have been implicated in the pathogenesis of demyelinating and neurodegenerative diseases, our results provide a potentially important mechanism whereby cinnamon metabolite NaB may ameliorate neural injury.
The signaling events required for the transcription of iNOS and proinflammatory cytokines are becoming clear. Although many transcription factors such as NF-κB, C/EBPβ, AP-1, STAT, IRF-1, etc., play a role in the expression of various proinflammatory molecules, activation of NF-κB seems essential for the transcription of most of the proinflammatory molecules (19–23, 26–28, 37, 38). Therefore, for a drug to exhibit an anti-inflammatory effect, it is almost mandatory to attenuate the activation of NF-κB. Although we did not see complete abrogation of NF-κB activation, NaB significantly decreased the activation of NF-κB in microglia. However, it was unknown by which mechanisms NaB suppressed the activation of NF-κB. p21ras, a membrane-associated small guanine nucleotide-binding protein, plays a central role in transmitting extracellular signals within the cell and in controlling cellular proliferation and differentiation (39, 40). Here we present evidence that NaB suppressed the activation of p21ras and thereby inhibited the activation of NF-κB and the expression iNOS in microglia. Our conclusion is based on the following observations. First, NaB reduced the level of cholesterol in vivo in mice at a level comparable with pravastatin suggesting that this drug may be used to lower cholesterol in patients with hypercholesterolemia. However, HMG-CoA, mevalonate and farnesyl pyrophosphate, but not cholesterol, reversed the inhibitory effect of NaB on the expression of iNOS and the activation of NF-κB, suggesting that depletion of intermediates, but not end products, of the mevalonate pathway is involved in the anti-inflammatory effect of NaB. Second, FPT inhibitor II, capable of inhibiting farnesylation of p21ras, inhibited the activation of NF-κB and the expression of iNOS. Third, NaB, but not NaFO, inhibited the activation of p21ras in LPS-stimulated microglial cells. Fourth, activation of p21ras alone was sufficient for the activation of NF-κB and the expression of iNOS in microglial cells underlining the importance p21ras activation in the activation of microglia.
The Ras proto-oncogene proteins, a family of GTP-binding proteins, function by binding to the cytoplasmic surface of the plasma membrane. This membrane localization of p21 ras involves prenylation of cysteine in a CAAX motif present at the C terminus, proteolytic removal of AAX tripeptide, and then carboxymethylation of the C-terminal cysteine (39). The activation of p21ras by receptor tyrosine kinase occurs through conversion of the GDP-bound inactive form to the GTP-bound active form by Sos and Grb2 and then transduction of signal to downstream effector molecules (40). The GTP-bound form is converted to the inactive form by the intrinsic GTPase activity, which is accelerated by GTPase-activating proteins (41). NaB preferentially attenuates farnesylation of p21ras and thereby inhibits the signal transmission to the downstream signaling molecules (42, 43). One such downstream candidate is Raf-1 (serine-threonine kinase). The p21ras interacts directly with Raf-1 and is believed to function by positioning Raf-1 at the plasma membrane in the vicinity of its activator, and tyrosine phosphorylation of Raf-1 seems to be essential for p21ras-induced activation of Raf-1 (42, 43). Raf-1, in turn, phosphorylates and activates MEKs and ERKs (members of the MAPK cascade). Therefore, the observed inhibition of NF-κB activation and induction of iNOS by NaB may be due to decrease and/or lack of signal transmission from receptor tyrosine kinase to Raf/MAPK cascade via p21ras.
There are several advantages of NaB over other proposed antineurodegenerative therapies. First, NaB is fairly nontoxic. Cinnamon has been widely used as flavoring material and spice throughout the world for centuries. Cinnamon is metabolized to NaB. NaB is excreted through the urine, if in excess. NaB is an FDA-approved drug against urea cycle disorders in children. Second, NaB can be taken orally, the least painful route. Recently, we have demonstrated that NaB treatment of mice with relapsing-remitting experimental allergic encephalomyelitis, an animal model of MS, via drinking water suppressed the disease process of experimental allergic encephalomyelitis (35). Third, NaB is very economical compared with other existing antineurodegenerative therapies. Fourth, although here we have not tested the premise, NaB as a lipophilic molecule is most likely able to diffuse through the blood-brain barrier. For example, glycine toxicity is a problem in urea cycle disorders. After treatment of patients with urea cycle disorders, NaB combines with glycine to produce hippurate, a compound that is readily excreted in the urine. Simultaneous serum and CSF sampling in those patients showed comparable levels of NaB and hippurate in the CSF (13–15), suggesting that NaB is capable of crossing the blood-brain barrier.
In summary, we have demonstrated that NaB (a metabolite of cinnamon, commonly used food additive and a FDA-approved drug for urea cycle disorders) inhibited glial activation of NF-κB and expression of iNOS and cytokines by modulating the mevalonate pathway and Ras activation. Because NaB suppressed the mevalonate pathway, it was also able to lower cholesterol in vivo in mice at a level comparable with that of pravastatin, a cholesterol-lowering drug. These results highlight undiscovered properties of NaB and indicate that this drug may be used for therapeutic intervention in neurodegenerative disorders as primary or adjunct therapy.
Acknowledgments
We thank Xiaojuan Liu for her help in transfection experiments.
Footnotes
This study was supported by grants from the National Institutes of Health (NS39940), Alzheimer's Association (IIRG-07-58684), and Michael J. Fox Foundation for Parkinson's Research.
Abbreviations used in this paper: AD, Alzheimer's disease; HAD, HIV-associated dementia; MS, multiple sclerosis; NaB, sodium benzoate; FDA, Food and Drug Administration; iNOS, inducible NO synthase; HMG, CoA, hydroxymethylglutaryl-CoA; NaFO, sodium formate; poly(IC), polyinosinic-polycytidylic acid; MPP, 1-methyl-4-phenylpyridinium; p402, p40 homodimer; GFAP, glial fibrillary acidic protein; PBST, PBS-Tween 20; Aβ, amyloid β; PrP, prion peptide; wtNBD, wild-type IKKγ NEMO-binding domain peptide; FPT inhibitor II, inhibitor of p21ras farnesyl protein transferase; FPP, farnesyl pyrophosphate; IKK-γ, IκB kinase-γ.
Disclosures: The authors have no financial conflict of interest.
References
- 1.Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 2.Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC, Wegiel J. Contribution of glial cells to the development of amyloid plaques in Alzheimer's disease. Neurobiol Aging. 2004;25:663–674. doi: 10.1016/j.neurobiolaging.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Barcia C, Sanchez Bahillo A, Fernandez-Villalba E, Bautista V, Poza Y Poza M, Fernandez-Barreiro A, Hirsch EC, Herrero MT. Evidence of active microglia in substantia nigra pars compacta of parkinsonian monkeys 1 year after MPTP exposure. Glia. 2004;46:402–409. doi: 10.1002/glia.20015. [DOI] [PubMed] [Google Scholar]
- 4.Carson MJ. Microglia as liaisons between the immune and central nervous systems: functional implications for multiple sclerosis. Glia. 2002;40:218–231. doi: 10.1002/glia.10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 6.Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Mosely RL, Ghosh S, Gendelman HE, Pahan K. Selective inhibition of NF-κB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson's disease. Proc Natl Acad Sci USA. 2007;104:18754–18759. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bo L, Dawson TM, Wesselingh S, Mork S, Choi S, Kong PA, Hanley D, Trapp BD. Induction of nitric oxide synthase in demyelinating regions of multiple sclerosis brains. Ann Neurol. 1994;36:778–786. doi: 10.1002/ana.410360515. [DOI] [PubMed] [Google Scholar]
- 9.Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J Immunol. 1993;151:2132–2141. [PubMed] [Google Scholar]
- 10.Abd El-Mawla AM, Schmidt W, Beerhues L. Cinnamic acid is a precursor of benzoic acids in cell cultures of Hypericum androsaemum L. but not in cell cultures of Centaurium erythraea RAFN. Planta. 2001;212:288–293. doi: 10.1007/s004250000394. [DOI] [PubMed] [Google Scholar]
- 11.Bridges JW, French MR, Smith RL, Williams RT. The fate of benzoic acid in various species. Biochem J. 1970;118:47–51. doi: 10.1042/bj1180047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kubota K, Ishizaki T. Dose-dependent pharmacokinetics of benzoic acid following oral administration of sodium benzoate to humans. Eur J Clin Pharmacol. 1991;41:363–368. doi: 10.1007/BF00314969. [DOI] [PubMed] [Google Scholar]
- 13.Scaglia F, Carter S, O'Brien WE, Lee B. Effect of alternative pathway therapy on branched chain amino acid metabolism in urea cycle disorder patients. Mol Genet Metab. 2004;81(Suppl. 1):S79–S85. doi: 10.1016/j.ymgme.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 14.Leonard JV, Morris AA. Urea cycle disorders. Semin Neonatol. 2002;7:27–35. doi: 10.1053/siny.2001.0085. [DOI] [PubMed] [Google Scholar]
- 15.Nair B. Final report on the safety assessment of benzyl alcohol, benzoic acid, and sodium benzoate. Int J Toxicol. 2001;20(Suppl. 3):23–50. doi: 10.1080/10915810152630729. [DOI] [PubMed] [Google Scholar]
- 16.Toth B. Lack of tumorigenicity of sodium benzoate in mice. Fundam Appl Toxicol. 1984;4(3 Pt 1):494–496. doi: 10.1016/0272-0590(84)90208-2. [DOI] [PubMed] [Google Scholar]
- 17.Giulian D, Baker TJ. Characterization of amoebid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jana M, Jana A, Pal U, Pahan K. A simplified method for isolating highly purified neurons, oligodendrocytes, astrocytes, and microglia from the same human fetal brain tissue. Neurochem Res. 2007;32:2015–2022. doi: 10.1007/s11064-007-9340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saha RN, Jana M, Pahan K. MAPK p38 regulates transcriptional activity of NF-κB in primary human astrocytes via acetylation of p65. J Immunol. 2007;179:7101–7109. doi: 10.4049/jimmunol.179.10.7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jana M, Anderson JA, Saha RN, Liu X, Pahan K. Regulation of inducible nitric oxide synthase in proinflammatory cytokine-stimulated human primary astrocytes. Free Radic Biol Med. 2005;38:655–664. doi: 10.1016/j.freeradbiomed.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 21.Pahan K, Sheikh FG, Liu X, Hilger S, McKinney M, Petro TM. Induction of nitric-oxide synthase and activation of NF-κB by interleukin-12 p40 in microglial cells. J Biol Chem. 2001;276:7899–7905. doi: 10.1074/jbc.M008262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jana M, Liu X, Koka S, Ghosh S, Petro TM, Pahan K. Ligation of CD40 stimulates the induction of nitric-oxide synthase in microglial cells. J Biol Chem. 2001;276:44527–44533. doi: 10.1074/jbc.M106771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K. Human immunodeficiency virus type 1 (HIV-1) Tat induces nitric-oxide synthase in human astroglia. J Biol Chem. 2002;277:39312–39319. doi: 10.1074/jbc.M205107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dasgupta S, Jana M, Liu X, Pahan K. Myelin basic protein-primed T cells induce nitric oxide synthase in microglial cells: implications for multiple sclerosis. J Biol Chem. 2002;277:39327–39333. doi: 10.1074/jbc.M111841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 26.Pahan K, Jana M, Liu X, Taylor BS, Wood C, Fischer SM. Gemfibrozil, a lipid-lowering drug, inhibits the induction of nitric-oxide synthase in human astrocytes. J Biol Chem. 2002;277:45984–45991. doi: 10.1074/jbc.M200250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jana M, Liu X, Pahan K. Involvement of phosphatidylinositol-3 kinase-mediated up-regulation of IκBα in anti-inflammatory effect of gemfibrozil in microglia. J Immunol. 2007;179:4142–4152. doi: 10.4049/jimmunol.179.6.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy A, Liu X, Pahan K. Myelin basic protein-primed T cells induce neurotrophins in glial cells via αVβ3 integrin. J Biol Chem. 2007;282:32222–32232. doi: 10.1074/jbc.M702899200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Clin Microbiol Rev. 2004;17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ling EA, Wong WC. The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia. 1993;7:9–18. doi: 10.1002/glia.440070105. [DOI] [PubMed] [Google Scholar]
- 31.Schwarz M, Nordt T, Bode C, Peter K. The GP IIb/IIIa inhibitor abciximab (c7E3) inhibits the binding of various ligands to the leukocyte integrin Mac-1 (CD11b/CD18, αMβ2) Thromb Res. 2002;107:121–128. doi: 10.1016/s0049-3848(02)00207-4. [DOI] [PubMed] [Google Scholar]
- 32.Pahan K, Sheikh FG, Namboodiri AMS, Singh I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia and macrophages. J Clin Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manne V, Ricca CS, Brown JG, Tuomari AV, Yan N, Patel D, Schmidt R, Lynch MJ, Ciosek CP, Jr, Carboni JM, et al. Ras farnesylation as a target for novel anti-tumor agent: potent and selective farnesyl diphosphate analog inhibitors of farnesyl transferase. Drug Dev Res. 1995;34:121–137. [Google Scholar]
- 34.Pahan K, Liu X, McKinney MJ, Wood C, Sheikh FG, Raymond JR. Expression of a dominant-negative mutant of p21ras inhibits induction of nitric oxide synthase and activation of NF-κB and in primary astrocytes. J Neurochem. 2000;74:2288–2295. doi: 10.1046/j.1471-4159.2000.0742288.x. [DOI] [PubMed] [Google Scholar]
- 35.Brahmachari S, Pahan K. Sodium benzoate, a food additive and a metabolite of cinnamon, modifies T cells at multiple steps and suppresses experimental allergic encephalomyelitis. J Immunol. 2007;179:275–283. doi: 10.4049/jimmunol.179.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao H, Urban JF, Anderson RA. Cinnamon polyphenol extract affects immune responses by regulating anti- and proinflammatory and glucose transporter gene expression in mouse macrophages. J Nutr. 2008;138:833–840. doi: 10.1093/jn/138.5.833. [DOI] [PubMed] [Google Scholar]
- 37.Xie QW, Kashiwabara Y, Nathan C. J Biol Chem. 1994;269:4705–4708. [PubMed] [Google Scholar]
- 38.Kleinert H, Wallerath T, Fritz G, Ihrig-Biedert I, Rodriguez-Pascual F, Geller DA, Forstermann U. Cytokine induction of NO synthase II in human DLD-1 cells: roles of the JAK-STAT, AP-1 and NF-κB-signaling pathways. Br J Pharmacol. 1998;125:193–201. doi: 10.1038/sj.bjp.0702039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hancock JF, Cadwaller K, Marshall CJ. Methylation and proteolysis are essential for efficient membrane binding of prenylated p21k-ras(B) EMBO J. 1991;10:641–646. doi: 10.1002/j.1460-2075.1991.tb07992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kikuchi A, Williams LT. The post-translational modification of ras p21 is important for raf-1 activation. J Biol Chem. 1994;269:20054–20059. [PubMed] [Google Scholar]
- 41.Boguski MS, McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- 42.Garnovskaya MN, van Biesen T, Hawe B, Casanas Ramos S, Lefkowitz RJ, Raymond JR. Ras-dependent activation of fibroblast mitogen-activated protein kinase by 5-HT1A receptor via a G protein βγ-subunit-initiated pathway. Biochemistry. 1996;35:13716–13722. doi: 10.1021/bi961764n. [DOI] [PubMed] [Google Scholar]
- 43.Qiu R, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature. 1995;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]