

Abstract
Background & Aims
Mutations in the DNA mismatch repair (MMR) gene MSH2 cause Lynch Syndromes I & II, and sporadic colorectal cancers (CRCs). Msh2null mice predominantly develop lymphoma and do not accurately recapitulate the CRC phenotype.
Methods
We generated and examined mice with a conditional Msh2 disruption (Msh2LoxP), permitting tissue-specific gene inactivation. ECMsh2LoxP/LoxP mice carried an EIIa-Cre transgene and VCMsh2LoxP/LoxP mice carried a Villin-Cre transgene. We combined the VCMsh2LoxP allele with either Msh2Δ7null (VCMsh2LoxP/null) or Msh2G674D mutations (VCMsh2LoxP/G674D) to create allelic phase mutants. These mice were given cisplatin, or 5-fluorouracil/leucovorin and oxaliplatin (FOLFOX) and their tumors were measured by magnetic resonance imaging.
Results
Embryonic fibroblasts from ECMsh2LoxP/LoxP mice do not express MSH2 and are MMR-deficient. Reverse transcription, PCR, and immunohistochemistry from VCMsh2LoxP/LoxP mice demonstrated specific loss of Msh2 mRNA and protein from epithelial cells of the intestinal tract. Microsatellite instability (MSI) was observed in all VCMsh2 strains and limited to the intestinal mucosa. Resulting adenomas and adenocarcinomas had somatic Apc truncation mutations. VCMsh2LoxP/LoxP mice did not develop lymphoma. Comparison of allelic phase tumors revealed significant differences in multiplicity and size. When treated with cisplatin or FOLFOX, tumor size was reduced in VCMsh2LoxP/G674D but not VCMsh2LoxP/null tumors. The apoptotic response to FOLFOX was partially sustained in the intestinal mucosa of VCMsh2LoxP/G674D animals.
Conclusion
Msh2LoxP/LoxP mice in combination with appropriate Cre recombinase transgenes have excellent potential for preclinical modeling of Lynch Syndrome, MMR deficient tumors of other tissue types, and use in drug development.
Keywords: MMR, Msh2, mouse, tumorigenesis, chemotherapy
Background & Aims
Approximately 150,000 new cases of colorectal cancer (CRC) are diagnosed per year in the United States. More than 50,000 patients die from it yearly. Generally classified into familial predisposition syndromes and sporadic cancers, several critical genes involved in both have been identified. Familial adenomatous polyposis (FAP) is caused by mutations in the APC gene. Lynch Syndromes I & II are caused by mutations in the Mismatch Repair (MMR) genes. MSH2 was found to be one of the most commonly mutated MMR genes 1-3. Msh2 is necessary for repair of base-base as well as insertional deletion mismatches and its absence results in increased mutation levels. Mice lacking MSH2 have a tumor disposition phenotype.
To develop mouse models for Lynch Syndrome three Msh2null knockout mouse lines have been generated, two by targeted disruption of Msh2 exon 12 4, 5 and one by disruption of exon 7 6. Homozygous mutant mice of all three Msh2null knockouts are MMR-deficient and display a highly increased predisposition to lymphoma. A proportion of older animals also develop intestinal neoplasms that are associated with Apc inactivation 7. However, the predominance of the lymphoma phenotype has limited the use of these animals as preclinical models.
We report a novel conditional knockout mouse model for the tissue-specific inactivation of Msh2 (Msh2LoxP). In this model, MMR can be inactivated by Cre-LoxP-mediated inactivation of Msh2 in different tissues by the expression of various Cre-recombinase transgenes. To constitutively inactivate MMR similar to Msh2null knockout mice we mated Msh2LoxP mice with EIIa-Cre recombinase transgenic mice (termed ECMsh2LoxP). To specifically inactivate MMR in the intestinal mucosa we combined the Msh2LoxP allele with the Villin-Cre transgene (VCMsh2LoxP). ECMsh2LoxP/LoxP mice display complete MMR deficiency and have a cancer phenotype similar to Msh2null knockout mice. In contrast, in VCMsh2LoxP/LoxP mice MMR deficiency is limited to the intestinal epithelium and the mice develop exclusively intestinal neoplasms. These data show that Msh2LoxP mice in combination with specific Cre recombinase transgenes allow the tissue-specific inactivation of MMR and the development of suitable mouse models for Lynch Syndrome.
We also demonstrate that it is possible to study allelic effects of different Msh2 mutations on intestinal tumorigenesis in VCMsh2LoxP mice by combining the Msh2LoxP allele with either a Lynch Syndrome related missense mutation (Msh2G674D) or an Msh2Δ7null mutation (Msh2null). Tumors from these allelic phase mutants have also been tested for their response to two chemotherapeutic regimens, cisplatin and FOLFOX, and their growth recorded by Magnetic Resonance Imaging (MRI). Although some tumors in VCMsh2LoxP/null mice were responsive to the two drugs, the majority were resistant to both chemotherapies. In contrast, almost all VCMsh2LoxP/G674D tumors were found to generally respond well to cisplatin and FOLFOX. The differences in responsiveness of tumors correlated with the absence of a significant DNA damage response in VCMsh2LoxP/null mice, and partial retention of this response in VCMsh2LoxP/G674D mice.
Methods
Generation of Msh2LoxP Mice
The targeting vector for the Msh2LoxP mouse was made by recombinogenic methods 8, 9. An Msh2 genomic fragment spanning exon 10 through intron 18 was PCR amplified from BAC clone 183K13 (RP-22 library) and subcloned. A LoxP site was introduced into Msh2 intron 12-13 followed by introduction of a LoxP-FRT-PGKneor-FRT selection cassette into Msh2 intron 11-12. The vector was linearized and transfected into WW6 ES cells 10. Male chimeric mice were generated and bred to C57BL/6J females to generate Msh2neoLoxP-FRT neo/+ F1 offspring. The PGKneor cassette was subsequently deleted in vivo by crossing Msh2neoLoxP-FRT neo/+ heterozygotes to FLP- deleter mice 11. Offspring from these crosses were genotyped by PCR, Southern Blot and sequence analyses (data not shown), to confirm the integrity of the Msh2LoxP allele. All procedures were in accordance with Institutional Animal Care and Use Committee Protocols.
Generation of Msh2LoxP Cre Recombinase Transgenic Mouse Lines
Msh2LoxP/+ mice were crossed with EIIa-Cre recombinase transgenic animals to generate ECMsh2LoxP/+ 12. Heterozygotes were intercrossed to generate ECMsh2LoxP/LoxP mice.
Msh2Lox/p/+ mice were mated with B6;D2-Tg(Vil-Cre) to create VCMsh2LoxP/+ mice, then intercrossed to create VCMsh2LoxP/LoxP mice 13. VCMsh2LoxP/+ mice were also mated to animals carrying the Msh2Δ7 knockout allele (termed Msh2null) 6 and the Msh2G674D knock-in allele. Offspring with one floxed Msh2 allele and one mutant allele, VCMsh2LoxP/null or VCMsh2LoxP/G674D respectively were obtained.
PCR Genotyping Msh2LoxP mice
Tail DNA was isolated using the DNAeasy kit (Qiagen, Valencia, CA) from ten day old mice. PCR primers used for genotyping were 184F (TACTGATGCGGGTTGAAGG), 184R (AACCAGAGCCTCAACTAGC), and 165R (GGCAAACTCCTCAAATCACG). Cycling conditions will be given upon request.
MMR analysis in ECMsh2LoxP/LoxP MEF cell lines
Cytosolic extracts were prepared from MEF cells as described in Thomas et al. 14. A heteroduplex G-G substrate was prepared and DNA repair reactions were performed as previously described 14, 15.
Western Blotting and Immunohistochemistry
MEF cell extracts were separated by SDS-PAGE and blotted onto Polyvinylidene fluoride membranes and probed with rabbit anti mouse MSH2 polyclonal antibody (MSH2 N-20:sc494, Santa Cruz, CA), an Msh6 monoclonal antibody (BD Biosciences, Franklin, USA) or a GADPH monoclonal antibody (Ambion for GADPH, Austin, TX).
For immunohistochemical analysis (IHC), monoclonal antibodies directed against Msh2 (N-20:sc494, Santa Cruz) and Apc (GTX15270, GeneTex, Inc.) were used.
Generation of Kaplan-Meier Survival Plots
Prism 3.0 software (Graphpad) was used to calculate percent survival of animals.
Histopathologic analysis
Mice were euthanized and the GI tract was removed, opened longitudinally, and fixed in 10% neutral-buffered formalin or Bouins solution. The number of tumors and their location was recorded under a dissecting microscope. For histological analysis, tumors were embedded in paraffin, sectioned to 5 μm and stained with hematoxylin and eosin. Relative tumor size was measured using a Vernier Caliper with fine adjustment.
MSI Analysis
Genomic DNAs from tail, spleen and flat mucosa were subjected to PCR amplification using a dilution assay as previously described 16. We screened for instability using a dinucleotide repeat marker, D17Mit123 17. In intestinal tumors two dinulceotide markers were studied in undiluted DNA, D7Mit91 and TG27. PCR products were separated on denaturing 6% polyacrylamide gels and autoradiographed for analysis.
Apc Truncation Mutations
The analysis of truncation mutations to the Apc gene was performed as described earlier 18.
Drug Treatment
VCMsh2LoxP/null and VCMsh2LoxP/G674D animals were divided into 3 groups. The first group received an intraperitoneal injection (i.p.) with cisplatin (20 mg/kg body weight), five times every second day with a total dose of 100 mg/kg body weight. The second group received an i.p. injection with FOLFOX (5-fluorouracil/leucovorin, five sequential days (20 mg and 10 mg/kg body weight respectively); Oxaliplatin (1 mg/kg body weight) was injected i.p. once. The third group was a control group injected i.p. with Phosphate Buffered Saline (PBS) five times every second day. All drugs were purchased from Sigma-Aldrich Corp. (St. Louis, MO).
Magnetic Resonance Imaging (MRI) & 3-D Reconstitution of Images
Tumor sizes were measured before and after treatment by in vivo MRI imaging. Animals were positioned in a 40 mm “bird cage” MRI coil in a 9.4 T GE Omega vertical bore imaging system. A 51.2 mm field of view with a 256 × 256 pixel image matrix was used. Image slices were 1 mm thick with no gap between slices. Series of routine spinecho images along all three planes were acquired to reconstruct 3D images of the mouse. Typical parameters for GI tract studies were used with echo time of 30 ms, repetition time of 400 ms, and signal averaging 4 scans. Typically 24 images were acquired along each plane. Image data was analyzed using MATLAB based software. 3D reconstructions of the digestive tract were created using Amira 3.1 software.
DNA Damage Response
TUNEL assays were conducted on intestinal and spleen tissue from VCMsh2LoxP/null and VCMsh2LoxP/G674D mice, (Promega DeadEnd (TM) Fluorometric TUNEL System).
Results
The Generation of Msh2LoxP Mice, ECMsh2LoxP/LoxP Cell Lines, and MMR Measurement
We have generated a conditional knockout mouse line for Msh2 by flanking exon 12, encoding a portion of the essential ATPase domain of MSH2, with LoxP sites (Fig. 1A, B, & C). To constitutively delete exon 12, Msh2LoxP/+ were mated with EIIa-Cre recombinase mice. In these mice, the Msh2LoxP allele was transmitted in a normal Mendelian ratio, and ECMsh2LoxP/LoxP mice obtained upon heterozygote intercrosses also developed normally. Western blot analysis of ECMsh2LoxP/LoxP MEFs revealed that deletion of exon 12 resulted in the complete loss of MSH2 protein, and also reduced levels of MSH6 protein (Fig. 2D).
Figure 1.

Strategy for the production of Msh2 conditional knockout mutant mice, and PCR genotyping of offspring. (A) Gene targeting strategy. (B) PCR genotyping strategy. 1) pGEM markers, 2) Wildtype mouse, 3) Msh2LoxP/LoxP, exon 12 deleted, 4) Msh2LoxP/+. (C) Where Msh2 exon 12 has been deleted, primer pair 184F/184R amplifies no product, whereas primer pair 184F/165R amplifies a 340 bp product. Wildtype animals without the conditional allele amplify a 210 bp PCR product using primers 184F/184R.
Figure 2.

Molecular characterization of VCMsh2LoxP/LoxP and ECMsh2LoxP/LoxP mice. (A) Specific rearrangement of the Msh2 gene in the small and large intestines of VCMsh2LoxP/LoxP animals, is absent in the kidney heart and spleen. Primer pairs A/C amplify a 340 bp product when exon 12 is deleted (intestine) and a 983 bp product when exon 12 is intact (kidney heart and spleen). (B) Measurement of MMR in ECMsh2LoxP/LoxP MEFs. Cell lines EM2-1 and EM2-2 are MMR deficient and compare to the Exo1-/- MMR deficient cell line, EM2 and Exo1-/- cell lines complement each other. (C) MSI in VCMsh2LoxP/LoxP mice (for MSI in C57B1/6J mice see 19). (D) Western blot analysis of EM2 MEF cell lines. (Δ12), MSH2 is absent from an EM2 cell line and shows reduced amounts of its complex partner, Msh6. WT, wildtype mouse embryonic fibroblast cell line. (E) IHC on VCMsh2LoxP/LoxP small intestine (top) compared to wildtype intestine (bottom). (F) RT-PCR using Msh2 primers with GI tissues 1) wildtype, 2) VCMsh2LoxP/LoxP.
To investigate the effect of Msh2 exon 12 deletion on MMR, cell extracts from two MEF lines (ECMsh2LoxP/LoxP lines 1 and 2) were screened for their ability to repair a G-G mismatch from a 3′ nick. Neither extract repaired this single base mismatch. Repair deficiencies in both ECMsh2LoxP/LoxP cell extracts were similar to repair deficiencies in an Exo1Δ6/Δ6 (exon 6 deletion) extract, previously found defective for repair of single base mismatches 19. The MMR defect in both ECMsh2LoxP/LoxP extracts was complemented by adding Exo1Δ6/Δ6 extract, demonstrating that it was caused by the specific loss of MSH2. After complementation of the ECMsh2LoxP/LoxP extracts by the Exo1Δ6/Δ6 extract, a reduction in the blue/white plaque color ratios was found, demonstrating that repair activity in the complemented extracts is directed to the nicked strand, indicating strand-specific MMR (Fig.2B).
Inactivation of Msh2 by Villin-Cre in VCMsh2LoxP Mice
To inactivate Msh2 in the intestinal mucosa, Msh2LoxP/+ animals were intercrossed with Villin-Cre transgenic mice 13. Msh2 inactivation was confirmed by PCR (Fig. 2A) and RT-PCR (Fig. 2F). Genomic DNA isolated from kidney, heart and spleen displayed either minimal or no exon 12 deletion.
MSH2 expression was examined by immunohistochemistry (Fig. 2E). It was detectable in the cytoplasm and nucleus of epithelial cells in wild-type mice, but was absent in the intestinal mucosa of VCMsh2LoxP/LoxP mice.
To study the effect of exon 12 deletion on MMR in vivo we analyzed MSI in different tissues of VCMsh2LoxP/LoxP mice (Fig. 2C). At the D17Mit123 locus only 5 of 77 (6.5%) alleles were unstable in genomic tail DNA of VCMsh2LoxP/LoxP mice and 6 of 82 (7.3%) were unstable in spleen genomic DNA which is comparable to the levels of MSI in wild-type mice for this marker19. However, epithelial cells in the intestinal mucosa of VCMsh2LoxP/LoxP mice displayed a significant increase in MSI with 20 of 96 unstable alleles (20.8%) indicating that the MMR-deficiency is highly restricted to the intestinal epithelium in VCMsh2LoxP/LoxP mice.
Survival and Tumor Development in ECMsh2LoxP/LoxP and VCMsh2LoxP/LoxP Mice
Both ECMsh2LoxP/LoxP and VCMsh2LoxP/LoxP mice were viable and fertile. However, a significant difference in survival was observed between the two mouse lines (p < 0.0001) (Fig. 3B). The median survival for VCMsh2LoxP/LoxP animals was 12 months and all animals expired at 17 months. In contrast, ECMsh2LoxP/LoxP mice had a reduced median survival of 6 months and all animals died by 11 months similar to Msh2null/null knockout mice 4-6.
Figure 3.

VCMsh2LoxP/LoxP mice have decreased median survival. (A) An intestinal adenocarcinoma from a VCMsh2LoxP/LoxP mouse. (B) Survival curves: wildtype, (green); VCMsh2LoxP/LoxP, (red); ECMsh2LoxP/LoxP, (blue, solid); Msh2null (black, dashed). (C) An intestinal adenocarcinoma from a VCMsh2LoxP/LoxP mouse stained with rabbit anti mouse E cadherin -24E10 (Cell Signaling Technology, Danvers, MA). The black square on the lower right shows tumor invasion of the muscularis and is enlarged in panel D). The smaller square on the lower right is enlarged in panel E) and shows multiple mitotic figures indicated by black arrows. F) IHC using antibody to Apc on a VCMsh2LoxP/LoxP intestinal polyp (top), specific staining of Apc in macrophages of the intestinal epithelium (lower left), wildtype control (lower right).
The analysis of several moribund ECMsh2LoxP/LoxP mice at 6 to 7 months of age revealed a high incidence of lymphoma (67% of animals) and a lower incidence of small intestinal tumors (33% of animals) indicating that the cancer predisposition phenotype in ECMsh2LoxP/LoxP mice is comparable to that of Msh2null/null knockout mice 4, 5 (Table 1).
Table 1. VCMsh2 LoxP/LoxP Tumor Incidence and Multiplicity.
| Genotype | N | Age (mo) Mean ± SD |
Sex (M:F) |
Overall | Intestinal tumors n (%) | Intestinal Tumor Multiplicity (Mean ± SEM) |
|
|---|---|---|---|---|---|---|---|
| Lymphoma | Intestine | ||||||
| VCMsh2 LoxP/LoxP | 18 | 9.0 ± 1.1 | 1:0.8 | 16 (89)b | 0 (0) | 16 (89)b | 1.61 ±0.30c |
| VCMsh2 LoxP/+ | 15 | 11.3 ± 1.8 | 1:1.5 | 0 (0)b | 0 (0) | 0 (0)b | 0.00±0.00c |
| Msh2+/+ a | 17 | 14.2 ± 2.7 | 1:04 | l (6)b | 0 (0) | l (6)b | 0.06±0.00c |
N, number of mice studied; n, number of mice with intestinal tumors.
Msh2 +/+ mice, VC positive and VC negative combined.
Tumor incidence compared by Fisher exact probability: P<0.0001.
Tumor multiplicity compared by Mann-Whitney or binomial calculation: P<0.0001.
In contrast, the cancer phenotype differed significantly between VCMsh2LoxP/LoxP mice and Msh2null/null knockout mice. A cohort of VCMsh2LoxP/LoxP mice (n=18) was sacrificed at 9.0±1.1 months of age and analyzed for the presence of tumors. 89% of VCMsh2LoxP/LoxP mice developed tumors in the small intestine at this age with a tumor multiplicity of 1.6±0.3 (Table 1). Histopathological analysis of 14 tumors showed that 50% were at the adenomas and the other 50% were highly invasive adenocarcinomas (Fig. 3A, C, D, & E). None of these animals developed lymphoma and only one in 150 VCMsh2LoxP/LoxP mice developed lymphoma after 12 months of life. VCMsh2LoxP/+ mice (n=15) at 11.3±1.8 months of age developed no tumors and 1 of 17 (6%) wild-type mice at 14.2±2.7 months of age developed an intestinal tumor (Table 1). We also analyzed the MSI status in the genomic DNA of the intestinal tumors in VCMsh2LoxP/LoxP mice at two dinucleotide markers. We found that at the D7Mit123 marker 7 of 12 (58%) tumors tested were unstable, while at the TG marker 16 11 of 12 (92%) tumors were unstable. All tumors showed MSI of at least one of the two markers tested.
Apc is Mutated in VCMsh2LoxP/LoxP GI Tumors
Immunohistochemistry of intestinal tumors from VCMsh2LoxP/LoxP mice were negative for Apc antibody staining (Fig. 3F).
Truncation mutations in VCMsh2LoxP/LoxP GI tumors were found by IVTT analysis indicating somatic mutations to both Apc alleles may have occurred for tumor initiation (Table 2). Twenty out of thirty-one (64.5%) VCM2LoxP/LoxP tumors screened were positive. Ten fragments were cloned and sequenced. Mutation types included mono and di-nucleotide deletions and C to T transitions leading to the formation of stop codons. These mutations are consistent with the types of mutations caused by loss of MMR function 18.
Table 2. Truncation mutations to Apc in tumors from VCMsh2 LoxP/LoxP.
 |
Allelic Phasing of Intestinal Tumor Development in VCMsh2LoxP Mice
The Msh2LoxP allele provided an opportunity to examine the impact of different Msh2 mutations on intestinal tumor development. We generated VCMsh2LoxP mice that carried the Msh2LoxP allele in combination with either an Msh2null (12) allele or the Msh2G674D allele previously found in an HNPCC patient 20 (Supplemental data). Deletion of exon 12 in the Msh2LoxP allele by Villin-Cre expression leads to intestinal epithelial cells containing two Msh2null alleles (that express no MSH2) in VCMsh2LoxP/null mice or one Msh2null and one Msh2G674D allele (that only express MSH2G674D) in VCMsh2LoxP/G674D mice. The analysis of tumorigenesis in these mice revealed remarkable differences (Fig. 4 A & B). Mice of both lines displayed a strong predisposition to intestinal cancers. However, the tumor onset was delayed in VCMsh2LoxP/G674D mice as compared to VCMsh2LoxP/null mice. In VCMsh2LoxP/null mice intestinal tumors were found beginning at 6 months of age and 50% of animals carried intestinal tumors at 10 months of age. In contrast, intestinal tumors were first detected in VCMsh2LoxP/G674D mice at 10 months of age and 50% of animals carried tumors at 13 months of age (p<0.0001). In addition, the tumor number and size differed significantly between the VCMsh2LoxP/null and VCMsh2LoxP/G674D allelic phase mutants. While VCMsh2LoxP/null mice developed 1.40±0.11 intestinal tumors, the VCMsh2LoxP/G674D mice developed 3.43±0.42 tumors (p<0.0001). There was also a significant difference in tumor size. Tumors in VCMsh2LoxP/null mice had an average diameter of 6.48±0.58 mm, while the tumors in VCMsh2LoxP/G674D mice were significantly smaller with a size of 3.25±0.49 mm (P<0.003). In VCMsh2LoxP/null mice 18% (3/17) of the intestinal tumors were adenomas and 82% (14/17) of tumors had progressed to adenocarcinoma. In VCMsh2LoxP/G674D mice 37% (7/19) of intestinal tumors were adenomas, while 63% (12/19) were adenocarcinomas.
Figure 4.

Tumor size measurements by caliper and tumor number count, from the intestines of allelic phase mutants. (A) The average number of tumors between VCMsh2LoxP/null and VCMsh2LoxP/G674D mice varied significantly, as did the size (B). (C) Intestinal tumors for chemotherapy are visualized and measured by MRI. A six month old VCMsh2LoxP mouse testing positive for occult blood, was subjected to MRI, successfully revealing one tumor. Two weeks later a second tumor was detected during a second MRI at the original location.
VCMsh2LoxP/null and VCMsh2LoxP/G674D Response to Cisplatin and FOLFOX
Allelic phase mutant mice were subjected to chemotherapy with either cisplatin or FOLFOX, and tumor response was analyzed by MRI (Fig. 4C). Tumors in both VCMsh2LoxP/null and VC Msh2LoxP/G674D mice continued growing after receiving Phosphate Buffered Saline (PBS) injections (i.p.). The two groups, however, differed in response to cisplatin or FOLFOX treatment. VCMsh2LoxP/null tumors were predominantly resistant to both chemotherapies, and only a small number of tumors showed growth retardation. In contrast almost all tumors in VCMsh2LoxP/G674D mice responded well to either cisplatin or FOLFOX treatment regimen (Fig. 5).
Figure 5.

Intestinal tumors from VCMsh2LoxP/null and VCMsh2LoxP/G674D mice, respond to chemotherapy. Tumors are measured in terms of relativity of MRI measurements. The tumor size at day 0 is 1, and the relative tumor growth or retardation is scored on the basis of percentage. Red lines indicate growth, green lines indicate retardation. The number of tumors for each treatment is as follows. VCMsh2LoxP/null: PBS-7, cisplatin-8, FOLFOX-11. For VCMsh2LoxP/G674D: PBS-7, cisplatin-9, FOLFOX-11.
To determine the molecular basis underlying the differences in intestinal tumorigenesis and drug response in VCMsh2LoxP/null and VCMsh2LoxP/G674D mice we performed in vivo analyses of MMR and the DNA damage response in the intestinal epithelium. While genomic DNA in the intestinal mucosa of wild type mice was previously shown to contain 4% unstable alleles at the dinucleotide marker D7Mit9121, genomic DNA in the mucosa of VCMsh2LoxP/null and VCMsh2LoxP/G674D mice displayed a significant increase in MSI at this marker that was comparable between both mouse lines (26/81 (32%) unstable alleles in VCMsh2LoxP/null mice; 30/89 (31%) unstable alleles in VCMsh2LoxP/G674D mice).
To determine whether VCMsh2LoxP/G674D mice sustain the MMR-dependent DNA damage response function post chemotherapy, the number of apoptotic cells in intestinal crypts was analyzed 18 hours after FOLFOX injection (Fig. 6). These measurements showed a significant reduction in the apoptotic response in VCMsh2LoxP/null mice as compared to Msh2LoxP/LoxP (wild-type) mice (p<0.001). Although the apoptotic response in the mucosal epithelium of VCMsh2LoxP/G674D mice was somewhat reduced in comparison to wild type mice, it was significantly higher than in VCMsh2LoxP/null mice (p=0.0118). As expected the apoptotic response appeared normal in the spleen in all three groups of mice (p=0.2929). Overall, these studies indicate that although the intestinal epithelial cells in VCMsh2LoxP/G674D mice display only a partial DNA damage response, it is still sufficient to significantly impact tumor growth after FOLFOX treatment.
Figure 6.
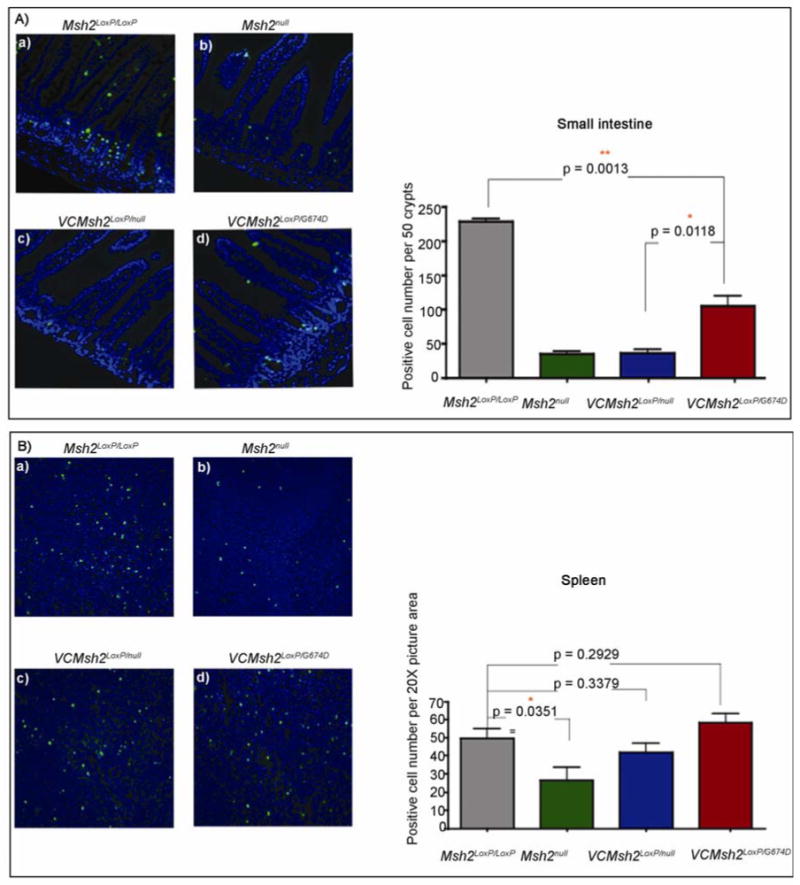
Apoptosis measured in intestinal mucosa of VCMsh2LoxP/null and VCMsh2LoxP/G674D mice, after 18 hours of FOLFOX treatment. (A) TUNEL staining (green fluorescence) in the intestinal mucosa of a. Msh2LoxP/LoxP, b. Msh2null, c. VCMsh2LoxP/null, d. VCMsh2LoxP/G674D mice with graphic analysis of the number of positive apoptotic cells per fifty crypts. (B) Apoptosis measured in the spleen of VCMsh2LoxP/null and VCMsh2LoxP/G674D mice, after 18 hours of FOLFOX treatment. A. TUNEL staining (green fluorescence) in the intestinal mucosa of a. Msh2LoxP/LoxP, b. Msh2null, c. VCMsh2LoxP/null, d. VCMsh2LoxP/G674D mice with graphic analysis of the number of positive apoptotic cells per fifty crypts.
Conclusions
We have generated a conditional Msh2LoxP knockout allele in mice that permits better modeling of the intestinal cancer features of Lynch Syndrome than previous models. Although these earlier mouse models are prone to a variety of cancers, they predominantly develop aggressive lymphomas early in life which has limited their use as preclinical models 4-6. Using the Msh2LoxP allele in combination with the Villin-Cre transgene we show that the lymphoma phenotype of constitutional Msh2 knockout mice can be avoided and tumorigenesis restricted to the intestinal tract.
VCMsh2LoxP/LoxP intestinal tissues display a high degree of MSI and tumors carry Apc mutations. A few documented Lynch Syndrome families exist with biallelic mutations in MMR genes 22-24. These patients display severe reduction in life span and hematological malignancies, a phenotype that resembles neurofibromatosis, as well as CRCs. Since MSI can also be found in many solid tumors 25, the Msh2LoxP allele should be helpful in modeling these types of MSI positive cancers. We have found, for instance, that the Msh2LoxP allele in combination with the Cre transgene under control of the human epithelial keratin 14 promoter (K14-cre) permits the generation of skin tumors (data not shown). The identification of tissue specific pathways altered in these tumors may be useful in identifying tissue specific cancer genes.
Similar to other mouse models of colorectal cancer, VCMsh2LoxP/LoxP mice develop tumors predominantly in the small intestine, in contrast to Lynch Syndrome patients who typically develop tumors of the colon. Inactivation of an ApcLoxP allele by Villin-cre also resulted mainly in small intestinal tumors. However, the tumor location could be shifted to the large intestine by colonic infection of ApcLoxP/LoxP mice with Adenoviral-Cre (personal communication, Dr. Kenneth Hung). We are currently infecting the intestines of Msh2LoxP/LoxP mice with Adenoviral-Cre to determine if this method may also be useful in permitting development of CRC without prior mutation to the Apc gene. Genetic approaches may also be useful in these experiments such as the introduction of retinoblastoma deficiency into Msh2LoxP/LoxP mice, followed by infection with Adenoviral-Cre. While Rb deficiency by itself does not cause CRC 26, it has been demonstrated to expand the compartment that tumors occur in to include the cecum and distal colon in Apc deficient mice 27. We also combined the Msh2LoxP allele with the Cdx2P-NLS-Cre transgene28 and observed a shift of tumors to the large intestine. However, like Msh2null mice, these animals also developed lymphomas (Kyeryoung Lee and Winfried Edelmann, unpublished observations).
Despite the tumor location in the small intestine, VCMsh2LoxP mice have been highly useful in determining the effect of different Msh2 alleles on intestinal tumorigenesis. In this study, we introduced either an Msh2null knockout allele that causes complete loss of MSH2, or the Msh2G674D allele representing a Lynch Syndrome missense mutation into VCMsh2LoxP mice. Interestingly, VCMsh2LoxP/G674D mice developed a higher number of intestinal tumors. However, the tumors developed at a later age and their size was reduced compared to VCMsh2LoxP/null mice. It is possible that the accelerated tumorigenesis and larger tumor size in VCMsh2LoxP/null mice led to intestinal obstruction and early death, preventing higher tumor multiplicity later in life. These data indicate that Msh2 missense mutations can have distinct effects on intestinal tumorigenesis.
The Msh2G674D mutation is located within the MSH2 ATPase domain at the identical amino acid residue as the Msh2G674A mutation previously studied 29. Neither mutation affect the DNA damage response function, and in vitro analysis using Msh2G674D/G674D MEFs showed a normal apoptotic response to FOLFOX exposure as did MEFs from wildtype mice. Msh2-/- MEFs displayed increased resistance (data not shown). Our in vivo analysis further showed that the Msh2G674D mutation caused complete MMR deficiency in the intestinal mucosa of VCMsh2LoxP/G674D mice but left the MMR-dependent DNA damage response function partially intact. These results support the idea that the DNA damage response function of MMR is important for the suppression of the early steps of intestinal tumorigenesis 30.
Retention of the DNA damage response function in VCMsh2G674D mice also had a significant impact on tumor response to chemotherapy. MRI based in vivo measurements of tumor growth showed that the intestinal tumors in VCMsh2LoxP/G674D animals were sensitive to both cisplatin and FOLFOX, while tumors from VCMsh2LoxP/null mice were generally resistant to both drugs. While cisplatin is commonly used to treat solid tumors 31 and sometimes GI tumors in combination treatment regimen (esophagus, stomach, anus, and sometimes small intestine), it is not routinely used to treat CRC. It is a platinum based drug that introduces GpG cross-links into DNA. These lesions are recognized by MutSα with high specificity; and MSH2-deficient cells display low level resistance to the drug 32, 33. In vitro, MutSα binds to cross links, recruits MutLα, and interacts with the helicase domain of FANC-J (Fanconi Anemia protein) for cross link repair. The response of VCMsh2LoxP/null and VCMsh2LoxP/G674D tumors to cisplatin is consistent with the notion that complete loss of MSH2 diminishes cross link repair, and that less severe mutations might still be able to recruit repair factors for the removal of such lesions.
FOLFOX (5-fluoroacil (5-FU), leucovorin, and oxaliplatin) is commonly used for the treatment of late stage colorectal cancers. 5-FU-based adjuvant chemotherapy benefits patients with MSS tumors, but not patients with MSI positive tumors 34. Our finding that VCMsh2LoxP/null tumors are resistant to FOLFOX is consistent with these studies. There is significant in vitro evidence that MMR defective colon cancer cell lines are resistant to killing by 5-FU, and that re-expression of MMR genes in these cell lines reverses the phenotype 35, 36. 5-FU acts to disrupt RNA synthesis and inactivate thymidylate synthase. It is also incorporated into DNA, and therefore could be processed by MMR. 5-FU substrates are recognized by MutSα, suggesting CRC chemosensitivity may in part be due to MMR protein recognition, leading to cell death either by futile repair cycles or signaling of cell cycle arrest and apoptosis 37. VCMsh2LoxP/G674D tumor sensitivity to FOLFOX implies that MutSα complexes interact with 5-FU adducts and mediate a cytotoxic response. It also raises the possibility that some tumors in human patients carrying MSH2 missense mutations that cause MSI without affecting DNA damage response will remain responsive to fluorouracil-based adjuvant chemotherapy.
Supplementary Material
Acknowledgments
We would like to thank Larissa Georgeon Richard for E-cadherin IHC on VCMsh2LoxP/LoxP adenocarcinomas and Dr. Kenneth Hung for discussions of work with Adenoviral-Cre on Apc conditional knockout mice.
Grant support: This work was supported by NIH Grants ES11040 (R.K.) CA084301 (R.K.) and CA76329 (W.E.) and CA93484 (W.E.). Center grant CA13330 (Albert Einstein College of Medicine), Project Z01 ES065089, Division of Intramural Research of the NIH, NIEHS (T.A.K.).
Footnotes
Disclosures: N/A
Transcript profiling: N/A
Writing assistance: N/A
Study Concept and design: MHK, KL, RK, WE
Acquisition of data: MHK, KL, AN, AC, HH, AR, KY, KF, ML, RB, LJ
Analysis and interpretation of data: MHK, KL, AC, KY, RB, LJ, TK, RK, WE
Drafting of the manuscript: MHK, RK, WE
Critical revision of manuscript, important intellectual content: MHK, RB, KY, RK, WE
Statistical analysis: KL, KF, KY, WE
Obtained funding: ML, TK, RK, WE
Technical or material support: MHK, KL, AN, AC, HH, AR, KY, KF, ML, RB, LJ
Study supervisor: MHK, KL, RK, WE
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–38. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 2.Lothe RA, Peltomaki P, Meling GI, et al. Genomic instability in colorectal cancer: relationship to clinicopathological variables and family history. Cancer Res. 1993;53:5849–52. [PubMed] [Google Scholar]
- 3.Marsischky GT, Filosi N, Kane MF, et al. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996;10:407–20. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- 4.de Wind N, Dekker M, Berns A, et al. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–30. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 5.Reitmair AH, Schmits R, Ewel A, et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet. 1995;11:64–70. doi: 10.1038/ng0995-64. [DOI] [PubMed] [Google Scholar]
- 6.Smits R, Hofland N, Edelmann W, et al. Somatic Apc mutations are selected upon their capacity to inactivate the beta-catenin downregulating activity. Genes Chromosomes Cancer. 2000;29:229–39. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1033>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 7.Reitmair AH, Cai JC, Bjerknes M, et al. MSH2 deficiency contributes to accelerated APC-mediated intestinal tumorigenesis. Cancer Res. 1996;56:2922–6. [PubMed] [Google Scholar]
- 8.Lee EC, Yu D, Martinez de Velasco J, et al. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73:56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- 9.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–84. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ioffe E, Liu Y, Bhaumik M, et al. WW6: an embryonic stem cell line with an inert genetic marker that can be traced in chimeras. Proc Natl Acad Sci U S A. 1995;92:7357–61. doi: 10.1073/pnas.92.16.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez CI, Buchholz F, Galloway J, et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–40. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- 12.Lakso M, Pichel JG, Gorman JR, et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860–5. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.el Marjou F, Janssen KP, Chang BH, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–93. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 14.Thomas DC, Umar A, Kunkel TA. Measurement of heteroduplex repair in human cell extracts. Methods:A companion to Methods in Enzymology. 1995;7:187–197. [Google Scholar]
- 15.Thomas DC, Roberts JD, Kunkel TA. Heteroduplex repair in extracts of human HeLa cells. J Biol Chem. 1991;266:3744–51. [PubMed] [Google Scholar]
- 16.Kabbarah O, Mallon MA, Pfeifer JD, et al. A panel of repeat markers for detection of microsatellite instability in murine tumors. Mol Carcinog. 2003;38:155–9. doi: 10.1002/mc.10157. [DOI] [PubMed] [Google Scholar]
- 17.Dietrich WF, Miller JC, Steen RG, et al. A genetic map of the mouse with 4,006 simple sequence length polymorphisms. Nat Genet. 1994;7:220–45. doi: 10.1038/ng0694supp-220. [DOI] [PubMed] [Google Scholar]
- 18.Kuraguchi M, Edelmann W, Yang K, et al. Tumor-associated Apc mutations in Mlh1-/- Apc1638N mice reveal a mutational signature of Mlh1 deficiency. Oncogene. 2000;19:5755–63. doi: 10.1038/sj.onc.1203962. [DOI] [PubMed] [Google Scholar]
- 19.Wei K, Clark AB, Wong E, et al. Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 2003;17:603–14. doi: 10.1101/gad.1060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andreutti-Zaugg C, Scott RJ, Iggo R. Inhibition of nonsense-mediated messenger RNA decay in clinical samples facilitates detection of human MSH2 mutations with an in vivo fusion protein assay and conventional techniques. Cancer Res. 1997;57:3288–93. [PubMed] [Google Scholar]
- 21.Avdievich E, Reiss C, Scherer SJ, et al. Distinct effects of the recurrent Mlh1G67R mutation on MMR functions, cancer, and meiosis. Proc Natl Acad Sci U S A. 2008;105:4247–52. doi: 10.1073/pnas.0800276105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menko FH, Kaspers GL, Meijer GA, et al. A homozygous MSH6 mutation in a child with cafe-au-lait spots, oligodendroglioma and rectal cancer. Fam Cancer. 2004;3:123–7. doi: 10.1023/B:FAME.0000039893.19289.18. [DOI] [PubMed] [Google Scholar]
- 23.Raevaara TE, Gerdes AM, Lonnqvist KE, et al. HNPCC mutation MLH1 P648S makes the functional protein unstable, and homozygosity predisposes to mild neurofibromatosis type 1. Genes Chromosomes Cancer. 2004;40:261–5. doi: 10.1002/gcc.20040. [DOI] [PubMed] [Google Scholar]
- 24.Rey JM, Noruzinia M, Brouillet JP, et al. Six novel heterozygous MLH1, MSH2, and MSH6 and one homozygous MLH1 germline mutations in hereditary nonpolyposis colorectal cancer. Cancer Genet Cytogenet. 2004;155:149–51. doi: 10.1016/j.cancergencyto.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 25.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57. [PubMed] [Google Scholar]
- 26.Kucherlapati MH, Nguyen AA, Bronson RT, et al. Inactivation of conditional Rb by Villin-Cre leads to aggressive tumors outside the gastrointestinal tract. Cancer Res. 2006;66:3576–83. doi: 10.1158/0008-5472.CAN-05-2699. [DOI] [PubMed] [Google Scholar]
- 27.Kucherlapati MH, Yang K, Fan K, et al. Loss of Rb1 in the gastrointestinal tract of Apc1638N mice promotes tumors of the cecum and proximal colon. Proc Natl Acad Sci U S A. 2008;105:15493–8. doi: 10.1073/pnas.0802933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hinoi T, Akyol A, Theisen BK, et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67:9721–30. doi: 10.1158/0008-5472.CAN-07-2735. [DOI] [PubMed] [Google Scholar]
- 29.Lin DP, Wang Y, Scherer SJ, et al. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004;64:517–22. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- 30.Fishel R. The selection for mismatch repair defects in hereditary nonpolyposis colorectal cancer: revising the mutator hypothesis. Cancer Res. 2001;61:7369–74. [PubMed] [Google Scholar]
- 31.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 32.Yamada M, O'Regan E, Brown R, et al. Selective recognition of a cisplatin-DNA adduct by human mismatch repair proteins. Nucleic Acids Res. 1997;25:491–6. doi: 10.1093/nar/25.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signalling. DNA Repair (Amst) 2004;3:1091–101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–57. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carethers JM, Chauhan DP, Fink D, et al. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117:123–31. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyers M, Wagner MW, Hwang HS, et al. Role of the hMLH1 DNA mismatch repair protein in fluoropyrimidine-mediated cell death and cell cycle responses. Cancer Res. 2001;61:5193–201. [PubMed] [Google Scholar]
- 37.Tajima A, Hess MT, Cabrera BL, et al. The mismatch repair complex hMutS alpha recognizes 5-fluorouracil-modified DNA: implications for chemosensitivity and resistance. Gastroenterology. 2004;127:1678–84. doi: 10.1053/j.gastro.2004.10.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.